
Changes in Gene Expression Profiles in Developing B Cells of Murine Bone Marrow
Abstract
Gene expression profiles of five consecutive stages of mouse B cell development were generated with high-density oligonucleotide arrays from as few as 2 × 104 ex vivo isolated and flow-cytometrically purified cells. Between 2.8% and 6.8% of all genes change on differentiation from one cellular stage to the next by at least twofold. The entire pathway involves differential expression of 10.7% of all genes. Previously known expression patterns of 15 genes (like surrogate light chain, RAG-1/2, MHC class II, mel-14 antigen) are confirmed. The gene expression patterns of the proliferating pre-BI and large pre-BII cells on the one hand, and the resting immature and mature B cells on the other hand, are most similar to each other. Small pre-BII cells display a pattern that is transitional between these two groups. Most of the genes expressed in early precursors are involved in general processes, like protein folding or cell cycle regulation, whereas more mature precursors express genes involved in more specific molecular programs (cell surface receptors, secreted factors, and adhesion molecules, among others). Between 19 and 139 genes share a given expression pattern. Combining knowledge about gene function and expression pattern allows identification of novel candidate genes potentially involved in self-maintenance of pre-BI cells, allelic exclusion and pre-B cell receptor signaling in large pre BII cells, cell-cycle arrest of small pre-BII cells, propensity toward apoptosis or anergization in immature B cells, propensity toward cell division and activation in mature B cells, and stage-specific interactions with stromal cells in the bone marrow.
[The sequence data described in this paper have been submitted to the Gene Expression Omnibus (GEO) at the National Center for Biotechnology Information (NCBI) under accession number GSE13. Online supplementary material available at www.genome.org.]
Mouse B-lymphocytes develop from progenitors and precursors in bone marrow in a sequence that can be ordered by changing status of their immunoglobulin gene rearrangements (Tonegawa 1983; ten Boekel et al. 1995). Cell cycle status and the differential surface expression of c-kit, CD25, IgM, and IgD on B220+ cells distinguish five consecutive stages of development (Rolink et al. 1994). Therefore, fluorescence-activated cell sorting (FACS) can be used to purify five cell populations that follow each other in progressive differentiation: (1) DH-JH rearranged, c-kit+CD25-cycling pre-BI cells; (2) VHDHJH-rearranged, c-kit-CD25+-cycling large pre-BII cells; (3) VHDHJH- and VLJL-rearranged c-kit-CD25+ resting small pre-BII cells; (4) sIgM+ resting immature; and (5) sIgM+IgD+ resting mature B-cells (Melchers and Rolink 1999).
In this developmental sequence of cells, pre-BI cells also express surrogate light chain encoded by VpreB and λ5 genes (Karasuyama et al. 1994), and the rearrangement machinery encoded by the RAG-1, RAG-2 (Grawunder et al. 1995), and TdT (Melchers and Rolink 1999) genes. As soon as one allele has been rearranged productively, somatic recombination is stopped, preventing additional rearrangements on the second allele. This process is termed allelic exclusion (Melchers and Rolink 1999). The μ heavy chain derived from a productively VHDHJH-rearranged IgH chain locus has to pair with the surrogate light chain to form a pre-BCR on the surface of large pre-BII cell (ten Boekel et al. 1997). Expression of the surrogate light chain and of the rearrangement machinery is then turned off (Grawunder et al. 1995). The pre-BCR induces two to five divisions of large pre-BII cells (Rolink et al. 2000). As the pre-BCR is diluted by these divisions, the cells come to rest as small pre-BII cells, the expression of the rearrangement machinery is turned on again and VL segments are rearranged to JL segments on the κL and λL chain gene loci. As soon as an L chain has paired with the pre-existing μ heavy chain, IgM can be deposited on the surface to give the cell the status of an immature B cell. Autoantigens select the emerging repertoire of immature B cells negatively to delete high-affinity autoreactive cells and may also select positively to differentiate low-affinity autoreactive cells into the B1 cell compartment (Nemazee et al. 2000). Immature B cells keep the rearrangement machinery up-regulated to allow for secondary rearrangements at the IgL chain gene loci with which they can change, thereby edit the specificity of autoreactive cells (Yu et al. 1999). During this differentiation program in the bone marrow, B-cell precursors interact with different cell types (osteoblasts, osteoclasts, reticular stromal cells, dendritic cells, and others) in a possibly stage-specific manner (Melchers and Rolink 1999). Immature B cells, finally, leave the bone marrow for the spleen where they mature to sIgM+ sIgD+ B cells.
These cellular stages of B-cell differentiation have been described in detail. Very little, however, is known about the molecular mechanisms controlling the various functions of B-lineage cells in this development. The self-renewing capacity of pre-BI cells with the ability to long-term proliferate on stromal cells with IL-7, the signal transduction from the pre-BCR for proliferative expansion and possibly for allelic exclusion (both taking place in large pre-BII cells), cell-cycle exit in small pre-BII cells, and susceptibility to autoantigen signaling in immature B cells all are processes lacking a precise molecular description to date. Some of the genes involved in such mechanisms can be expected, however, to show specific changes in expression during B-cell development. Such genes appear to be good candidate genes that could control this development.
High-density oligonucleotide arrays allow the analysis of a large number of genes expressed as mRNA in parallel (Lockhart et al. 1996). Using higher-level data analysis algorithms, expression profiles of different cellular stages can be compared, and genes potentially involved in biological processes of interest can be identified by their expression patterns (Eisen et al. 1998; Tamayo et al. 1999). Previously, a large number of cells has been required to perform such analysis, essentially excluding the use of this technology for cells that are too infrequent in vivo, as in this case for the development of B cells in mouse bone marrow. Here, we describe the modification of an in-vitro-transcription-based RNA amplification procedure (Eberwine et al. 1992; Luo et al. 1999) that now allows us to perform array-based expression profiling from as few as 5 × 104 cells or less, lowering the amount of necessary input material by a factor of 1000. This has enabled us, for the first time, to analyze gene expression patterns of five consecutive stages of a mammalian developmental program using ex vivo isolated cells.
RESULTS
To study gene expression profiles in murine B-cell development, total cellular RNA was extracted from 5 × 104 to 1.5 × 105 cells of each of five consecutive B-lymphocyte lineage subpopulations (Fig. 1). mRNA was amplified by two subsequent cycles of cDNA synthesis and in vitro transcription (Eberwine et al. 1992; Luo et al. 1999). The RNA samples were hybridized to high-density oligonucleotide arrays interrogating 13,026 transcripts [5231 known genes and 7795 expressed sequence tags (ESTs)]. Five independent replicate experiments were performed, and differential expression was assessed on the basis of statistical algorithms (see Methods). The raw, non-normalized gene expression values for each of the individual experiments are presented in supplementary Table 1 (available as an on-line supplement athttp://www.genome.org).
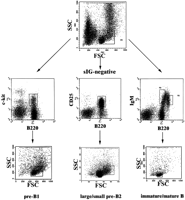
Flow cytometric purification of mouse B-cell precursors in three steps. Step 1 (top row): Enrichment of lymphoid cells. Shown is a forward–sideward scatter plot of total femoral bone marrow cells. The boxed area indicates the lymphoid gate. Second step (middlerow): Enrichment of c-kit+B220+ pre-BI cells, CD25 +B220+ pre-BII cells, and sIgM+B220+ immature/mature B cells. Surface marker staining of three aliquots of lymphoid-gated bone marrow cells is shown. Pre-BI and immature/mature B cells were sorted as indicated by the gates displayed as boxes in the top and middle rows. Third step (bottom row): Separation of pre-BII cells according to cell size. Shown are forward–sideward scatter plots of cells gated as in the middle row. Pre-BII cells were separated according to cell size into the large pre-BII cells (right box) and small pre-BII cells (left box). Note that pre-BI cells consist of a small and a large subpopulation, which were not separated. Immature and mature B cells were sorted according to the gates shown in themiddle row, right panel. As both populations consist of homogeneous small cells, only one back-gated forward–sideward scatter plot is shown in the right panel of thebottom row.
We first determined how many genes change on differentiation from one cellular stage to the next. Between pre-BI and large pre-BII, large pre-BII and small pre-BII, small pre-BII and immature B cells, and immature and mature B-cells, a total of 1028, 1604, 996, and 873 genes have a t-test P value of 0.02 or less. Of these genes, 488, 885, 462, and 362 genes change at least twofold; 133, 321, 154, and 121 genes change at least fivefold; and 39, 110, 58, and 43 genes change at least 10-fold, respectively. Therefore, between 2.8% and 6.8% of all genes present on the arrays change on differentiation from one stage to the next. A complete list of genes changing at least fivefold together with the expression level values and associatedt-test P values is available as supplementary Table 2 and supplementary Table 3 contains the information for all probe sets represented on the arrays (tables available as an on-line supplements at http://www.genome.org).
Because only ∼45% of the genes reaching statistical significance at the 98% confidence level also fulfill the fold change and absolute difference criteria, the type I error rate (number of genes falsely detected to be differentially expressed) can be expected to be well below the 2% that the confidence criterion alone allows.
Employing analysis of variance (ANOVA), 1734 genes reach statistical significance at the 98% level. Of these, 1406 genes were identified as differentially expressed by a factor of two or more during mouse B-cell development. The entire information about the ANOVA analysis is presented in supplementary Table 4 (available as an on-line supplement at http://www.genome.org). A hierarchical cluster analysis (Eisen et al. 1998) performed on this subset of genes is shown in Figure2. Except for large pre-BII cells, all B-cell precursor stages up-regulate a specific set of genes. This is indicated on the left side of the plot. Genes up-regulated in large pre-BII cells are already expressed in pre-BI cells. Those genes are often cell-cycle related (cyclins, brca-1, ki-67, PCNA), involved in DNA replication (Topoisomerase II, helicase, DNA polymerase, ligase, and primase), or cytoskeleton components (tubulins)(see the bottom two clusters in Fig. 2, right). This is consistent with the fact that pre-BI and large pre-BII cells are cycling while the subsequent three cellular stages are resting. In Figure 2, functionally related genes tend to show similar expression patterns and come to lie in close proximity to each other. Examples include parts of immunoglobulin genes (expression in immature and mature B cells, top cluster in Fig. 2, right) and MHC class II sequences (expressed specifically in mature B cells, second from top in Fig. 2, right).
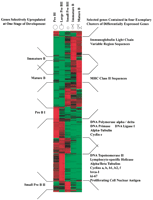
Hierarchical cluster analysis of 1406 differentially expressed genes as detected by ANOVA with 98% confidence (measured by Kruskal-Wallis statistics) with a change of at least twofold and a difference of at least 100 average difference units (for details see Methods). Genes are organized in rows, whereas the five columns represent the five developmental stages from the most immature, pre-BI compartment on theleft to the most mature compartment, mature B cells on theright. The expression level of every gene for every developmental stage has been normalized. Green denotes a normalized expression level below, black near to, and red above the mean. Developmental stage-specific clusters are indicated on theleft and clusters of selected functional classes of genes are indicated on the right side of the plot.
Comparing the gene expression patterns across the five cellular stages, cycling pre-BI and large pre-BII on the one hand, and resting immature and mature B cells on the other hand, appear to be most similar to each other (Fig. 2). Small pre-BII cells appear to be intermediate between these two groups, expressing some genes expressed in pre-BI and large pre-BII cells (bottom half in Fig. 2) and some others expressed in immature and mature B cells (top half in Fig. 2). Therefore, a major change in gene expression takes place after the large pre-BII cell stage, probably because proliferation ceases and new differentiation programs are initiated in resting cells.
Figure 3 shows a tree diagram representing correlations between gene expression profiles from individual replicate experiments of the five developmental stages examined. All of the stagewise replicate experiments form tight clusters, indicating small intra-stage variability as compared with variability between stages. The tree reflects the relationships between stage-specific gene expression profiles as mentioned above, as the pre-BI and large pre-BII cells on the one side and the immature and mature B cells on the other cluster together on distal branches of the tree, respectively.
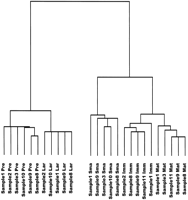
Tree diagram displaying correlations between gene expression patterns from individual replicate experiments of the five stages of B-cell differentiation, based on the set of 1406 differentially expressed genes. Shorter branches indicate more similar gene expression profiles. (Sample) Number of the sample (consisting of cells pooled from four mice) from which the cell populations have been purified by FACS, numbered consecutively from the beginning of the study; (Pre) pre-BI cells; (Lar) large pre BII cells; (Sma) small pre-BII cells; (Imm) immature B cells; (Mat) mature B cells.
Next, the 1406 differentially expressed transcripts were grouped according to their expression pattern by self-organizing maps (Tamayo et al. 1999). The entire data set is available as supplementary Table 5 (available as an on-line supplement at http://www.genome.org). Twenty different, though sometimes closely related, patterns could be identified (Fig. 4). We compared these patterns with published data for genes whose expression patterns during mouse B-cell development have been determined earlier (Grawunder et al. 1995; Melchers and Rolink 1999). Fifteen such known genes [VpreB, λ5, terminal deoxynucleotidyltransferase, c-kit (all c1), RAG-1/2 (c10 and c8 in Fig. 4, respectively), μ heavy chain constant region (c11), CD40 (c14), b29 (Igβ), mb-1 (Igα), κ light chain constant region (all c15), CD19 (c16), Igδ constant region, MHC class II sequences, mel-14 antigen (all c17)] were examined, and all the shown array-based expression patterns are in concordance with the published data (Table 1).
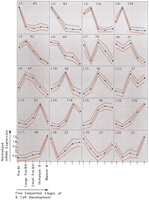
Gene expression patterns identified by self-organizing maps. The cluster numbers are indicated on the top left of each cluster diagram, and the number of genes plus ESTs belonging to every cluster is indicated on the top right. Expression levels are shown on y-axis and developmental stages on x-axis. Dots indicate developmental stages, with the most immature, pre-BI cells on the left and the most mature, that is, mature B cells, on the right. Expression level of each gene was normalized to have mean = 0 and SD = 1 across developmental stages. Blue and red lines indicate expression level means and standard deviations, respectively.
Array-Based Gene Expression Values of Genes with Pre-Established Expression Patterns During Mouse B Cell Development
A total of 656 of the differentially expressed transcripts were genes; the remainders were ESTs. After review of published data, a definitive or putative function could be assigned to 625 of the 656 known genes. The 31 remaining genes were often cloned as growth factor inducible genes, and no follow-up studies have been performed.
A synopsis of functional classifications of genes correlated with their cluster membership is shown in Table 2. It shows that early precursors express genes involved in V(D)J-recombination, protein folding and degradation, general and redox metabolism, DNA replication, chromatin and cytoskeleton structure, RNA processing, cell cycle regulation, and molecular transport (Table 2; clusters 1–4 in Fig. 4). In contrast, more mature precursors express genes involved in intercellular communication (cell surface receptors and secreted factors), transcriptional regulation, immunoglobulins, complement components, cytoskeleton modification, intercellular contact and adhesion, MHC components, and apoptosis (Table 2; clusters 14–17 in Fig. 4). Therefore, the majority of genes up-regulated by more immature precursors are involved in general metabolic processes, whereas more mature precursors up-regulate genes involved in specific molecular programs.
Numbers of Genes with Known Functions Differentially Expressed in the Five Developmental Stages of B Cells with the Same Patterns of Changes
The correlation of gene expression pattern with biochemical function of the respective protein product also allowed us to identify candidate genes that could be involved in functional properties of B-cells above.
Pre-BI cells, for example, have self-renewing capacity and are capable of long-term proliferation in the presence of bone marrow stromal cells and IL-7 or IL-3 (Winkler et al. 1995). These capacities are lost on differentiation to the pre-BII cell stage. Therefore, genes involved in these processes are probably specifically expressed in pre-BI cells, that is, in clusters 1 or 2. As can be seen in Table 2, these clusters contain a high proportion of genes involved in general metabolism and DNA replication, but four signaling molecules (p116RIP, T-cell specific clone U2, TIAM-1, and SOCS-2), five cell-surface receptors (thromboxane A2 receptor, FcɛRI, endoglin, common beta chain of IL-3/IL-5/GM-CSF receptors, and c-kit) and three genes involved in cell–cell contact or adhesion (gp-70, alpha-catenin, and lectin L14) (Table3A) are candidate genes potentially involved in the pre-BI specific processes.
Genes Potentially Involved in Stage-Specific Properties of B Cells Table 3A. Genes Up-Regulated in Pre-BI Cells
Genes Potentially Involved in Specific Properties of Large Pre BII Cells (pre-BCR Mediated Proliferative Burst and Allelic Exclusion)
Genes Potentially Responsible for Cell Cycle Arrest after the Large Pre BII Cell Stage
Genes Distinguishing Immature from Mature Cells
Expression of Cell Adhesion Molecules During Mouse B Cell Development
In large pre-BII cells, V(D)J-recombination on the heavy chain locus is inhibited after one allele has been productively rearranged, a process termed allelic exclusion (Melchers and Rolink 1999). After surface expression of a pre-BCR consisting of the surrogate light chain components λ5 and VpreB and the newly rearranged heavy chains, these cells undergo a proliferative burst (Rolink et al. 2000). Allelic exclusion and proliferative burst are likely to require specific signals. Genes involved in these processes are likely to be found in clusters 3, 4, 5, or 9. Cluster 3 contains many genes involved in DNA replication, and cluster 4 contains many genes involved in cell-cycle regulation. In addition, however, these clusters also contain many genes encoding signaling molecules, with a total of 23 signaling components specifically up-regulated in large pre-BII cells (Table 3B and supplementary Table 5, available as an on-line supplement athttp://www.genome.org). A number of these (CDC25, ect2, STK-1, sak-a, nek-2, and ran) are known to transmit signals that are important for cell-cycle progression (Coutavas et al. 1994; Fode et al. 1994;Wickramasinghe et al. 1995; Niwa et al. 1996; Tanaka et al. 1997;Tatsumoto et al. 1999). Two of the signaling components up-regulated in pre-BII cells (calmodulin and the calmodulin synthesis cDNA) are involved in calcium signaling (Chin and Means 2000), whereas the rest of the signaling molecules (PP2Abalpha3, citron, fug1, pLK, ayk1, pMELK, MKP-1, and STAM) remain rather poorly characterized. Therefore, signal transduction from the preBCR might involve some of these genes.
For allelic exclusion, one possible part of the total mechanism is structural reorganization of chromatin that would make the immunoglobulin heavy chain loci inaccessible for further recombination. In fact, the clusters 3, 4, and 5 contain seven, six, and three genes, respectively, involved in chromatin structure (Table 3B). Five of these genes code for histones, two more for chromosomal structural proteins; it is likely that the expression pattern of these genes simply reflects the cycling status of large pre-BII cells. Four genes, however, are involved in transcriptional regulation via structural changes: the modifier-1 protein is involved in heterochromatin formation (Singh et al. 1991), whereas SRG-3 is associated with the SWI–SNF complex to modify locus accessibility (Jeon et al. 1997). The retinoblastoma-binding proteins RbAp46 and RbAp48 are involved in histone acetylation-dependent transcriptional regulation (Zhang et al. 2000).
Small pre-BII cells are characterized by cell-cycle arrest and induction of immunoglobulin light chain rearrangements. For cell-cycle arrest, at least two mechanisms can be deduced from the chip data (Table 3C). The major cell-cycle regulator, PCNA, is highly expressed in pre-BI and large pre-BII cells, decreasing in more mature cell stages (cluster 3). MyD118, known to inhibit cell cycle progression by heterodimerizing with PCNA (Vairapandi et al. 2000), shows a peak of expression in small pre-BII cells (cluster 11). A second PCNA-interacting partner with similar function (EAT/MCL-1) (Fujise et al. 2000) is up-regulated from immature B cells onward (cluster 14). Together, these two genes might be involved in sustained cell-cycle arrest after exit from the large pre-BII cell stage. A second cell cycle regulatory pathway involves chk-1 (clusters 2, 3), phosphorylating cdc25 (clusters 4, 5) in response to DNA damage signals (Sanchez et al. 1997), possibly generated during aberrant heavy chain V(D)J-recombination. This renders cdc25 inactive and generates a binding site for 14-3-3 (cluster 7), maintaining cdc25 in an inactive state and effectively blocking cell-cycle progression (Fu et al. 2000). The clusters 10 and 11, however, containing genes with peak expression in small pre-BII cells, contain four more cell-cycle repressing genes (gadd45, gas7, c-jun, and tis-21).
The pool of immature B cells is exposed to selective pressures by autoantigens. These undergoing negative selection are likely to be on their way toward apoptotic cell death or anergization, and are prevented of entering the cell cycle on BCR crosslinking. Other immature B cells are expected to be in the process of positive selection, possibly also initiated by autoantigens. BCR crosslinking should signal cell survival and proliferation in these cells (Melchers and Rolink 1999). Genes involved here are likely to be found most prominently in cluster 13, but also in cluster 14 (Table 3D). Cluster 13 contains CD72, a negative regulator for B-cell responsiveness (Parnes and Pan 2000); c-fes, a non-receptor tyrosine kinase involved in negative regulation of cytokine-induced activation in macrophages (Hackenmiller et al. 2000); and PAC-1, a tyrosine phosphatase that specifically dephosphorylates the MAP-kinases erk-1 and erk-2 (Gerondakis et al. 1994). Cluster 14 includes genes for the ITIM-bearing inhibitory receptor PIRB1 (Maeda et al. 1998); the inhibitory BCR coreceptor FcγRIIB; calcineurin, a Ca++-dependent protein phosphatase (Rusnak and Mertz 2000); and two genes involved in inhibition of cAMP-derived signals, the cyclic nucleotide phosphodiesterase PDE7A2 and the inhibitor of cAMP-dependent protein kinase. Susceptibility to apoptotic stimuli, most prominently differing between immature and mature B cells, appears to be guided by the differential expression of survival-mediating bcl-2 homologs. Therefore, although bcl-x peaks in small Pre-BII cells, PS-2 and the inhibitor of apoptosis-1 are expressed from small pre-BII cells onward (cluster15), and EAT/MCL-1 is up-regulated in immature B cells with some sustained expression in mature B cells (cluster 14), bcl-2 beta is specifically upregulated in mature B cells (cluster 17). This reconfirms that bcl-2 might be the major regulator of the difference in response to apoptotic stimuli between immature and mature B cells (Lang et al. 1997).
In contrast to immature B cells, mature B cells proliferate in response to stimulation by foreign antigens (Melchers and Rolink 1999). Although this might in part be attributable to the down-regulation of the cell-cycle inhibitors (mentioned above), as well as to the expression of bcl-2 (preventing apoptotic cell death), genes contained in cluster 17 provide additional candidate molecules potentially involved in keeping mature B cells in an activation-prone status (Table 3D). Cluster 17 contains 10 signaling molecules, for example annexin V (a collagen-regulated calcium channel; von der Mark and Mollenhauer 1997), type 1 ryanodine receptor (an intracellular calcium release channel;Giannini et al. 1995), the mitogen activated protein kinase kinase kinase 8 (activating NFκB; Lin et al. 1999), TANK (another NFκB activator downstream of TRAF in both CD40 and TNF-receptor II pathways;Cheng and Baltimore 1996), the cytokine-induced tyrosine kinase jak-2, and the cAMP synthesizing adenylyl-cyclase type VII. Also in this cluster are the surface receptors for Interferon-gamma, the chemokine receptors CCR7 and CXCR5, and the T-cell activating protein TAP. The latter has been shown previously to be decreased in anergic B cells (Glynne et al. 2000). Mature B cells also express immunosuppressive receptors, however, like the IL-10 receptor (cluster 17), and immature B cells also express activating receptors like PIRA6 (cluster 14). Therefore, these signal-transducing molecules might be wired differently in these functionally heterogeneous B cell populations to signal either for activation or for silencing.
During differentiation in the bone marrow, B-cell precursors interact with various cell types in a stage-specific manner. Immature B cells are ready to leave the bone marrow to migrate to the spleen, subsequently maturing into mature, recirculating B cells (Melchers and Rolink 1999). These processes are likely to be guided by the differential expression of adhesion molecules. Table 3E lists genes expressed differentially throughout the five stages of B cell development that are involved in cell-to-cell contact and adhesion. Cluster 8 contains adhesion molecules that might be involved in retaining the B-cell precursors in the bone marrow (DGCR-6 and P-selectin glycoprotein ligand 1). The different intramedullary cellular interactions of B-cell precursors might be governed by molecules contained in clusters 1 and 2 (pre-BI-specific: Ig-related glycoprotein-70, alpha-catenin, and L14 lectin), cluster 5 (up-regulated in large pre-BII cells: fertilization antigen-1), and cluster 10 (up-regulated in small pre-BII cells: reelin). Cluster 12 and 15 contain adhesion molecules (galectin-3, Stra1/ ephrin B1, ICAM-2) expressed in small pre-BII, immature and mature B cells that are about to, or have already left the bone marrow. In addition, some other adhesion molecules appear to be expressed more specifically in only one of these three cellular stages. In immature B cells, these are CD37 and ICAM-1 (clusters 13 and 14). Mature B cells specifically upregulate C33/R2/IA4, CD22, LFA-1, VCAM-1, novel stromal cell protein, P-selectin/MEL-14 antigen, and integrin beta-7 subunit (clusters 16 and 17). However, there are also a number of cell adhesion molecules that are expressed both in pre-BI as well as in immature and mature B cells (clusters 18 and 20: Ly-6C.2, clusterin, Cd73, sialic acid O-acetylesterase). It remains to be determined which adhesive properties should be preserved for different microenvironments of B lymphocyte development.
DISCUSSION
This study describes gene expression patterns of five consecutive cellular stages in mouse B-cell development. A large number of differentially expressed genes is identified, and a combination of previous knowledge about B-cell development, gene expression pattern analysis, and functional annotation allows us to identify genes potentially involved in specific processes taking place in mouse B-cell precursors. These candidate genes can be subjected to more detailed functional analysis by heterologous expression, by functional inactivation employing antisense technology, or by gene inactivation via targeted deletion.
Two factors underline the validity of the gene expression data set. First, genes whose expression patterns have been identified earlier are detected in our analysis in a manner concordant with previous results (Rolink et al. 1994; Grawunder et al. 1995; Melchers and Rolink 1999). Second, analysis of co-expressed genes groups together genes with similar function in a manner consistent with what is known about these cells; genes involved in cell-cycle regulation, for example, although never formally investigated in the setting of B-cell development, are highly expressed in the cycling pre-BI and large pre-BII cells only. A number of aspects, however, require special attention.
First, before the entire murine genome sequence is available, this type of analysis remains incomplete. Nevertheless, we consider the selection of genes analyzed here to be representative for the entire genome, since in every group of genes with similar expression pattern, the ratio of known genes to (randomly chosen) ESTs is ∼1:2, reflecting the proportion on the arrays.
Second, this analysis is based on differentially expressed genes that differ by a factor of two or more during B-cell differentiation. A number of examples demonstrate a functional significance of mono- versus bi-allelic expression (Nutt et al. 1999). Different genes, however, might require different changes in expression level to become functionally relevant. Therefore, not all of the genes analyzed here might actually contribute to the phenotypes of mouse B-cell precursors.
Third, many genes appear to exert different functions in different cellular contexts. Therefore, biochemical function of a gene product has to be seen in the context of the respective interaction partners present in a particular cell type. This context can signify different cellular effects of a given gene product in different cells. The functional associations presented here can only be a first evidence that these genes function in our given cellular setting, but not how exactly they exert their function.
Fourth, whereas the cells examined here have been ordered as consecutive stages of an ordered differentiation pathway, there might be additional, small, intermediate, transitional populations not distinguishable by current surface marker analysis. Moreover, a given cell might have distinct ways to react to external stimuli. Immature B cells, for example, are known to react to BCR crosslinking in a ligand-dependent manner. Although high-avidity antigens lead to negative selection, low- and medium-avidity antigens might lead to anergization or even positive selection (Hayakawa et al. 1999; Nemazee et al. 2000). The pool of immature B cells examined here might actually consist of cells before and at different points during the selection process.
A surprisingly high number of genes, that is, almost 11% of all, are differentially expressed in this single mammalian developmental pathway. This indicates that there must be substantial redundancy in usage of genes between different cellular differentiation programs. Additional gene expression studies of other lymphoid and nonlymphoid cellular differentiation programs shall identify genes that are used in many developmental pathways and distinguish them from those that are truly pathway-specific. These unique genes appear to be the best candidates for genetic modification in germline or somatic cells, leading to interpretable functional changes.
METHODS
Purification of B-Cell Precursors
Total femoral bone marrow cells of five- to six-week-old C57/BL6 mice (n = 4 per experiment) were aliquotted into three parts, stained, and sorted as shown in Figure 1. A total of 50,000 (pre-BI, large pre-BII) or 150,000 (small pre-BII, immature and mature B cells) cells were sorted directly into TRIzol RNA isolation reagent (Life Technologies) at 50,000 cells/500 μL TRIzol. A cell purity of ≥98% was routinely achieved.
RNA Amplification
Total cellular RNA was precipitated with 5 μg Escherichia coli tRNA as coprecipitant, and pellets were resuspended in 9 μL DEPC-H2O. In vitro transcription-based RNA amplification was performed essentially as described earlier (Eberwine et al. 1992; Luo et al. 1999). Briefly, double-stranded cDNA was synthesized using the T7-tagged oligo-dT primer (5′-GGCCAGTGAATTGTAATACGACTCACTATAGGG AGGCGG(T)24-3′), and, after cleanup with Microcon YM-50 columns, a first in vitro transcription reaction was done using the Promega RiboMax T7 kit. RNA was cleaned up again, and a random-hexamer primed first strand cDNA synthesis was performed. The resulting RNA–DNA hybrid was treated with RNAseH, heat denatured, and the T7-oligo-d(T)-Primer was annealed. Second-strand cDNA synthesis was the performed with a mix of Klenow and T4 DNA polymerases. After cleanup, a second in vitro transcription was performed, incorporating biotinylated CTP and UTP, respectively. RNA was purified with RNeasy columns (QIAGEN).
Hybridization of Affymetrix GeneChip Probe arrays
Affymetrix Mu11k GeneChips, interrogating 13,104 genes and ESTs on two different arrays, were processed according to the manufacturer's recommendations. Approximately 20 pairs of oligonucleotides (probe pairs) represent every gene or EST in a probe set, with one sequence being complementary to the target sequence and the other having a 1-bp mismatch in a central position. This serves as an internal control for hybridization specificity. The relative abundance is reported as the averaged difference of the fluorescence intensity values between the perfectly matched and the mismatched oligonucleotides, resulting in the so-called “average difference” value (Lockhart et al. 1996; Wodicka et al. 1997).
Data Analysis
To enable statistically valid data analysis, five independent replicate experiments were performed. Analysis of this data set consisted of three steps. First, normalization of the individual arrays; second, identification of differentially expressed genes between adjacent developmental stages using pair-wise comparisons; and third, identification of sets of differentially expressed genes sharing similar changes across the sequence of five B-cell developmental stages.
First, individual GeneChip expression results were normalized as follows. All average difference values of every chip were summed up, and the mean of these sums across all chips of the same layout was calculated. The ratio of the actual average difference sum for any given chip and the mean of all average difference sums across all chips with the same layout served as a correction factor for this chip, with which all the average difference values were multiplied. This process was performed separately for the two different array types used (Mu11k sub A and sub B arrays, respectively). It results in normalized average difference values, so that all chips of the same array layout have the same overall average difference.
Second, differentially expressed genes between developmentally adjacent stages were identified as follows. In this analysis, average difference values beyond the values of most other individual hybridization values seen in five replicate experiments were removed using the Nalimov outlier test at 95% confidence level (Kaiser and Gottschalk 1972). Means and standard deviations of the remaining values were calculated for every gene separately for every developmental stage. Probability of differential expression was calculated with unpaired t-test statistics. Genes were considered differentially expressed if thet-test results in a confidence level of at least 98%, and if they changed at least twofold with a difference in mean average difference value of at least 100.
Third, sets of genes were identified that shared similar changes in their expression patterns during the five B-cell developmental stages. For this multicomparison analysis, ANOVA with H-(Kruskal-Wallis) test statistics was performed to identify genes with a statistically significant differential expression in at least one of all the possible pairwise comparisons. Threshold criteria were set as above. Hierarchical Cluster Analysis was performed according to the method ofEisen et al. (1998) after row normalization to a mean of 0 and a sum of value squares of 1. Genes with similar expression patterns were identified with self-organizing maps (Tamayo et al. 1999).
As an additional quality criterion, the homogeneity of the hybridization signal across the 20 gene-specific probe pairs was assessed for all genes mentioned in this paper as follows. First, individual probe pairs are scored as “positive” (PM intensity > MM intensity) or “negative” (PM intensity < MM intensity) according to standard Affymetrix procedures. Next, the “purity” value was calculated as follows.
Purity = [(number of pairs in the probe set) − least (number of positive pairs, number of negative pairs) × 2)]/number of pairs in the probe set.
This value indicates how well probe pairs agree in their signal. In our analyses, the purity value had to be ≥0.7 in at least four replicate experiments of the population with the highest expression level to be considered significant.
Acknowledgments
We are indebted to Mark Dressing, Annette Pickert, Hubertus Kohler, and Tracy Hayden for cell sorter operation. The Basel Institute for Immunology was founded and supported by F. Hoffmann-LaRoche Ltd.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Present address: Max-von-Pettenhofer-Institut, Pettenhofer Str. 9A, 80336 Munich, Germany.
-
↵4 Corresponding authors.
-
E-MAIL r_hoffmann{at}m3401.mpk.med.uni-muenchen.de; FAX 49-89-5160-5202.
-
F. Melchers FAX 41-61-605-1300.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.201501.
-
- Received June 19, 2001.
- Accepted October 16, 2001.
- Cold Spring Harbor Laboratory Press








