
Abstract
Homologs of the eukaryotic DNA-end-binding protein Ku were identified in several bacterial and one archeal genome using iterative database searches with sequence profiles. Identification of prokaryotic Ku homologs allowed the dissection of the Ku protein sequences into three distinct domains, the Ku core that is conserved in eukaryotes and prokaryotes, a derived von Willebrand A domain that is fused to the amino terminus of the core in eukaryotic Ku proteins, and the newly recognized helix–extension–helix (HEH) domain that is fused to the carboxyl terminus of the core in eukaryotes and in one of the Ku homologs from the Actinomycete Streptomyces coelicolor. The version of the HEH domain present in eukaryotic Ku proteins represents the previously described DNA-binding domain called SAP. The Ku homolog from S. coelicolor contains a distinct version of the HEH domain that belongs to a previously unnoticed family of nucleic-acid-binding domains, which also includes HEH domains from the bacterial transcription termination factor Rho, bacterial and eukaryotic lysyl-tRNA synthetases, bacteriophage T4 endonuclease VII, and several uncharacterized proteins. The distribution of the Ku homologs in bacteria coincides with that of the archeal-eukaryotic-type DNA primase and genes for prokaryotic Ku homologs form predicted operons with genes coding for an ATP-dependent DNA ligase and/or archeal-eukaryotic-type DNA primase. Some of these operons additionally encode an uncharacterized protein that may function as nuclease or an Slx1p-like predicted nuclease containing a URI domain. A hypothesis is proposed that the Ku homolog, together with the associated gene products, comprise a previously unrecognized prokaryotic system for repair of double-strand breaks in DNA.
The multifunctional eukaryotic protein Ku binds to discontinuities in double-stranded (ds) DNA such as double-strand breaks, single-strand gaps, and noncomplementary segments. The repair of double-strand breaks in eukaryotes occurs via a pathway of nonhomologous end-joining, or illegitimate recombination, that depends on the Ku protein (Critchlow and Jackson 1998; Featherstone and Jackson 1999). The Ku protein consists of two tightly associated subunits, Ku70 and Ku80, which bind DNA ends and transiently bring them together (Blier et al. 1993; Ramsden and Gellert 1998). In vertebrates, Ku has been shown to recruit the catalytic subunits of the DNA-dependent protein kinase to initiate a phosphorylation and protein–protein interaction cascade that, in turn, leads to the recruitment of repair enzymes including DNA ligase IV, whose activity the Ku protein stimulates in vitro (Gottlieb and Jackson 1993; Teo and Jackson 1997,2000; Ramsden and Gellert 1998). Ku is also a part of the telomere-binding complex and is required for the perinuclear localization of the telomeres (Hsu et al. 1999, 2000; Mishra and Shore 1999; Galy et al. 2000). In addition, Ku forms complexes with numerous other chromosomal proteins such as HP1α, Werner syndrome helicase and poly(ADP-ribose)-polymerase, along with which it binds to chromosomal matrix-attachment regions (MARs) (Galande and Kohwi-Shigematsu 1999; Li and Comai 2000, 2001; Song et al. 2000).
Ku70 and Ku80 are paralogs (Gell and Jackson 1999) and are both conserved throughout the eukaryotic crown group as well as in early-branching eukaryotes such as trypanosomes. This suggests that Ku is an ancient component of the DNA repair and chromatin integrity system, with the duplication that gave rise to Ku70 and Ku80 probably predating the divergence of most, if not all, extant eukaryotes. Prokaryotic counterparts of Ku and the entire illegitimate-recombination-dependent double-strand break repair system, of which Ku is a central component, have not been identified.
We have reported previously the presence of a homolog of the small, catalytic subunit of the eukaryotic-archeal DNA primase (EP) in several bacteria including Bacillus, Mycobacterium, andStreptomyces (Koonin et al. 2000). The gene for this predicted primase is fused to or juxtaposed with a gene for a eukaryotic-archeal ATP-dependent DNA ligase (ADDL), which suggests a functional association between the two enzymes and the presence of a previously undetected, eukaryotic-type DNA repair mechanism in bacteria. Here, we report the first prokaryotic homologs of the DNA-binding protein Ku and discuss evidence that they are part of the same DNA repair system with EP and ADDL. This analysis also reveals the modular architecture of the Ku proteins and allows us to define ancient protein modules involved in DNA repair and other aspects of nucleic acid metabolism.
RESULTS AND DISCUSSION
Bacterial and Archeal Ku Homologs
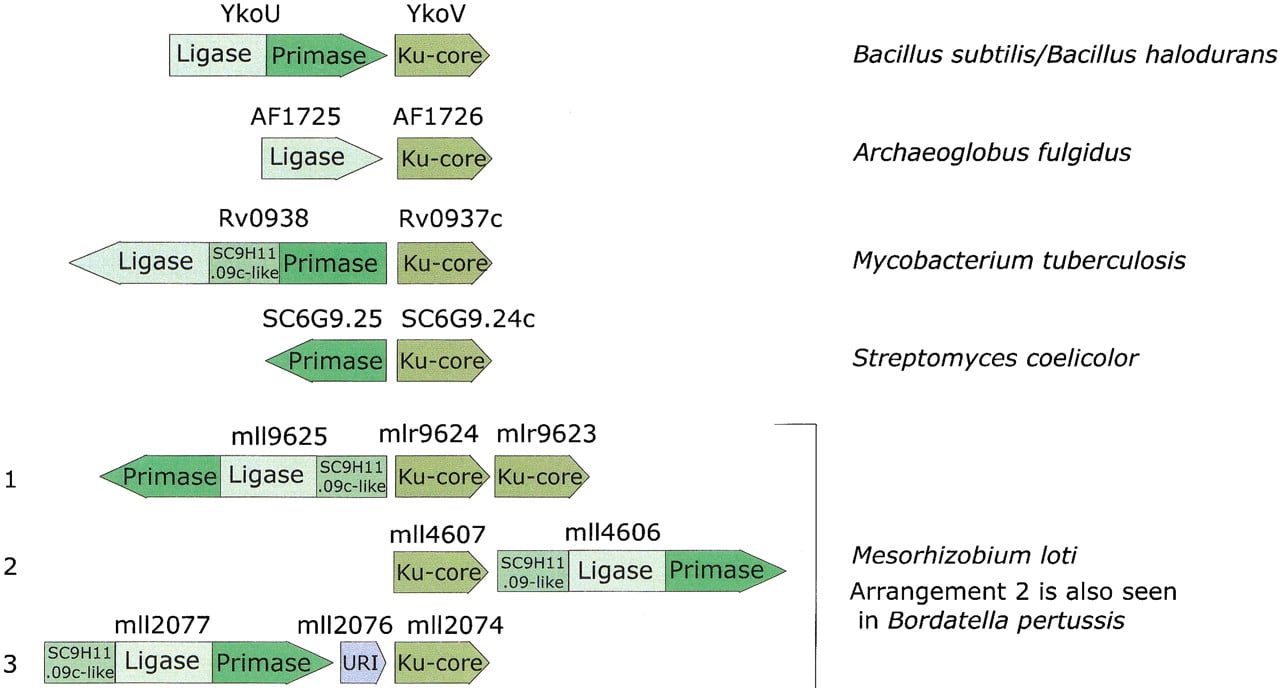
To gain further insight into the functions of the eukaryote-type DNA ligases and primases in bacterial DNA repair, we searched the gene neighborhood of the genes encoding these proteins for other conserved genes. The Bacillus subtilis gene ykoV is adjacent to the ykoU gene, which encodes a two-domain protein with fused EP and ADDL domains; the YkoV protein is highly conserved in all bacteria that encode an EP, but is not detectable in any other bacterial species. Furthermore, the juxtaposition of the ykoVorthologs and the genes coding for EP or ADDL is maintained in phylogenetically diverse bacteria, including Mycobacterium tuberculosis, Streptomyces coelicolor, Mesorhizobium loti, and Bordatella pertussis, and the archeonArchaeoglobus fulgidus. This strong preservation of gene neighborhood of the EP, ADDL, and YkoV orthologs suggests that these genes belong to the same operon, although the exact gene arrangement is variable (Fig. 1). Gene neighborhood or operonic cooccurrence of genes is conserved between multiple, distantly related prokaryotic genomes only when the products of the corresponding genes interact functionally, and often, also physically (Dandekar et al. 1998; Wolf et al. 2001). Hence, it appears most likely that YkoV and its orthologs form a functional complex with EP, ADDL, and another predicted conserved protein (SC9H11.09c and its orthologs) that is also associated with these predicted operons (Fig. 1).
Gene organization in the predicted operons encoding components of the postulated novel double-strand-break repair system. The direction of an arrow indicates the direction of transcription. Distinct regions of each gene encoding separate domains in the protein, such as primase and ligase, are indicated in different shades.
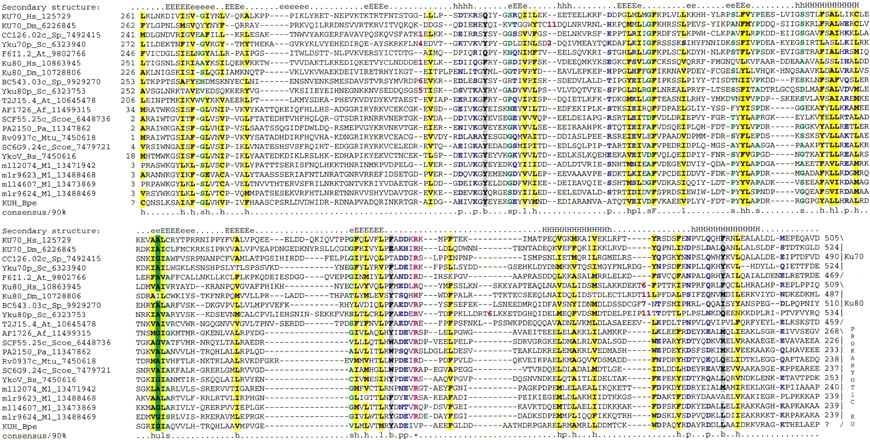
To identify potential distant homologs of the YkoV protein and thus possibly predict its function, we performed iterativePSI-BLAST database searches (Altschul et al. 1997) using the sequences of YkoV and its orthologs as queries; the searches were run to convergence, with a profile inclusion threshold of expect (E) value of 0.01. Most of these searches detected eukaryotic Ku proteins in the second iteration. For example, the search with the sequence of AF1726, the A. fulgidus ortholog of YkoV, detects the central region of the fission yeast Ku70 subunit with E = 10−4 in iteration two and, at convergence, retrieves only the eukaryotic Ku70 and Ku80 proteins. Similarly, reverse searches with the corresponding regions of Ku70 sequences retrieve first the eukaryotic orthologs, then the paralogous Ku80 sequences and, finally, the prokaryotic YkoV-like proteins (e.g., in a search initiated with the sequence of the central region of human Ku70, B. subtilisYkoV is detected in iteration two with E = 10−3). This shows that the YkoV-like proteins are the prokaryotic homologs of the Ku70 and Ku80 proteins. The region of similarity shared by these proteins covers almost the entire length of most prokaryotic proteins. In contrast, the eukaryotic Ku proteins are much larger and contain a conserved amino-terminal extension and, in the case of Ku70, also a conserved carboxy-terminal extension. Thus, the prokaryotic Ku homologs described here appear to define a distinct, previously unnoticed domain that forms the ancient core of these proteins. The conserved blocks, identified previously in the eukaryotic Ku proteins and termed ‘primary homology regions’ (PHR) 3–5 (Gell and Jackson 1999), map entirely within this domain shared by the eukaryotic and prokaryotic Ku homologs. In contrast, PHR 2 and 3 (Gell and Jackson 1999) map to the amino-terminal region exclusively shared by the eukaryotic Ku proteins.
This core domain shared by the prokaryotic YkoV-like proteins and the eukaryotic Ku70 and Ku80 (hereinafter Ku core; Fig.2) is ∼234–280 amino acids long and is larger than most common globular domains. The multiple-alignment-based secondary structure prediction using the PHD program (Rost and Sander 1993) shows that the Ku-core domain is likely to form two distinct substructures. The amino-terminal region (∼85 residues) is poorly conserved and is predicted to form a β-strand-rich subdomain. The remaining portion is more strongly conserved and is predicted to form an α/β structure ending in a strongly predicted bihelical hairpin (Fig. 2). This complex fold is consistent with the functions associated with this region as demonstrated by experimental studies on the eukaryotic Ku70 and Ku80. The principal determinants of heterodimerization (Osipovich et al. 1997; Cary et al. 1998; Koike et al. 1998; Gell and Jackson 1999) and DNA-binding (Wu and Lieber 1996;Wang et al. 1998; Osipovich et al. 1999) of these proteins map to the Ku-core domain as defined by the present sequence comparisons with the prokaryotic Ku homologs. This region also mediates the interactions of the eukaryotic Ku proteins with other chromosomal proteins (Song et al. 2000). Thus, the prokaryotic Ku homologs are predicted to form a homodimer that binds DNA and also associates with other proteins via the conserved Ku core.
Multiple sequence alignment of the Ku-core domains. The secondary structure predicted using the PHD program is shownabove the alignment. E indicates a β-strand and H indicates an α-helix, with the uppercase used to denote the most confident prediction (>82% accuracy). The 90% consensus shown belowthe alignment was derived using the following amino acid classes: polar (p: KRHEDQNST) colored blue; hydrophobic (h: ALICVMYFW) and the aliphatic subset of these are (l: ALIVMC) all shaded yellow; small (s: ACDGNPSTV) colored green, charged (c: DEHKR) colored pink, big (b: Q,E,R,K,Y,M,F,W,L,I) shaded gray. The limits of the domains are indicated by the position numbers on each side of the alignment. The subclasses of Ku-core domains are indicated to the right of the alignment. The sequences are denoted by their gene names followed by the species abbreviations and GenBank identifiers. Subsequent to the submission of this manuscript, the prokaryotic KU homologs were identified in the SMART database (Schultz et al. 1998). The species abbreviations are: At, Arabidopsis thaliana; Hs, Homo sapiens; Mm, Mus musculus; Dm, Drosophila melanogaster; Sc, Saccharomyces cerevisiae; Sp,Schizosaccharomyces pombe; Af,Archaeoglobus fulgidus; Pa, Pseudomonas aeruginosa; Bs, Bacillus subtilis; Scoe, Streptomyces coelicolor; Bpe, Bordatella pertussis; Mtu, Mycobacterium tuberculosis; Ml,Mesorhizobium loti.
The common ancestor of the prokaryotic and eukaryotic Ku proteins might have resembled the extant prokaryotic version, with the essential functions of dimeric DNA-end-binding and interactions with other components of the DNA repair complex. The conserved structure of the predicted operons that encode Ku homologs (Koonin et al. 2000) strongly suggests that these proteins function together as subunits of a protein complex with a possible role in DNA repair or replication. The Ku homologs, EP, and the associated ADDL show sporadic distribution in prokaryotes, as is typically the case with DNA repair systems (Aravind et al. 1999). This is in sharp contrast to the DNA replication components such as, for example, bacterial-type DnaG-primase or NAD-dependent DNA ligase, which show a practically universal distribution among bacteria, including those that possess the Ku–EP–ADDL operons. These observations support a function for these proteins in a DNA repair system, most likely one involved in correction of double-strand breaks in DNA, similar to their eukaryotic counterparts.
In addition to the EP and ADDL, other potential components of the predicted prokaryotic, Ku-associated DNA repair system are revealed by examination of the operons encoding these proteins. Rv0938 contains a conserved domain between its ADDL and EP domains (Fig. 1) that occurs as a stand-alone protein (SC9H11.09c) in S. coelicolor. A homologous domain is also present amino-terminal of the ADDL and EP domains in the ligase-primase proteins from Pseudomonas aeruginosa, B. pertussis, and M. loti, which in the latter two organisms cooccur in a predicted operon with the genes coding for Ku homologs. Thus, this uncharacterized domain is only found in those organisms that also encode Ku and EP, and, given the predicted operonic organization, probably interacts with them functionally. A multiple alignment of this domain reveals conserved histidine and aspartate residues that could form a metal-coordinating cluster within an all β-strand fold (Fig. 3A). This strongly suggests a catalytic function, most probably that of a DNAse, for this conserved domain.
Multiple sequence alignment of predicted nucleases associated with the hypothetical EP-ADDL-Ku repair system. The novel potential nuclease (A) and The URI-domain-containing nuclease (B). The potential metal chelating and active site residues are shown in reverse red shading. (B) The K7M2.9 from Arabidopsis is fused to a MutS domain at the amino terminus, whereas the other eukaryotic forms show carboxy-terminal fusion to a PHD fingers. The species abbreviations are as in Figure 2; the additional abbreviations not present in Figure 2 are: Nc, Neurospora crassa; AcNPV,Autographa californica Nuclear polyhedrosis virus; Ngo,Neisseria gonorrhea; Bs, Bacillus subtilis; Ec,Escherichia coli; Ccr, Caulobacter cresentus.

One of the predicted Ku-encoding operons from M. lotiincludes a small gene (Msl2076) between the genes coding for the EP–ADDL fusion protein and the Ku homolog (Fig. 1). This gene shows the same direction of transcription as the two other genes and probably is a part of the operon. Sequence profile searches with the Msl2076 showed that it belongs to a distinct family of UvrC-Intron-type (URI) endonucleases (Aravind et al. 1999) typified by theEscherichia coli YhbQ, B. subtilis YazA, and yeast Slx1p. This family of URI nucleases (Fig. 3B) is represented widely in single or duplicate copies in bacteria, eukaryotes, DNA viruses, and, so far, in a single archeon, Halobacterium salinarium. The prokaryotic members of this family are characterized by their distinct, small size (typically, <100 amino acids); thus, they represent stand-alone forms of the URI endonuclease domain. The eukaryotic members typified by the yeast DNA repair protein Slx1p (Mullen et al. 2001) contain an additional, carboxy-terminal PHD-finger domain, whereas one of the paralogs in Arabidopsis is fused to the MutS DNA-repair ATPase (Fig. 3B). Yeast Slx1p functionally interacts with the yeast RecQ-like helicase Sgs1p and is likely to function in resolution of recombination intermediates in DNA repair (Mullen et al. 2001), which is consistent with its predicted nuclease activity. Thus the Slx1p-YhbQ family of proteins is likely to define a highly conserved repair-recombination pathway present in both eukaryotes and bacteria. This hypothetical repair pathway might interact with the predicted Ku–EP–ADDL-dependent pathway.
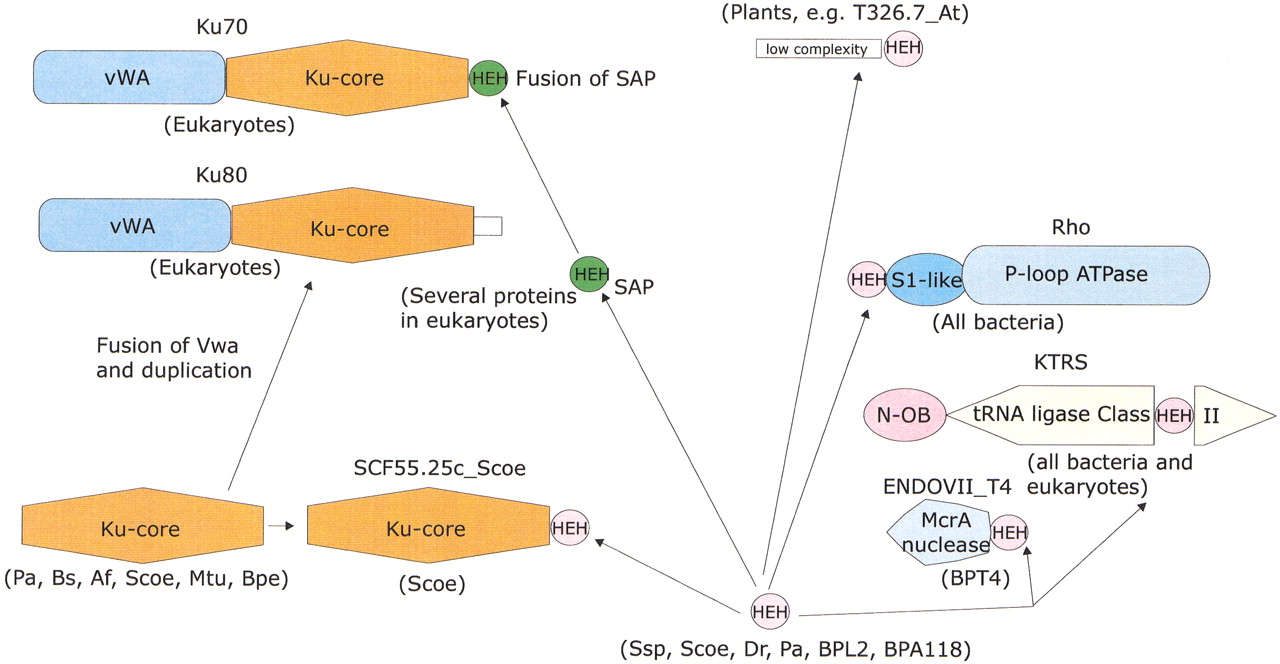
The Helix–Extension–Helix Fold and its Association with the Carboxyl Terminus of the Ku-Core Domain in Bacteria and Eukaryotes
In an attempt to glean more details of the functional interactions and evolution of the Ku proteins, we analyzed the domains that are associated with the Ku core in eukaryotes and prokaryotes. All prokaryotic Ku homologs, with the exception of SCF55.25c fromS. coelicolor, consist of the Ku-core domain alone. SCF55.25c contains a carboxy-terminal extension of ∼40 amino acid residues that show significant sequence similarity to several small, uncharacterized proteins from bacteria, bacteriophages, andArabidopsis thaliana. Iterative PSI-BLASTsearches resulted in the detection of the same region of similarity in the bacterial transcription terminator Rho, where it occurs at the extreme amino terminus, immediately upstream of the OB-fold domain. The conserved region precisely corresponds to the amino-terminal α-helical domain of Rho (hereinafter Rho-N) as defined by its X-ray and NMR structures (Allison et al. 1998; Bogden et al. 1999). To further investigate the distribution of this small domain, we searched the protein structure database using the DALI search tool (Holm and Sander 1998). This resulted in the detection of two structures with high similarity to Rho-N, namely the carboxy-terminal domain of Endonuclease VII (Raaijmakers et al. 1999) (a nuclease and Holliday junction resolvase from bacteriophage T4) and the small α-helical domain inserted into the catalytic domain of bacterial and eukaryotic lysyl-tRNA synthetases (KTRS) (Onesti et al. 2000). InPSI-BLAST searches initiated with the Rho-N domain sequence, these proteins were detected with borderlineE-values. A structure-based sequence alignment of the Rho-N domain with the α-helical domains of KTRS and Endonuclease VII shows that, in addition to the structural similarity, they contain the conserved residues characteristic of Rho-N and its homologs that were detected in sequence searches (Fig.4A). Thus, we conclude that these α-helical domains have a common evolutionary origin and define a novel superfamily of ancient mobile domains that are found in various contexts related to nucleic acid metabolism.
(A) Multiple sequence alignment of different classes of HEH domains. Each of the alignments is colored according to a separate consensus using the rules described in the legend to Figure 2. The secondary structure shown above the alignment was derived from the structures of Rho, Endo-VII and K-TRS. For the SAP domains, the structure was predicted using the PHD program. The species abbreviations are the same as in Figure 2; those not present in Figure2 are : Ec, Escherichia coli; Ssp, Synechocystis sp.; Tma, Thermotoga maritima; Dr, Deinococcus radiodurans; Aae, Aquifex aeolicus; BPL2, lactococcal Bacteriophage L2; BPA118, Listeria bacteriophage A118; T4, Bacteriophage T4; Miclu, Micrococcus luteus; Ce,Caenorhabditis elegans; Ct, Chlamydia trachomatis; Hp, Helicobacter pylori; Bst, Bacillus stearothermophilus. (B) Structures and models of different forms of the HEH domain shown in the alignment. The NH2 (N) and COOH (C) termini of the HEH domains are indicated.

Examination of the multiple alignment and the three structural prototypes of this superfamily (Rho-N, Endonuclease VII carboxy-terminal domain, and KTRS-insert domain) shows that these domains share a novel fold (Fig. 4B) that is distinct from other α-helical folds found in small, primarily nucleic-acid-binding domains such as Helix–turn–Helix, Helix–loop–Helix, and Helix–hairpin–Helix (Doherty et al. 1996; Aravind and Koonin 1999;Massari and Murre 2000). In this newly detected fold, the first short, mobile helix almost immediately leads to the second helix that is separated from the parallel third helix by a prominent extended segment (Fig. 4B). We designated this fold the helix–extended-region–helix (HEH) domain after this unique structural pattern. The fusion of the HEH to the Ku-core and Endonuclease VII is suggestive of DNA-binding, whereas the presence of this domain in Rho and the KTRS is more consistent with RNA-binding. In KTRS, the HEH domain undergoes movement on lysine-binding and might facilitate recognition of specific structural features of tRNALys (Onesti et al. 2000).
Additional extensive PSI-BLAST searches with the HEH domain sequences detected, with a moderately significantE-value of 0.09, a previously identified nucleic-acid-binding domain, the SAP domain (Aravind and Koonin 2000). Reciprocal searches with the SAP domain sequences also retrieved from the database some of the HEH domain sequences with significant E-values (e.g., a stand-alone HEH-domain protein from Listeria phage A118 was recovered in a search with the ArabidopsisAP-endonuclease SAP domain in iteration 8 withE = 4 × 10−4), suggesting a potential evolutionary relationship. SAP is a small domain of approximately the same size as the HEH domain, and its core is strongly predicted to contain two helices separated by a relatively long extended region, similar to helix-2 and helix-3 of the HEH domain (Aravind and Koonin 2000). The amino acid residue conservation pattern in these helices and the extended region between them is also similar in the HEH and SAP domains (Fig. 4A). The only noticeable difference in the sequence pattern between these two domains is the presence of an insert of two amino acids at the end of the extended region in the HEH domains. A homology model of the SAP domain from the Acinus protein (Sahara et al. 1999; Aravind and Koonin 2000) built using the HEH from E. coli Rho as the structural template showed that the absence of these two residues is unlikely to disrupt the extended region characteristic of this fold (Fig. 4B). Thus, it appears likely that SAP domain is a derived, eukaryote-specific version of the HEH fold, which interacts with DNA via the charged surfaces of the helices.
Notably, the SAP domain is present in the eukaryotic Ku70 proteins as a conserved carboxy-terminal extension (Aravind and Koonin 2000). Based on the functions of the characterized SAP domains, it has been predicted that this domain binds the MARs and participates in tethering chromosomal proteins to these sites (Aravind and Koonin 2000); the SAP domain is probably responsible for MAR-binding by the Ku protein (Galande and Kohwi-Shigematsu 1999). It appears that HEH-fold domains, namely Rho-N and SAP, have been fused to the carboxyl termini of the Ku-core domain on two independent occasions, in bacteria and eukaryotes, respectively. This may point to a specific cooperation between the HEH and Ku-core domains in binding unusual DNA structures including MARs and their analogs in bacteria.
The Amino-Terminal Region of the Eukaryotic Ku Proteins Contains a Divergent Von Willebrand Factor A Domain
The bacterial Ku homologs contain no counterpart of the conserved amino-terminal extension that is present in the eukaryotic Ku70 and Ku80. To determine the origin of this extension, we performedPSI-BLAST searches with these regions from the eukaryotic Ku proteins. At convergence, these searches retrieved from the database, in addition to the Ku proteins, several Von Willebrand factor A (vWA) domains from various organisms (e.g., the sequence of the Serum opacity factor from Streptococcus pyogenes was retrieved in iteration 4, E = 10−2, and the YwmC protein fromB. subtilis in iteration 8, E = 10−3, and chicken Collagen α2 in iteration 11,E = 10−3). To further assess these observations, we constructed a multiple alignment of all amino-terminal extensions from diverse Ku70s and Ku80s and predicted the secondary structure of this domain using the PHD program (Rost and Sander 1993). The predicted structural elements exactly matched the pattern characteristic of the vWA domain for which experimentally determined structures are available (Lee et al. 1995; Leitinger and Hogg 2000). Furthermore, the two Mg2+-binding aspartates located at the ends of strands 1 and 4 of the vWA domains are typically conserved in the Ku proteins (Fig. 5) (Lee et al. 1995). Sequence-structure threading using the hybrid fold recognition method (Fischer 2000) with the human Ku70 protein as a query recovers the vWA domain of integrin (PDB:1ido) as the best hit. Thus, the amino-terminal extension of the eukaryotic Ku proteins appears to be a divergent version of the vWA domain.
Multiple sequence alignment of the vWA domains of Ku70 and Ku80. The secondary structure shown above the figure was based on the solved structures of vWA domains; the same consensus-based coloring scheme as in Figures 2 and 3 is used. The species abbreviation Spy is for Streptococcus pyogenes, whereas the rest are the same as in Figures 2 and 3.
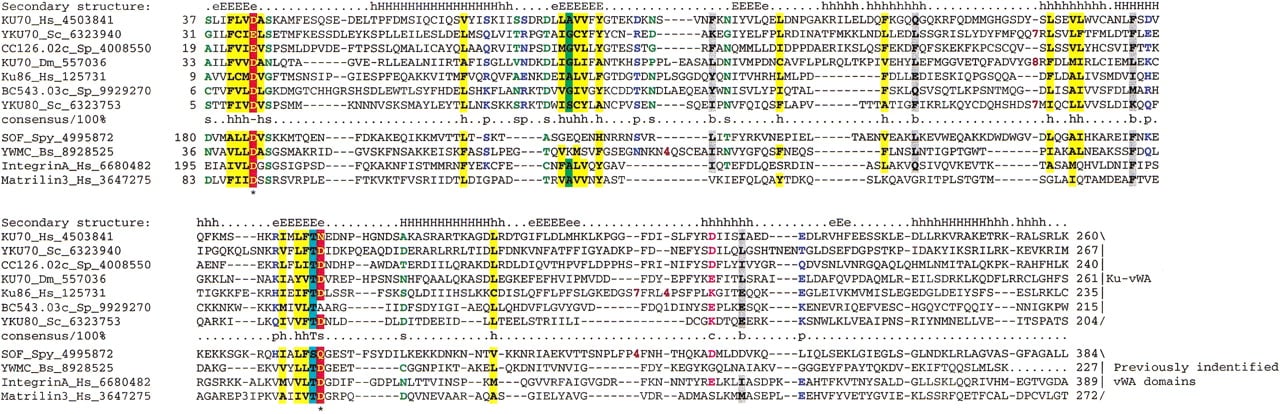
The vWA domain, although originally discovered in several animal extracellular adhesion molecules (Lee et al. 1995), has been subsequently detected in intracellular contexts in both prokaryotes and eukaryotes (Ponting et al. 1999). In addition to Ku, at least one other protein with a function in DNA repair and transcription, the TFIIH subunit p44, contains a vWA domain (Ponting et al. 1999). In Ku70 and Ku80, the region encompassing the vWA domain is the second determinant of heterodimerization, which is consistent with the role of vWA in protein–protein interactions (Singleton et al. 1997; Wang et al. 1998). The conservation of the Mg+2-binding aspartates in most sequences of the vWA domains from the Ku proteins (Fig. 5) suggests that they probably function as cation-dependent interaction modules similar to vWA domains in other contexts. Some of the numerous protein–protein interactions demonstrated for the eukaryotic Ku proteins, in addition to heterodimerization, probably depend on the vWA domains of Ku70 and Ku80. To our knowledge the experiments to address the Mg+2 dependence of these interactions has not been performed.
Evolutionary Implications and Conclusions
The dissection of the Ku protein from eukaryotes and prokaryotes into individual domains described above suggests an evolutionary scenario for these proteins. The Ku core is an ancient domain that was probably present in bacteria and archea even before the advent of the eukaryotes. There are clear indications that, in these organisms, the Ku homologs are functionally associated with the ATP-dependent DNA ligase and the eukaryotic-type primase, probably as components of a double-strand break repair system. Because ADDL and EP are ubiquitous in archea, but are present only sporadically in bacteria, it seems plausible that this hypothetical repair system, including the Ku-core domain, has originally evolved within the archeal lineage and subsequently has been disseminated among bacteria through multiple horizontal transfers. However, the difficulty with this scheme is that a Ku homolog so far was identified in only one archeon, A. fulgidus, whereas the mobile Ku–EP–ADDL operon so far is widely represented only in bacteria. Hence, a bacterial origin for this mobile operon, with a subsequent transfer of the gene coding for Ku to A. fulgidus is also equally possible. In Streptomyces, there was a duplication of the Ku-core-encoding gene, and one of the paralogs fused with another ancient nucleic-acid-binding domain, the HEH (Fig.6), whereas, in M. loti, the entire Ku–EP–ADDL operon was duplicated (Fig. 1).
Protein domain architectures and possible evolutionary trajectories for the constituent domains of the Ku proteins. The domains are indicated by different shapes; the two distinct forms of the HEH are indicated by differential coloring. For each domain architecture, the phyletic distribution is shown in parentheses; the species name abbreviations are as in Figures 2 and 3. The arrows indicate probable evolutionary events such as derivation of a new form of a particular domain (SAP from ancestral HEH) and domain fusion. The connection shown between prokaryotic and eukaryotic forms of the Ku protein does not differentiate between two evolutionary scenarios discussed in the text.
Eukaryotes might have vertically inherited the Ku-core protein, along with the primase and the ATP-dependent ligase, from a common ancestor shared with a certain archeal lineage or through horizontal transfer from a bacterial lineage such as the mitochondrial precursor. Under this scenario, early in the evolution of eukaryotes, the Ku-core domain underwent an amino-terminal fusion with the vWA domain, followed by a duplication giving rise to the paralogous Ku70 and Ku80 proteins (Fig.6). Subsequently, but prior to the radiation of the major crown-group lineages, the Ku70 protein fused with the eukaryote-specific version of the HEH fold, the DNA-binding SAP domain (Fig. 6). Ku80 evolved its own unique distinct carboxy-terminal extension resulting in the acquisition of distinct functions by the eukaryotic Ku paralogs. These fusions conferred several new interactive abilities on Ku70 and Ku80 that allowed them to associate with various eukaryote-specific protein complexes involved in DNA repair, telomere formation, and chromatin remodeling. This scenario seems plausible because prokaryotic Ku homologs that contain the Ku-core domain alone seem to be the best candidates for the role of the primitive form of this protein. An alternative scenario would hold that the Ku-core domain evolved at an early stage of eukaryotic evolution and was horizontally acquired by a bacterium or an archeon, probably prior to the fusion with the vWA domain, followed by horizontal dissemination among the prokaryotes. Sequencing of additional archeal genomes and those of early-branching eukaryotes help in resolving these alternative hypotheses.
Regardless of the exact evolutionary scenario, the detection of Ku homologs in prokaryotes and dissection of the Ku protein into previously undetected, distinct domains will allow experimental exploration of simpler model systems to understand the essential functions of these important proteins.
METHODS
The archeal and bacterial genome sequences were retrieved from the Genomes division of the Entrez system (Tatusova et al. 1999). The nonredundant database of protein sequences at the National Center for Biotechnology Information (NIH, Bethesda) was iteratively searched using the PSI-BLAST program (Altschul et al. 1997). The cut-off of E < 0.01 was typically employed for inclusion of sequences in the position-specific weight matrices. Nucleotide sequences of unfinished bacterial and archeal genomes translated in all six reading frames were searched using the TBLASTN program (Altschul et al. 1997). Multiple alignments of protein sequences were constructed using the ClustalW (Thompson et al. 1994) program and corrected on the basis of PSI-BLAST results. Protein secondary structure was predicted using the PHDprogram, with a multiple alignment submitted as the query (Rost and Sander 1993; Rost et al. 1997). Sequence-structure threading was performed using the hybrid fold recognition method that incorporates both structural and evolutionary information in sequence comparisons into a single algorithm (Fischer 2000). Homology modeling of protein structures was performed by using the SWISS-MODEL server (Guex and Peitsch 1997). The target was threaded through the template using theSWISS-PDBviewer software and the alignment with the template was manually adjusted to minimize the clashes of the protein backbones. The energy minimization was carried out using theGROMOS program that employs a Sippl-like force field (Guex and Peitsch 1997). The ribbon diagrams of the structures were generated using the MOLSCRIPT program (Kraulis 1991).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Notes
[1] Corresponding author.
Notes
[2] E-MAIL [email protected]; FAX (301) 480-9241.
[3] Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.181001.
REFERENCES
- ↵T.J. AllisonT.C. WoodD.M. BriercheckF. RastinejadJ.P. RichardsonG.S. Rule(1998) Crystal structure of the RNA-binding domain from transcription termination factor rho [letter]. Nat. Struct. Biol. 5:352–356.
- ↵S.F. AltschulT.L. MaddenA.A. SchafferJ. ZhangZ. ZhangW. MillerD.J. Lipman(1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402.
- ↵L. AravindE.V. Koonin(1999) DNA-binding proteins and evolution of transcription regulation in the archaea. Nucleic Acids Res. 27:4658–70.
- ↵(2000) SAP — a putative DNA-binding motif involved in chromosomal organization. Trends Biochem. Sci. 25:112–114, ibid.
- ↵L. AravindD.R. WalkerE.V. Koonin(1999) Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 27:1223–1242.
- ↵P.R. BlierA.J. GriffithJ. CraftJ.A. Hardin(1993) Binding of Ku protein to DNA. Measurement of affinity for ends and demonstration of binding to nicks. J. Biol. Chem. 268:7594–7601.
- ↵C.E. BogdenD. FassN. BergmanM.D. NicholsJ.M. Berger(1999) The structural basis for terminator recognition by the Rho transcription termination factor. Mol. Cell 3:487–493.
- ↵R.B. CaryF. ChenZ. ShenD.J. Chen(1998) A central region of Ku80 mediates interaction with Ku70 in vivo. Nucleic Acids Res. 26:974–979.
- ↵S.E. CritchlowS.P. Jackson(1998) DNA end-joining: From yeast to man. Trends Biochem. Sci. 23:394–398.
- ↵T. DandekarB. SnelM. HuynenP. Bork(1998) Conservation of gene order: A fingerprint of proteins that physically interact. Trends Biochem. Sci. 23:324–328.
- ↵A.J. DohertyL.C. SerpellC.P. Ponting(1996) The helix–hairpin–helix DNA-binding motif: A structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res. 24:2488–2497.
- ↵C. FeatherstoneS.P. Jackson(1999) Ku, a DNA repair protein with multiple cellular functions? Mutat. Res. 434:3–15.
- ↵D. Fischer(2000) Hybrid fold recognition: Combining sequence derived properties with evolutionary information. Pac. Symp. Biocomput . pp 119–130.
- ↵S. GalandeT. Kohwi-Shigematsu(1999) Poly(ADP-ribose) polymerase and Ku autoantigen form a complex and synergistically bind to matrix attachment sequences. J. Biol. Chem. 274:20521–20528.
- ↵V. GalyJ.C. Olivo-MarinH. ScherthanV. DoyeN. RascalouU. Nehrbass(2000) Nuclear pore complexes in the organization of silent telomeric chromatin. Nature 403:108–112.
- ↵D. GellS.P. Jackson(1999) Mapping of protein–protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Res. 27:3494–3502.
- ↵T.M. GottliebS.P. Jackson(1993) The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell 72:131–142.
- ↵N. GuexM.C. Peitsch(1997) SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18:2714–2723.
- ↵L. HolmC. Sander(1998) Touring protein fold space with Dali/FSSP. Nucleic Acids Res. 26:316–319.
- ↵H.L. HsuD. GilleyE.H. BlackburnD.J. Chen(1999) Ku is associated with the telomere in mammals. Proc. Natl. Acad. Sci. 96:12454–12458.
- ↵H.L. HsuD. GilleyS.A. GalandeM.P. HandeB. AllenS.H. KimG.C. LiJ. CampisiT. Kohwi-ShigematsuD.J. Chen(2000) Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes & Dev. 14:2807–2812.
- ↵M. KoikeT. MiyasakaT. MimoriT. Shiomi(1998) Subcellular localization and protein–protein interaction regions of Ku proteins. Biochem. Biophys. Res. Commun. 252:679–685.
- ↵E.V. KooninY.I. WolfA.S. KondrashovL. Aravind(2000) Bacterial homologs of the small subunit of eukaryotic DNA primase. J. Mol. Microbiol. Biotechnol. 2:509–512.
- ↵P.J. Kraulis(1991) Molscript. J. Appl. Cryst. 24:946–950.
- ↵J.O. LeeP. RieuM.A. ArnaoutR. Liddington(1995) Crystal structure of the A domain from the alpha subunit of integrin CR3 (CD11b/CD18). Cell 80:631–638.
- ↵B. LeitingerN. Hogg(2000) From crystal clear ligand binding to designer I domains. Nat. Struct. Biol. 7:614–616.
- ↵B. LiL. Comai(2000) Functional interaction between Ku and the Werner syndrome protein in DNA end processing. J. Biol. Chem. 275:28349–28352.
- ↵(2001) Requirements for the nucleolytic processing of DNA ends by the Werner syndrome protein-ku70/80 complex. J. Biol. Chem. 276:9896–9902, ibid.
- ↵M.E. MassariC. Murre(2000) Helix–loop–helix proteins: Regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 20:429–440.
- ↵K. MishraD. Shore(1999) Yeast Ku protein plays a direct role in telomeric silencing and counteracts inhibition by rif proteins. Curr. Biol. 9:1123–1126.
- ↵J.R. MullenV. KaliramanS.S. IbrahimS.J. Brill(2001) Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 157:103–118.
- ↵S. OnestiG. DesogusA. BrevetJ. ChenP. PlateauS. BlanquetP. Brick(2000) Structural studies of lysyl-tRNA synthetase: Conformational changes induced by substrate binding. Biochemistry 39:12853–12861.
- ↵O. OsipovichS.K. DurumK. Muegge(1997) Defining the minimal domain of Ku80 for interaction with Ku70. J. Biol. Chem. 272:27259–27265.
- ↵O. OsipovichR.J. DuheP. HastyS.K. DurumK. Muegge(1999) Defining functional domains of Ku80: DNA end binding and survival after radiation. Biochem. Biophys. Res. Commun. 261:802–807.
- ↵C.P. PontingL. AravindJ. SchultzP. BorkE.V. Koonin(1999) Eukaryotic signalling domain homologues in archaea and bacteria. Ancient ancestry and horizontal gene transfer. J. Mol. Biol. 289:729–745.
- ↵H. RaaijmakersO. VixI. ToroS. GolzB. KemperD. Suck(1999) X-ray structure of T4 endonuclease VII: A DNA junction resolvase with a novel fold and unusual domain-swapped dimer architecture. EMBO J. 18:1447–1458.
- ↵D.A. RamsdenM. Gellert(1998) Ku protein stimulates DNA end joining by mammalian DNA ligases: A direct role for Ku in repair of DNA double-strand breaks. EMBO J. 17:609–614.
- ↵B. RostC. Sander(1993) Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232:584–599.
- ↵B. RostR. SchneiderC. Sander(1997) Protein fold recognition by prediction-based threading. J. Mol. Biol. 270:471–480.
- ↵S. SaharaM. AotoY. EguchiN. ImamotoY. YonedaY. Tsujimoto(1999) Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature 401:168–173.
- ↵J. SchultzF. MilpetzP. BorkC.P. Ponting(1998) SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. 95:5857–5864.
- ↵B.K. SingletonA. PriestleyH. SteingrimsdottirD. GellT. BluntS.P. JacksonA.R. LehmannP.A. Jeggo(1997) Molecular and biochemical characterization of xrs mutants defective in Ku80. Mol. Cell. Biol. 17:1264–1273.
- ↵K. SongY. JungD. JungI. Lee(2000) Human Ku70 interacts with HP1alpha. J. Biol. Chem. 276:8321–8327.
- ↵T.A. TatusovaI. Karsch-MizrachiJ.A. Ostell(1999) Complete genomes in WWW Entrez: Data representation and analysis. Bioinformatics 15:536–543.
- ↵S.H. TeoS.P. Jackson(1997) Identification of Saccharomyces cerevisiae DNA ligase IV: Involvement in DNA double-strand break repair. EMBO J. 16:4788–4795.
- ↵(2000) Lif1p targets the DNA ligase Lig4p to sites of DNA double-strand breaks. Curr. Biol. 10:165–168, ibid.
- ↵J.D. ThompsonD.G. HigginsT.J. Gibson(1994) CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680.
- ↵J. WangX. DongK. MyungE.A. HendricksonW.H. Reeves(1998) Identification of two domains of the p70 Ku protein mediating dimerization with p80 and DNA binding. J. Biol. Chem. 273:842–848.
- ↵Y.I. WolfI.B. RogozinA.S. KondrashovE.V. Koonin(2001) Genome alignment, evolution of prokaryotic genome organization and prediction of gene function using genomic context. Genome Res. 11:356–372.
- ↵X. WuM.R. Lieber(1996) Protein–protein and protein–DNA interaction regions within the DNA end-binding protein Ku70-Ku86. Mol. Cell. Biol. 16:5186–5193.