
Changes in Gene Expression Associated with Developmental Arrest and Longevity in Caenorhabditis elegans
- Steven J.M. Jones1,7,
- Donald L. Riddle2,
- Anatoli T. Pouzyrev2,
- Victor E. Velculescu3,
- LaDeana Hillier4,
- Sean R. Eddy5,
- Shawn L. Stricklin5,
- David L. Baillie6,
- Robert Waterston4, and
- Marco A. Marra1
- 1Genome Sequence Centre, British Columbia Cancer Research Centre, Vancouver, British Columbia V5Z 4E6, Canada; 2Molecular Biology Program and Division of Biological Sciences, University of Missouri, Columbia, Missouri 65211, USA; 3Johns Hopkins Oncology Center, Baltimore, Maryland 21231, USA; 4The Genome Sequencing Center, Washington University School of Medicine, St. Louis, Missouri 63108, USA; 5Howard Hughes Medical Institute, Department of Genetics, Washington University School of Medicine, St. Louis Missouri 63110, USA; 6Institute of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, British Columbia V5A 1S6, Canada
Abstract
Gene expression in a developmentally arrested, long-lived dauer population of Caenorhabditis elegans was compared with a nondauer (mixed-stage) population by using serial analysis of gene expression (SAGE). Dauer (152,314) and nondauer (148,324) SAGE tags identified 11,130 of the predicted 19,100 C. elegans genes. Genes implicated previously in longevity were expressed abundantly in the dauer library, and new genes potentially important in dauer biology were discovered. Two thousand six hundred eighteen genes were detected only in the nondauer population, whereas 2016 genes were detected only in the dauer, showing that dauer larvae show a surprisingly complex gene expression profile. Evidence for differentially expressed gene transcript isoforms was obtained for 162 genes. H1 histones were differentially expressed, raising the possibility of alternative chromatin packaging. The most abundant tag from dauer larvae (20-fold more abundant than in the nondauer profile) corresponds to a new, unpredicted gene we have named tts-1 (transcribed telomere-like sequence), which may interact with telomeres or telomere-associated proteins. Abundant antisense mitochondrial transcripts (2% of all tags), suggest the existence of an antisense-mediated regulatory mechanism in C. elegansmitochondria. In addition to providing a robust tool for gene expression studies, the SAGE approach already has provided the advantage of new gene/transcript discovery in a metazoan.
The nematode Caenorhabditis elegans can enter the dauer diapause stage under conditions of high population density and limited food (Golden and Riddle 1984). In the soil, dauer larvae disperse to a fresh environment and resume development to the reproductive adult. Molecular genetic analyses have shown that the nervous system responds to environmental stimuli to regulate larval development via transforming growth factor (TGF)–β (Ren et al. 1996), cyclic guanosine monophosphate (cGMP) (Birnby et al. 2000), and insulin-like (Kimura et al. 1997) signaling pathways, which, in turn, act on a nuclear receptor (Antebi et al. 2000) to control dauer versus nondauer morphogenesis (Riddle and Albert 1997). Dauer larvae are long lived and resistant to environmental stress (Klass and Hirsh 1976). Mutations that reduce insulin-like signaling promote both dauer diapause and adult longevity (Kimura et al. 1997; Gems et al. 1998). As a step toward understanding the unique physiology of the long-lived dauer state, we compared the overall gene expression profiles of dauer larvae and nondauer, growing (mixed-stage) populations. Serial analysis of gene expression (SAGE) (Velculescu et al. 1995) was used to comprehensively survey the transcriptomes of the two populations and to search for previously unidentified transcripts. SAGE allows the detailed profiling of mRNA populations through the isolation of unique sequence tags from individual transcripts, typically 14 nucleotides in length. Detection and enumeration of the tags is conducted by the concatenation of the tags and the subsequent sequencing of concatemer clones. In the case of C. elegans, the identification of the cellular transcripts from the SAGE tag is aided greatly through the availability of a sequenced and annotated genome.
RESULTS AND DISCUSSION
Considering the arrested state of development, we suspected that the dauer transcriptome would not be complex. Transcription rates in the dauer stage previously were estimated to be approximately six- to seven-fold reduced relative to growing larvae (Dalley and Golomb 1992). Surprisingly, SAGE tags for 8449 transcripts were detected in the dauer stage. In total, SAGE tags were identified for 11,130 (58.8%) of the 18,931 C. elegans transcripts predicted to contain theNlaIII (CATG) restriction site required for detection by using the SAGE technology. Of these, 29.2% of the transcripts were identified by tags observed only once (singletons). Two thousand sixteen (18.1%) of the detected transcripts were found only in the dauer stage and 2681 (24.1%) only in the mixed stages, although many of these transcripts were detected at low levels. Dauer (358) and nondauer (533) -specific transcripts were detected at significance ofP ≤ 0.05. The dauer-specific genes (many of which encode novel proteins) are candidates for specifying functions associated with efficient life maintenance and longevity. The overall abundance of SAGE tags is shown in Figure1. Of the transcripts detected, 5765 (51%) previously had been confirmed by expressed sequence tag (EST) data (Kohara 1996).
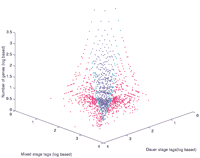
Expression profile comparing relative expression in dauer and mixed stages. Singleton tags were excluded. (blue) Tags for which no significant expression difference is observed; (green) significance is 95%–99%; (red) significance >99% confidence, as determined by the G test (Sokal and Rohlf 1995). The z-axis represents the number of transcripts with a specific mixed/dauer tag ratio.
Thirty-three thousand forty unique tag sequences (tag species) representing 129,813 observed tags could not be unambiguously assigned to a predicted transcript. Of these, 1397 tag species matched either ESTs (sense strand) or unfinished sections of the C. elegansgenomic sequence yet to be annotated. There were 23,628 (71.5%) singletons that lacked validating sequence data confirming sequence integrity. Unassigned tag species fell into three classes. (1) Two thousand seven hundred thirteen matched more than one gene and therefore lacked an unambiguous gene assignment. (2) Sixteen thousand three hundred seventy-nine could not be matched with either C. elegans genomic or cDNA sequence. These tags arose presumably because of sequencing errors, strain differences, or tags overlapping undiscovered splice junctions or poly(A) tails. (3) Twelve thousand five hundred fifty-one tag species (66.4% of these were singletons) matched genomic DNA sequence but could not be assigned to a predicted transcript. A subset of these tags derive from transcripts that currently are not predicted within the genomic annotation or where the gene structure is mispredicted. Of the 12,551 unmapped tag species, 6489 (63.4% of which were singletons) matched a transcript in the antisense orientation (Fig. 2). It is likely that most of the observed antisense tags are caused by mispriming of the oligo(dT) to internal poly(A) stretches after first-strand synthesis during SAGE library construction.
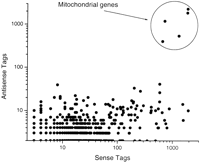
Comparison of sense and antisense tag frequencies. From 6489 tag sequences matching only an antisense transcript, 549 transcripts were studied in which both the antisense and sense SAGE tags could be unambiguously correlated. Only in 56 transcripts are the antisense transcripts in a higher abundance than the sense tags.
Mitochondrial transcripts were found to be highly represented in both SAGE libraries profiled. The five mitochondrial protein coding transcripts detectable by the SAGE methodology (cytochrome C oxidase subunits I, II, and III and NADH-ubiquinone oxidoreductase subunits 4 and 5) contributed 4247 observed tags (2.8%) in the dauer population and 2150 tags (1.4%) in the mixed stages. However, 3924 tags (2.6%) from the dauer library and 2302 (1.6%) from the mixed-stage profile could only be assigned to the antisense strand of mitochondrial genes (Table 1). The relative abundance of antisense and sense mitochondrial tags indicates that the generation of antisense tags from mitochondrial transcripts possibly is not an artifact caused by mispriming (Fig. 2). This observation is supported by the previous detection of stable polyadenylated antisense mRNA for both NADH-ubiquinone oxidoreductase in the rat (Tullo et al. 1994) and for cytochrome C oxidase in a human cell line (Shirafuji et al. 1997). As no proteins with RNAase activity currently are known to be imported into the C. elegans mitochondria (Costanzo et al. 2000), antisense RNA may serve to negatively regulate mitochondrial translation.
SAGE Tags Corresponding to Five Mitochondrial Sense and Antisense Transcripts
Genes previously implicated in longevity showed increased representation in the dauer expression profile (Table2) including two superoxide dismutase genes (C08A9.1/sod-3 and F55H2.1/sod-4) and two glutathione peroxidase genes adjacent to each other in the genome (C11E4.1 and C11E4.2). The tRNA dimethylallyltransferase (DMAPP transferase)gro-1 gene (Heikimi et al. 1997), mutations in which increase longevity in C. elegans, was not detected among dauer-derived tags. DMAPP transferase is required for correct activation of tRNA molecules whose anticodons begin with U, and therefore its reduced expression in the dauer form should decrease the rate of protein synthesis (Dihanich et al. 1987). A subsequent increase in levels of DMAPP transferase might allow for the rapid resumption of protein synthesis on exit from the dauer stage. The expression of a single poly(A)-binding (PABP) like protein, elevated levels of which are known to decrease message turnover (Stambuk and Moon 1992), has been shown previously by subtractive cDNA library approach to be increased in dauer (Cherkasova et al. 2000). Two C. elegans genes, Y106G6H.2 and F18H3.3, show the greatest degree of similarity to human PABP. Y106G6H.2 (dauer 217; mixed 80) and both alternative transcripts of F18H3.3 (F18H3.3a dauer 12, mixed 5; F18H3.3b dauer 15, mixed 4) show increased levels of expression in the dauer stage. In this instance, the SAGE methodology discriminated between two paralogous genes and detected alternative transcripts.
Expression of Genes Implicated Previously in Lifespan and Dauer
The most abundant tag in the dauer stage (dauer, 4329 tags; mixed, 215 tags) derives from a transcript lacking a long open reading frame or protein similarity (dbEST nos. 1280045 and 1300904; GenBank accession number U41749, positions 17209–17831), which we have namedtts-1 (transcribed telomere-like sequence). This transcript is enigmatic, but it shares characteristics with known telomerase RNAs, from ciliate through human (Chen et al. 2000). These include (1) a one-and-a-half-length repeat of the C. elegans telomeric template sequence 60 nucleotides from the 5′ end; (2) a 20-nucleotide region of base pair complementarity upstream and downstream of the telomeric template sequence; and (3) a canonical pseudoknot predicted within the region between the telomeric template and the 3′ end of the predicted 20 nucleotide helix. However, other characteristics oftts-1 differ from telomerase RNA genes. Alternative splicing was detected between the two ESTs representing this locus (Kohara 1996), resulting in the possible presence of an intron (52 nucleotides) within the central region of the transcript. Although the 3′ end of thetts-1 transcript can be folded in a manner consistent with the three terminal helices seen in the vertebrate model of telomerase RNA (and a degenerate H box sequence lies in the proper place), no matching ACA box can be found three nucleotides in from the 3′ end.
We would not expect high levels of telomerase activity in the dauer where the germ line remains undeveloped and hence predict no increased requirement for the telomerase template RNA. Accordingly, six tags were observed for the putative C. elegans telomerase reverse transcription protein DY3.4 (SWISS-PROT accession O45321) in the mixed stages and none in the dauer. This seems inconsistent withtts-1 being a telomerase component, but it remains possible that tts-1 has a function involving binding to the telomeres or telomere-associated proteins. It is possible thattts-1 may havea chromosome-protective function, especially in the dauer stage.
A second transcribed telomerase-like sequence, tts-2 (dauer 5; mixed 10), was identified through sequence similarity totts-1 (GenBank accession number Z48795 17488–18489). The tts-2 transcript also contains degenerate telomeric repeat sequences but lacks any of the structural signatures of other telomerase RNA subunits. The two cDNA clones deriving fromtts-2 also showed alternative splicing with one of the transcripts excluding the telomeric repeat sequence.
The most abundant dauer and mixed-stage–specific transcripts are listed in Table 3. Except where noted, these tags were not detected in the other library. The most highly expressed dauer-specific transcript is F38E11.2/hsp-12.6, encoding a small heat shock protein of the α-crystallin family. Small heat shock proteins have been implicated in oxidative and mechanical stress responses in many organisms, but in C. elegans hsp12.6 is not induced by biological stress through exposure to heat or chemical agents (Leroux et al. 1997). It has been detected at high levels in synchronized populations arrested at the L1 stage by starvation. Therefore, hsp12.6 may be specifically induced during times of developmental arrest. Other heat shock genes such asHsp90 (dauer 578: mixed 172) and Hsp70 (dauer 166: mixed 172) are also expressed within the dauer stage, although not in a dauer-speific manner, consistent with previous studies (Dalley and Golomb 1992; Cherkasova et al. 2000). The two highly abundant, dauer-specific G-protein coupled receptors (Table 3) are candidates for chemoreceptors involved in triggering dauer exit. It is striking that 15 of the 20 most abundant dauer-specific proteins are novel, lacking similarities with known proteins indicative of putative functions.
The Twenty Most Abundant Dauer and Mixed-Stage-Specific SAGE Tags that Have Been Correlated to a Transcript
Chromosomes may be packaged differently in the dauer stage. Tags for histone H1-like genes C18G1.5 and M163.3/his-24 were prominent in the mixed-stage profile. Histone H1-like genes C30G7.1 and F22F1.1 showed increased dauer expression whilst C18G1.5 was reduced (Fig.3A). The 40-fold enrichment of tags for the histone H1 variant C30G7.1 (only 33% and 31% identical to the histones encoded by M163.3 and C18G1.5, respectively) suggests that dauer chromatin is altered, possibly to reduce overall transcription rate or assume a structure less susceptible to damage. Two different M163.3/his-24 transcripts were detected, with the longer transcript more highly represented in the mixed-stage profile than in the dauer (dauer 10; mixed 31). This is consistent with a previous observation that a longer 1.3-kb transcript for M163.3 is present within male germ cells (Sanicola et al. 1990), a tissue represented only in the mixed-stage library. The D2096.8 nucleosome assembly protein transcripts were also present in two different forms, one of which displayed a 10-fold increase in tag abundance in the dauer profile. The differential mRNA forms may represent splice variants or alternative transcriptional termination.
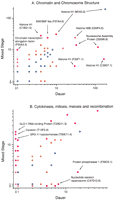
Relative abundance of dauer and mixed-stage tags for genes from specific cellular processes (Costanzo et al. 2000). (blue) No significant expression difference between the two sets. (orange) Significance of P ≤ 0.05; (red) significance of P ≤ 0.01, as determined by the G test (Sokal and Rohlf 1995).
Other genes more highly represented in the dauer SAGE library include the C47D12.8 DNA repair endonuclease, further indicating possible increased levels of proteins involved in maintaining DNA integrity during diapause (Fig. 3B). Tags for the F56C9.1 protein phosphatase I (PP1C) γ protein, also more prominent in the dauer profile, has been postulated to negatively regulate of entry into mitosis (Doonan and Morris 1989; Ohkura et al. 1989). A high level of activated PP1C protein would be antagonistic to the role of the DAF-2 insulin receptor family protein, which is a negative regulator of genes involved in oxidative stress (Honda and Honda 1999) and dauer entry. Thespo-11 gene (Dernburg et al. 1998), the gld-1 gene (Jan et al. 1999), and the gene for caveolin (Scheel et al. 1999), all involved in meiotic and germ-line function, were not observed within the dauer stage, where the germ line does not develop.
To explore the fecundity of the SAGE approach to detect novel expressed sequences, the 10 most abundant tag species that could not be correlated to the sense strand of a predicted transcript were examined. Three were antisense tag species from mitochondrial genes. Three other tag species were from genes represented in EST data sets but not currently in the annotated genomic data: the Ribosomal L27A protein (dauer 505; mixed 305), a nuclear cytochrome C oxidase (dauer 120; mixed 143), and a gene of unknown function (dauer 56; mixed 71). Two tag species were single-base variants of abundant tags, possibly representing sequence heterogeneity in the N2 populations. One species, CATGCGACTTCTGA (dauer 94; mixed 4), possessed a single mismatch with the tts-1 tag, and its relative abundance in the two profiles was consistent with that observed for tts-1. Another correlated with a single-base variant of the highly expressed F15A2.1 collagen gene. One tag species correlated with the mitochondrial small ribosomal RNA gene (dauer 102; mixed 6; Okimoto et al. 1992), and the remaining species, CATGCGCTAAAAAA, is most likely the result of the CATG restriction site being too close to the polyadenylation site for an unambiguous gene assignment. Therefore, we can account for highly expressed unmapped tags as deriving from the transcriptome.
We investigated whether SAGE data also could be used to detect alternative splicing. Different (15,596) tag species could be correlated to the 10,581 transcripts unambiguously assigned to SAGE tags (excluding the 549 transcripts from 326 genes known to have alternatively spliced transcripts (Durbin and Thierry-Mieg 1991). Two thousand six hundred sixty-five transcripts had two tag species present, 721 had three, 194 had four, and 76 transcripts had five or more tag species present (if singleton tags are ignored these numbers become 1161, 159, 74, and 24, respectively). Many of these tag variants may be resulting from incomplete NlaIII digestion during SAGE library construction. However, 153 genes were candidates for possessing alternate transcript structures as these showed an altered ratio of different tag species between the mixed and dauer populations, as determined by the Fisher exact test (P ≤ 0.05). In addition, nine of the genes known to have alternatively spliced transcripts had altered transcript representation between the dauer and mixed-stage profiles, for a total of 162 such genes.
Our findings represent the first large-scale investigation into gene expression during C. elegans diapause. The mixed-stage profile provides a reference point for future study of specific life stages or of mutant strains. Furthermore, our analysis has revealed some surprising and previously unsuspected differences leading to new hypotheses about the nature of the long-lived dauer form. The most abundant dauer-specific transcript shows strong similarity to the vertebrate α-B-crystalline/hsp20, which is predicted to function as a stabilizer of protein structure and cellular integrity (Leroux et al. 1997). At least three transcripts possibly affecting the structure or stability of chromatin (tts-1, a variant histone H1 gene and an isoform of a nucleosome assembly protein) are much more prominent in the dauer than in the nondauer expression profile, suggesting that chromatin structure may be altered to improve stability, to reduce overall gene expression, or possibly both. Finally, antisense RNA corresponding to mitochondrial gene transcripts is present in both populations and may play a role in preventing continuous translation of mitochondrial transcripts.
METHODS
Nematode Growth
Wild-type (N2) C. elegans were grown asynchronously in three 250-mL liquid cultures containing 4% Escherichia coliχ1666 in S medium, harvested by centrifugation, then allowed to settle for 30 min at 25°C to digest any E. coli present in the gut (Epstein Henry et al. 1995). The pooled, mixed-stage populations contained stages in the estimated ratio 20 L1 : 2 L2 : 1 L3 : 1 L4 : 1 adult. N2 dauer larvae were purified by the sucrose flotation method (Epstein Henry et al. 1995) from two starved liquid cultures grown at 25°C. The final preparation of dauer larvae did not contain more than one animal of any other stage per 500 dauer larvae. Settled worms were quick-frozen in small pellets by dropping into liquid nitrogen and stored at −80°C.
RNA Preparation
Frozen animals were crushed in liquid nitrogen using a mortar and pestle for 5–10 min. Total RNA was isolated by the guanidinium isothiocyanate : phenol method (Chomczynski and Sacchi 1987). The yields of total RNA from 5 mL of settled dauer larvae and from 3.5 mL of settled mixed-stage populations were ∼12 and 20 mg, respectively.
Generation and Analysis of SAGE Tags
SAGE libraries were produced as previously described (Velculescu et al. 1995) (detailed SAGE protocol available at www.sagenet.org), and the resultant clones were used to generate 15,371 successful sequencing reads performed on ABI 377 automated DNA sequencers. Quality assessment and clipping of the reads was performed by using PHRED(Ewing et al. 1998) and vector_clip (Staden et al. 2000). Sage tags derived from the linker sequence used in SAGE library construction were removed. Conceptual transcript sequences were predicted from the C. elegans genome sequence, obtained fromC. elegans ACEDB version WS9 and the mitochondrial genome (GenBank accession number X54252). Where possible, 5′ and 3′ untranslated region (UTR) sequences were derived from the coordinates of similarity to C. elegans EST sequences. In the absence of EST data, up to 270 bases were added to the 5′ end of genes and up to 460 bases added to the 3′ end of genes. No estimated UTR sequences extended into other gene predictions. Lengths added were sufficient to encompass the UTRs of 99% of the genes studied, as estimated from EST data. Conceptual SAGE tags were generated for all transcript sequences by determining the presence of the NlaIII restriction site and the subsequent 10 3′ base pairs. Observed tags were correlated to conceptual transcript sequences. Where a tag matched two or more transcripts, the tags corresponding to the most 3′ CATG site were used to resolve the ambiguity, where possible. Estimated UTR sequences were not used to map tags to antisense transcripts. The SAGE data and the gene correlations are available from http://elegans.bcgsc.bc.ca/SAGE.
When comparing the abundance of mRNA species corresponding to SAGE tags, we used the overall expression profile reflected in each library. For example, the tts-1 tag species in the dauer library was present 4329 times among the total of 152,314 tags (2.8%). In the mixed-stage library, this tag was present 215 times among a total of 148,324 (0.14%). Hence, when the two profiles are compared, this tag is 20 times more abundant in the dauer than in the mixed-stage profile. This is not to say that there is necessarily 20 times more of this RNA per cell in the dauer.
Acknowledgments
Under a licensing agreement between the Johns Hopkins University and Genzyme, the SAGE technology was licensed to Genzyme for commercial purposes, and V.E.V. is entitled to a share of royalty received by the University from sales of the licensed technology. The SAGE technology is freely available to academia for research purposes. V.E.V. is a consultant to Genzyme. The University and researchers (V.E.V.) own Genzyme stock, which is subject to certain restrictions under University policy. The terms of these arrangements are being managed by the University in accordance with its conflict of interest policies. This work was partially supported by DHHS grants GM60151 and AG12689 to D.L.R., grants from the Howard Hughes Medical Institute and NIH National Human Genome Research Institute for S.R.E., and an NIH genome sciences training grant to S.L.S. We thank Peter Candido for helpful discussions on heat shock protein data.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵7 Corresponding author.
-
E-MAIL sjones{at}bcgsc.bc.ca; FAX (604) 877-6085.
-
Article published on-line before print: Genome Res., 10.1101/gr.184401.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.184401.
-
- Received February 15, 2001.
- Accepted May 14, 2001.
- Cold Spring Harbor Laboratory Press








