
Detection of Spurious Interruptions of Protein-Coding Regions in Cloned cDNA Sequences by GeneMark Analysis
Abstract
cDNA is an artificial copy of mRNA and, therefore, no cDNA can be completely free from suspicion of cloning errors. Because overlooking these cloning errors results in serious misinterpretation of cDNA sequences, development of an alerting system targeting spurious sequences in cloned cDNAs is an urgent requirement for massive cDNA sequence analysis. We describe here the application of a modified GeneMark program, originally designed for prokaryotic gene finding, for detection of artifacts in cDNA clones. This program serves to provide a warning when any spurious split of protein-coding regions is detected through statistical analysis of cDNA sequences based on Markov models. In this study, 817 cDNA sequences deposited in public databases by us were subjected to analysis using this alerting system to assess its sensitivity and specificity. The results indicated that any spurious split of protein-coding regions in cloned cDNAs could be sensitively detected and systematically revised by means of this system after the experimental validation of the alerts. Furthermore, this study offered us, for the first time, statistical data regarding the rates and types of errors causing protein-coding splits in cloned cDNAs obtained by conventional cloning methods.
The draft sequencing of the human genome is almost completed, providing a wealth of information. Molecular biologists have long dreamed of establishing a complete catalog of the full complement of human proteins. However, decoding the human genome sequence to obtain the protein sequences is not always straightforward because of the presence of introns that divide protein-coding sequences (CDSs) into small pieces in the genome. Although many efforts have been made to develop reliable programs for prediction of CDSs from the genomic sequence alone, the prediction specificity and sensitivity are not sufficiently high to allow us solely to rely on them in the case of mammalian genes (Claverie 1997; Thanaraj 2000). Therefore, human cDNA sequencing, not collection of expressed sequence tags (ESTs) but sequencing of entire cDNAs, is accepted as an important aspect of human genome sequencing, especially for prediction of protein sequences. More important, these sequence-confirmed, expression-ready cDNA clones of human genes provide us with a set of versatile reagents indispensable for gene expression studies. Considering the importance of human cDNA analysis as described above, we have carried out a cDNA sequencing project for the prediction of protein sequences encoded by unidentified human genes during the past 5 yr (Nomura et al. 1994). Long cDNAs derived from brain RNA are current targets of our project; the total number of cDNAs sequenced exceeds 1500 (in terms of the number of nucleotide residues, > 7 Mb; Nagase et al. 2000). Through this project, we have come to realize that the prediction of protein primary structures from cDNA sequences is not so straightforward as it was supposed at the beginning. The problems we encountered originated from the fact that cDNA is nothing but an artificial copy of mRNA. For example, in vitro cDNA synthesis has risks of generating artifactual copies of mature mRNAs: cDNA synthesis can be primed not only from poly(A) tail at the 3′-extremity of the mRNA but also from an A-rich internal site such as the site of a repetitive element (Aaronson et al. 1996; Bonaldo et al. 1996); reverse transcription is known to be an error-prone process and thus might generate mutations in the cDNA (Bebenek et al. 1993); intron sequences may be reverse transcribed and retained in cDNA because total cellular poly(A)+ RNA usually contains a considerable amount of heteronuclear RNA including premature forms of cytoplasmic mRNA (Hillier et al. 1996; Wolfsberg and Landsman 1997). All of these problems can lead to serious misinterpretation of cDNA sequences with respect to the protein structures encoded by them. Although in conventional cDNA analysis these artifacts can be eliminated through analysis of multiple clones for a single gene, the same approach is not feasible for comprehensive analysis of a large number of cDNAs, primarily because of various practical limitations. Therefore, the matter of how to systematically detect and remove spurious sequences from a collection of cloned cDNA sequences is a matter of urgent concern in the implementation of comprehensive cDNA sequencing projects. Nevertheless, the matter of these possible artifacts being encountered in cDNA analysis has been only implicitly addressed to date, although it has been seriously anticipated.
In this context, as the first step, we focused our efforts on identification of spurious interruptions of CDSs in cloned cDNAs because they usually cause more serious problems than missense mutations. In this study, we developed an alerting system targeting spurious CDS interruptions based on GeneMark analysis, which was originally developed for assignment of CDSs along a prokaryotic genome (Borodovsky and McIninch 1993). We exploited this method of analysis in evaluation of cDNA sequences because the logical framework for assigning CDSs in cDNA sequences is almost identical to that for prokaryotic gene finding. In fact, Borodovsky et al. (1994) previously mentioned that GeneMark analysis is well suited to detect putative frame-shift sequence errors. To evaluate the prediction accuracy and sensitivity of this alerting system, 817 cDNA sequences deposited in public databases by us were subjected to GeneMark analysis. As a result, 198 sites in 159 cloned cDNAs were identified as sites suspected of having a CDS split. Of these, 133 sites suspected of having a CDS split were experimentally examined by reverse transcription-coupled polymerase chain reaction (RT-PCR) method. The results indicated that our alerting system based on GeneMark analysis serves as a reliable tool to detect cDNA clones for which sequence revision is required. More interesting, the results obtained in experimental verification of CDSs splits for the first time have clarified statistically the frequency of occurrence of various types of spurious CDS splits in human cDNAs obtained by conventional cloning methods.
RESULTS
Detection of Putative Coding Interruptions in Cloned cDNA Sequences by GeneMark Analysis
Interruptions of a CDS are classified into two types, split and truncation. In this study, we focused our main efforts on prediction of CDS split. Our alerting system targeting putative CDS splits in cloned cDNAs is based on the assumption that an authentic human mRNA carries only one CDS. Because a spurious CDS split always results in generation of multiple CDSs in a single cDNA, the occurrence of multiple CDSs in a cloned cDNA sequence can be taken as a warning sign of artifacts, although some mRNAs may carry multiple CDSs even in their authentic forms. In this alerting system, prediction of CDSs, not the open reading frame (ORF), serves as a critical step; ORFs can be unambiguously defined only on the basis of the nucleotide sequences, but ORFs do not always correspond to CDSs. Although long ORFs (> 500 nucleotide residues [nt]) are very likely CDSs, the likelihood of ORFs being CDSs greatly decreases with the decrease in their size, whereas short ORFs appear more frequently in cDNA sequences than do long ones. To detect possible CDSs among ORFs, we exploited the GeneMark program in this study. The GeneMark program identifies gene regions on the basis of statistical patterns of CDSs described in terms of Markov models (Borodovsky and MacIninch 1993), and a modified version of GeneMark analysis, termed recursive GeneMark analysis, could reduce the prediction error rate down to 1.7% in prediction of genes along the cyanobacterial genome (Hirosawa et al. 1997). It should be noted that the logical framework required for prediction of a possible CDS in human cDNA is rather similar to that for prokaryotic gene finding than that for exon prediction of eukaryotic genes because CDSs in both prokaryotic genes and human cDNAs are uninterrupted at least in principle. However, the application of GeneMark analysis for detection of a spurious CDS split required the following modifications to the original method: only the sense-strand sequences of cDNAs are used as a training data set; for prediction of CDSs, CAG, AAG, GAG, AGT, AGC, AGA, and AGG, besides ATG, are taken to be virtual start codons for convenience. The second modification was introduced so as not to miss split CDSs where a conventional initiation codon (ATG) is absent because GeneMark analysis requires a set of start and termination codons for identification of a CDS.
We applied GeneMark analysis in examining 817 cDNA sequences we had determined and deposited in public databases (Nomura et al. 1994;Nagase et al. 1998; Nakayama et al. 1998). The genes corresponding to these cDNAs were systematically designated KIAA plus a four-digit number. It must be noted that the cDNA clones analyzed here were selected on the basis of meeting three different criteria: the group I cDNAs (KIAA0001-KIAA0268, 268 cDNA sequences) were synthesized from cytoplasmic poly(A)+ RNA derived from a human immature myeloid cell line (KG-1 cells) and were selected on the basis of the coincidence of the cDNA sizes with the corresponding mRNA sizes (i.e., the cDNA size being larger than 90% of the corresponding mRNA size;Nomura et al. 1994); the group II cDNAs (KIAA0444-KIAA0481 and KIAA0483-KIAA0510, 66 cDNA sequences) originated from human brain poly(A)+ RNA and were selected on the basis of specific gene location on chromosome 1 (Seki et al. 1997); the group III cDNAs (KIAA0269-KIAA0443 and KIAA0511-KIAA0818, 483 cDNA sequences) were derived from human brain poly(A)+ RNA and were selected on the basis of their capability to produce large proteins in an in vitro system (Ohara et al. 1997; Nagase et al. 1998; Nakayama et al. 1998). Taking the sources and the criteria of clone selection for each group into consideration, we hereafter designate the groups I, II, and III as KG1/nearly full length (NFL), Brain/Chromosome 1 (Ch1), and Brain/long CDS (LCDS), respectively. Among these 817 cDNA sequences, 36 sequences (KIAA0097-KIAA0099, KIAA0169, KIAA0177, KIAA0242, KIAA0263, KIAA0302, KIAA0440-KIAA0443, KIAA0698-KIAA0710, and KIAA0799-KIAA0809) had already been revised to exclude possible artifacts before depositing these sequence data in the databases. Thus, in the case of these genes, the sequences of the cloned cDNA before revision were subjected to GeneMark analysis in this study. The results regarding prediction of CDS split are summarized in Table 1. The cDNA sequences carrying multiple predicted CDSs were considered to be candidates including spurious CDS split(s), and their rate of occurrence was 19.5% among these KIAA cDNAs. Notably, 52% (83/159) of these cDNAs with multiple predicted CDSs were predicted to include at least single small CDSs (< 500 nt) in spite of the fact that they were only ambiguously assigned as CDSs without using this program. Twenty-four cDNA sequences were predicted to contain no CDS, and in fact, there were no long ORFs (> 500 nt) in these cDNAs except for KIAA0033. Interestingly, all of these clones belonged to groups I and II, where the protein-coding capabilities of the cDNA clones had not been experimentally tested at all. As for the KIAA0033 clone, however, it contained a long ORF (about 1 kb), but the statistical characteristics of its nucleotide sequence deviated significantly from the shared characteristics of the CDSs in human cDNAs. Nevertheless, this ORF was considered to be an actual CDS because the predicted protein sequence of KIAA0033 showed significant homology to several entries in the OWL database and contained a known protein domain (Pfam entry, PF00597; Bateman et al. 1999). Thus, KIAA0033 was considered to be a rare case of false-negative detection of CDSs by GeneMark analysis.
Prediction of the Numbers of CDSs in KIAA cDNAs by GeneMark Analysis
According to the apparent positional relationship of the split CDSs predicted by GeneMark analysis, CDS interruption could be tentatively classified into the occurrence of a stop codon (i.e., a nonsense mutation), a frame-shift, or a large insertion (probably due to a retained intron or alternative splicing). Figure 1shows the typical GeneMark profiles of these three different types of CDS split. The typical GeneMark profile of a cDNA sequence with a large insertion also demonstrates how CDS truncation appeared in terms of the GeneMark profile; an alert of CDS truncation was issued if the 5′-most end of the cDNA sequence was predicted to be involved in a CDS because the complete CDS should be preceded by a 5′ noncoding sequence. Since GeneMark analysis specified possible CDSs in terms of the nucleotide residue numbers of the cDNA sequences, the predicted CDSs are indicated by gray bars in Figure 1 (Borodovsky et al. 1994). Although the original results of GeneMark analysis include GeneMark profiles in six reading frames (i.e., three reading frames in the sense and three in the antisense strand), only the profiles of the three reading frames in the sense strand are shown in Figure 1.
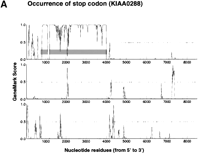
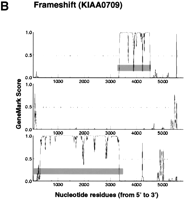
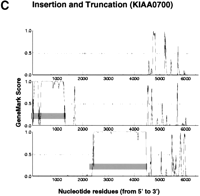
GeneMark profiles of typical CDS interruption. GeneMark profiles were generated according to the previous report by Borodovsky et al (1994). Small vertical bars in each profile indicate the positions of termination codons, and the predicted CDSs are highlighted by horizontal gray bars. The GeneMark profiles for three reading frames only along the sense strand are shown here.
Evaluation of the GeneMark Predictions by Comparison of Cloned KIAA cDNA Sequences with Their Homologous Ones Independently Determined
Among the 817 cDNA sequences analyzed in this study, 176 were found to be orthologous or identical to those independently determined by other groups and registered in OWL protein database (release 31.4). In this analysis, the cDNA sequences from other mammals were considered to be orthologous if the predicted protein sequence exhibited significant sequence identity (> 80%) to the entire KIAA sequences. Because the same criterion is used for assigning alias names to KIAA genes, KIAA genes with alias name(s) in the Human Unidentified Gene-Encoded (HUGE) protein database (http://www.kazusa.or.jp/huge; Kikuno et al. 2000) correspond to those having orthologous and/or identical gene entries in the database. Under the assumption that these independently determined cDNA sequences are authentic, we estimated the frequencies of occurrence of false-positive and false-negative alerts for CDS split by GeneMark analysis. For the estimation, we first checked the results of a homology search of KIAA gene products of interest in the Gene/Protein characteristic table in the HUGE database. When KIAA clones encoded shorter proteins than the entries in OWL database, we then compared the nucleotide sequences of the KIAA clones with those of their corresponding gene entries to check the number of CDSs encoded in cloned KIAA sequences. KIAA clones encoding the partial protein sequences in single CDSs probably missed 5′- and/or 3′-end portion of their full-length cDNAs because of incomplete reverse transcription and/or the presence of retained intron(s) at the 5′- and/or 3′-extreme end. Table 2 shows the number of estimated correct and false alerts for CDS split. The false-positive rate (the number of sites found to include no CDS split/the number of sites triggering an alert) was found to be 32.2% (10/31), while the false-negative rate (the number of actual CDS split sites/the number of clones that did not trigger an alert) was about 3.3% (5/150). Comparison of our cloned cDNA sequences with the corresponding sequences deposited in databases by others revealed that the failure to detect CDS interruption was mainly because of the small size of split CDSs. Interestingly, in addition to 5′-truncation (about 24% of the clones examined), we observed several cases in which 5′- and/or 3′-intron sequence interrupted authentic CDSs (about 9%). Since only a single CDS was present in each of these KIAA cDNAs, GeneMark analysis was unable to trigger an alert for this type of CDS interruption.
Prediction Accuracy and Sensitivity of GeneMark Analysis Estimated from Comparison of KIAA Sequences with Those Independently Determined by Others
Experimental Validation of Cloned KIAA cDNA Sequences by GeneMark Prediction
The results described above indicated that the alerting system assisted by GeneMark analysis is of great help for detection of any spurious split of CDSs in cDNA sequence data. However, we could not solely depend on GeneMark analysis because it is not completely free from false alerts. Therefore, we considered it reasonable to use this alerting system just for selecting cloned cDNA sequences to be experimentally examined. In practice, regions predicted to contain CDS split were analyzed by direct sequencing of the corresponding products obtained by RT-PCR (see Methods). In these experiments, we analyzed only the most predominant RT-PCR product(s) unless otherwise noted, and their structures were taken as authentic ones. This was based on the assumption that immature and/or biologically insignificant transcripts are always likely to be present in lower quantity as compared to mature and biologically significant ones.
The number of predicted CDS split sites among the 817 cDNA sequences analyzed was 198 in 159 cloned cDNA. All of these sites were subjected to experimental validation, and as a result, 100 sites in 89 clones were confirmed to require revision. However, the nucleotide sequences of predominant RT-PCR products obtained corresponding to 33 sites in 32 clones were proven to be identical to the cloned ones. All the gel images and the experimental conditions of the RT-PCR products are accessible at http://www.kazusa.or.jp/∼hirosawa/interruption/entrance.html. The remaining sites (65 sites) were not characterized yet because the purity and the amount of the RT-PCR products were not sufficient for direct sequencing. Comparison of the nucleotide sequences of the cDNA clones with those of the corresponding RT-PCR products revealed that the false-positive rate in the case of GeneMark prediction was 24.8% (33/133), which was slightly lower than that in the case of the method of analysis described in the above section.
Interestingly, RT-PCR products of several genes from KG-1 mRNA and human brain mRNA gave distinct band patterns in terms of both size and quantity, which suggested the presence of alternative forms in these mRNA sources (Fig. 2). Because the KIAA genes shown in Figure 2 were originally identified in a KG-1 cDNA library, it was no surprise that the KG-1 mRNA bands observed in gels were always more prominent than those observed in the case of brain mRNA. The KG-1 mRNA was prepared from the cytoplasm, whereas the brain mRNA was derived from whole cells in the brain. Nonetheless, the RT-PCR products from KG-1 cells sometimes contained larger bands than those derived from the brain mRNA, besides showing common major bands (Fig. 2). Therefore, the differences in gel patterns of the RT-PCR products appeared to be due to cell-type specific alternative splicing rather than contamination of intron-retaining mRNA.
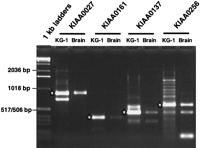
Gel images of RT-PCR products generated from two different mRNA sources. RT-PCR products obtained from KG-1 and brain mRNAs were run on a 1% agarose gel. Because clones for all of these KIAA genes were originally found in a KG-1 cDNA library, the RT-PCR products from KG-1 mRNA, which are marked by white stars in this figure, were directly sequenced.
Table 3 summarizes the results of direct sequencing of major RT-PCR products in regions containing a split CDS. All of the KIAA cDNA sequences revised by RT-PCR experiments are accessible through the HUGE database (http://www.kazusa.or.jp/huge; Kikuno et al. 2000). Importantly, although only two cDNA clones for KIAA0302 and KIAA0324 were found to trigger an alert of GeneMark analysis due to sequencing errors, the sites triggering alerts in other KIAA cDNAs did not contain errors in sequencing as far as we saw. Thus, we omitted KIAA0302 and KIAA0324 clones from a list of clones used for the subsequent analysis because this cloned cDNA did not contain a split CDS. The causes of the CDS splits can be classified as follows: insertion (> 3 bp), deletion (> 3 bp), frame-shift mutation (< 3 bp), nonsense mutation, and their combination (Table 3). The CDS splits in the last class resulted from a combination of more than two causes; while multiple insertions were observed frequently, it is interesting to note that clones for KIAA0007 and KIAA0443 included two frame-shift mutations and two nonsense mutations in their single CDS splits, respectively. Each insertion or deletion was further classified according to whether or not GT and AG dinucleotides were present at the upstream and downstream boundaries, respectively. An insertion having GT/AG at the boundaries was considered to be an intron (Mount 1982). Since the clones analyzed were selected on the basis of different criteria, the types of CDS splits are listed separately according to each of the cDNA groups in Table 3. Notably, no insertions in the KG1/NFL cDNAs were found to have boundary sequences following the GT-AG splicing rule. Table 4 shows the nucleotide sequences at the sites of frame-shift mutations found in this study. Interestingly, frame-shift mutations frequently occurred within regions where there were homopolymeric runs or dinucleotide repeats, essentially in agreement with previous observations regarding the hot spots for mutations caused by human immunodeficiency virus–derived reverse transcriptase (HIV-RT; Bebenek et al. 1993).
Classification of the Observed CDS Splits
Observed Sequences at the Sites of Frame-Shift Mutations
DISCUSSION
Comprehensive human cDNA analysis is considered to be important for identification of transcribed regions along the human genome, prediction of primary structures of human gene products, and preparation of a set of expression-ready human cDNA plasmids for future functional analyses. The first two reasons have been enthusiastically pursued as gene discovery games to date, and these lines of information are indispensable components of the human genome information. The practical importance of the third subject, however, is expected to increase after the complete genome structure becomes available. For all these purposes, the accuracy of cDNA sequence data is highly critical. Accordingly, we have made every effort to minimize sequencing errors and, consequently, have accumulated highly accurate cloned cDNA sequences. However, we have realized that it is impossible to completely eliminate spurious cDNA sequences from the cloned ones because comprehensive cDNA analysis is routinely done using randomly sampled cDNA clones on a single-clone-for-single-gene basis. Although multiple cDNA clones derived from the same gene are isolated and analyzed in order to eliminate artifacts and spurious sequences in the course of conventional cDNA sequencing, the same approach cannot be applied in comprehensive cDNA analysis mainly because of limitations of time, cost, and labor to spare. Although having been long anticipated, this problem has not been explicitly addressed to date and has yet to be solved. Direct sequencing of RT-PCR products enables us to determine cDNA sequences without spurious ones, as shown in this study, but it cannot provide us with sequence-verified cDNA clones to be used as reagents for functional analysis.
In this article, we have described an alerting system targeting CDS splits using an accurate CDS prediction program based on GeneMark analysis. For this purpose, exon prediction programs for human genes are obviously unsuitable because they are primarily designed to predict splice sites, and no splice sites should be found in cDNA at least in principle (Thanaraj 2000). However, statistical sequence analysis based on Markov models is widely accepted as the method of choice for assessing protein-coding potential along DNA sequence (Hirosawa et al. 1997; Audic and Claverie, 1998; Delcher et al. 1999), and the GeneMark program was used as a representative program in this study. The use of GeneMark analysis for inspection of sequencing errors is an idea already known (Borodovsky et al. 1994); however, here we present the first report of the actual application of GeneMark analysis for evaluation of sequence data obtained in comprehensive cDNA analysis. The results indicated that this alerting system faithfully detected CDS splits that could not be unambiguously identified unless GeneMark analysis was applied. At the same time, the results revealed that most of the failures in detection of a CDS interruption were due to missing short split CDSs or the presence of retained intron(s). This observation implies that there would be difficulty in further lowering the false-negative rate without raising the false-positive rate because discrimination of intron sequences from untranslated ones in mature mRNA and reliable detection of very short CDSs (< 100 nt) cannot be achieved by means of GeneMark analysis. In this respect, applying a combination of intrinsic and extrinsic approaches for detecting CDSs would hopefully be a way to further improve the sensitivity of the alerting system without sacrificing accuracy, as previously described (Borodovsky et al. 1994).
To our knowledge, this study is also the first one in which the causes of CDS splits in randomly sampled human cDNAs have been examined. When the causes of CDS splits were classified into four categories, that is, nonsense mutation, frame-shift mutation, and large insertion/deletion, 71% of the sites experimentally proven to have a CDS split (100 sequences) were those due to insertion/deletion. A possible origin of a large insertion is a retained intron in immature mRNA present in the nucleus. In fact, no insertions that followed the GT-AG rule for the splicing boundary were found in the KG1/NFL cDNAs because they were synthesized from cytoplasmic RNA. Thus, the insertions with the GT-AG boundary in the brain/chromosome 1 and the brain/long CDS cDNAs (23 out of 35 insertions, 65.7%) were likely to be retained introns, whereas, in case of the remaining insertions (12 out of 35, 34.3%) and all of the deletions, the CDS splits could not be ascribed to either retained introns or errors of reverse transcription. A plausible explanation is that these cDNAs were derived from minor alternative forms of the corresponding mRNAs. If this is the case, insertions or deletions in the cloned cDNA sequences, except for insertions following the GT-AG intron boundary rule, are not spurious but are naturally present in a minor population of mature brain mRNAs. In fact, mistakes in splicing of RNA from complex genes are considered not to be uncommon (Sharp 1994). However, as described by Kozak (1996), it should be also kept in mind that the predominance of a particular mRNA species might not always guarantee its authenticity as a biologically significant mature form. Accordingly, the biological meanings of these insertions/deletions, including the large insertions apparently regarded as introns, cannot be assigned conclusively without more careful studies in the future.
Our alerting system targeting spurious cDNA sequences was based on the assumption that there is a single CDS for a single cDNA. However, the results indicated that this assumption was not always true; as described above, we observed that alternative splicing sometimes controlled the number of possible CDSs in a single mRNA. In these cases, only the CDS most upstream will be translated in vivo, and thus, the C-terminal truncated product of the alternative form will be produced. Accordingly, in some instances, the alert obtained by GeneMark analysis may be regarded as an indication of the presence of alternative forms of mRNA rather than that of artifacts. Hanke et al. (1999) recently reported that alternative splicing of human genes is more the rule than the exception as determined through analysis of data in the human EST database. Surprisingly, Hanke et al. (1999) suggested that the level of alternative splicing indicated by EST analysis alone (alternative splicing occurs in 34% of the proteins studied) might be a significant underestimate. Since the number of exons included in a transcript increases with increasing size of the transcripts in general, KIAA cDNAs are likely to have more alternative forms than the conventional cDNAs because KIAA cDNAs are usually much longer than the EST cDNAs. Because alternative splicing frequently generates functional diversity in the case of the product of a single gene and since information on alternative splicing can be obtained only through cDNA analysis, characterization of alternative forms of transcripts of KIAA genes will become an important subject of our further cDNA studies. In this respect, the alerting system described in this report may be useful also for this purpose because it enables us to detect dormant CDSs in a particular transcript of interest.
In contrast to insertion/deletion, nonsense and frame-shift mutations were most likely artifacts generated during reverse transcription. If the error rate of the modified Moloney murine leukemia virus-derived reverse transcriptase (MMLV-RT) lacking RNase H activity used in construction of our cDNA library construction, Superscript II (Life Technologies), is almost identical to that of wild-type MMLV-RT (1/3.7 × 104; Ji and Loeb 1992), one mutation is expected to be present for every seven to eight cDNA sequences or for every 14–15 CDSs in our data, as the average lengths of our cDNAs and CDSs were about 5 kb and 2.5 kb, respectively. Although these copying errors in a CDS result in silent, missense, nonsense, and frame-shift mutations, we could detect only nonsense and frame-shift mutations in CDSs in this study. Taking into consideration the coverage of experimentally verified sites among the sites that triggered an alert (67%) and the alerting sensitivity (81%), the frequency of occurrence of nonsense and frame-shift mutations in CDS were estimated to be, on average, approximately 1/3 × 105 and 1/4 × 104, respectively. These results indicated that the rate of frame-shift error in the human cDNAs was very close to the overall error rate of MMLV-RT previously estimated by Kunkel's method, although the frame-shift mutations are reported to occur less frequently than single-base substitutions in the course of reverse transcription by HIV-RT (Ji and Loeb 1992). More interestingly, homopolymeric runs are reported to serve as hot spots of reverse transcription errors in the case of HIV-RT (Bebenek et al. 1993). Since 80% of the mutations detected in this study were located at homopolymeric runs (≥ 2 nt) or dinucleotide repeats (Table 4), this is also the case for the modified MMLV-RT. This observation suggests that the prevalence of these error-prone hot spots in mRNA greatly affects the error rate observed in cloned cDNA sequences.
In this study, we developed an alerting system targeting spurious CDS interruptions in cloned cDNA sequences and evaluated it through analysis of our own data. It must be noted that these data were obtained from a collection of large cDNAs (> 4 kb). In our experience, large cDNAs derived from total-brain RNAs contained more problematic sequences than shorter cDNAs: because cDNA clones carrying intermolecularly ligated inserts (i.e., chimeras) or retained intron(s) inevitably become larger than the authentic ones, the number of these artificial clones increases as cDNAs become larger. In addition, large poly(A)+ RNA prepared from tissues (> 4 kb) generally contained more premature heteronuclear poly(A)+ RNA than small ones, and therefore, the prevalence of cDNA clones retaining introns becomes higher in case of large cDNA clones. Thus, our sequence data for large cDNAs serves as a unique resource for retrieving information as to how often and what kind of spurious cDNA clones occur, as these lines of information are hard to obtain through analysis of small cDNA clones unless an extremely large number of them is entirely sequenced.
Once the complete human genome sequence becomes available, cDNA sequences will surely be used for interpretation of the genomic sequence. Comparison of cDNA sequences and the genome sequence will provide a wealth of information as to how human genes are organized and might uncover new types of regulatory mechanisms controlling gene expression. For example, posttranscriptional modification of the genomically encoded sequence of an mRNA, known as RNA editing, might be more common for mammalian genes than assumed previously as reported recently by Paul and Bass (1998). cDNA analysis is an important means by which to examine this issue, and thus the data must be as accurate as possible and hopefully free from spurious sequences. The alerting system developed in this study is expected to make a major contribution to the improvement of the quality of cDNA sequence data obtained through comprehensive approaches. In fact, we have been routinely using this alerting system in the Kazusa human cDNA sequencing project because there is a considerably high risk that large cDNA clones derived from human brain RNA, which are our current targets, may include spurious sequences. GeneMark analysis has been successfully used not only for detection of CDS split but also for warning of truncation of cloned cDNAs as briefly described in this study. Very recently, we also applied GeneMark analysis to clone selection based on 5′-single-pass sequence data in order to identify cDNA clones having protein-coding potential (Hirosawa et al. 1999). These results demonstrate that GeneMark analysis is an important tool for interpreting mammalian cDNA sequences, although it has been mainly used for interpretation of prokaryotic genomic sequences to date.
METHODS
The Alerting System Targeting CDS Splits Based on GeneMark Analysis
The alerting system targeting CDS splits was based on the prediction results of GeneMark analysis; when GeneMark analysis predicted multiple CDSs in a single cDNA sequence, a warning of a spurious CDS split was issued. GeneMark analysis of human cDNA sequences was carried out essentially as described previously (Hirosawa and Isono 1997; Hirosawa et al. 1997). The statistics necessary for assignment of CDSs were extracted from and optimized for a set of cDNA sequences, KIAA0001 to KIAA0817 (Nomura et al. 1994; Nagase et al. 1998).
An auxiliary program first enumerated candidates of coding regions (from ATG to any termination codon) longer than 500 nt. Regions outside of the candidate coding regions in cDNAs were taken as noncoding regions. As an exception, when multiple candidate coding regions were present in a single cDNA sequence, only the upstream and the downstream regions (> 100 nt) from the first and the last candidate coding regions, respectively, were considered as noncoding regions. Parameters of Markov models, transition probabilities, were derived as the three-phase-dependent and phase-independent oligomer statistics sampled from the nucleotide sequences of coding regions and noncoding regions, respectively. The iteration of finding a stationary set of candidate coding regions by recursive GeneMark application was performed as described previously (Hirosawa and Isono 1997; Hirosawa et al. 1997). Classes and class-specific statistics of coding sequences were derived as described previously (Hirosawa and Isono 1997; Hirosawa et al. 1997), and the derivation was repeated until the number of unclassified coding sequences became < 50. A GeneMark parameter, the order of Markov models, was 5. Another GeneMark parameter, the decision-making level, was 0.8 for core-class derivation, 0.5 for finding coding sequences in a specified class, and 0.4 for long CDSs (> 500 nt) on detection of spurious coding splits.
Experimental Examination of CDS Splits
A region predicted by GeneMark analysis to span two CDSs was amplified from cytoplasmic poly (A)+ RNA of a human immature myeloid cell line, KG-1 cells, or from poly (A)+ RNA of human adult brain by RT-PCR. RT-PCR was carried out essentially as described previously (Ishikawa et al. 1997). PCR primers were designed with the assistance of the Oligo 4.06 program (National Biosciences), and actual primer sequences and thermal cycling conditions for each of the genes are available upon request. The resultant RT-PCR products were run on agarose gels and only the major products, if multiple bands were seen, were recovered from the gels. The purified products were subjected to direct DNA sequencing using an ABI Big-dye terminator cycle sequencing kit and an ABI373 or 377 DNA sequencer. The regions suspected of containing CDS splits were sequenced in both strands and the determined sequences were compared to those of cDNA clones isolated previously. All the revised sequences described in this study are accessible in Gene/Protein characteristic tables in the HUGE database (http://www.kazusa.or.jp/huge; Kikuno et al. 2000).
Acknowledgments
This project was supported by grants from the Kazusa DNA Research Institute. We thank Tomomi Tajino, Keishi Ozawa, Tomomi Kato, Kazuhiro Sato, Kiyoe Sumi, Naoko Shibano, and Chie Mori for their technical assistance. We also thank Dr. Borodovsky for allowing us to use the GeneMark program at the Kazusa DNA Research Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL ohara{at}kazusa.or.jp; FAX 81-438-52-3914.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.129500.
-
- Received December 20, 1999.
- Accepted June 30, 2000.
- Cold Spring Harbor Laboratory Press








