
Conservation of DNA Regulatory Motifs and Discovery of New Motifs in Microbial Genomes
Abstract
Regulatory motifs can be found by local multiple alignment of upstream regions from coregulated sets of genes, or regulons. We searched for regulatory motifs using the program AlignACE together with a set of filters that helped us choose the motifs most likely to be biologically relevant in 17 complete microbial genomes. We searched the upstream regions of potentially coregulated genes grouped by three methods: (1) genes that make up functional pathways; (2) genes homologous to regulons from a well-studied species (Escherichia coli); and (3) groups of genes derived from conserved operons. This last group is based on the observation that genes making up homologous regulons in different species are often assorted into coregulated operons in different combinations. This allows partial reconstruction of regulons by looking at operon structure across several species. Unlike other methods for predicting regulons, this method does not depend on the availability of experimental data other than the genome sequence and the locations of genes. New, statistically significant motifs were found in the genome sequence of each organism using each grouping method. The most significant new motif was found upstream of genes in the methane-metabolism functional group inMethanobacterium thermoautotrophicum. We found that at least 27% of the known E. coli DNA-regulatory motifs are conserved in one or more distantly related eubacteria. We also observed significant motifs that differed from the E. coli motif in other organisms upstream of sets of genes homologous to known E. coli regulons, including Crp, LexA, and ArcA in Bacillus subtilis; four anaerobic regulons in Archaeoglobus fulgidus (NarL, NarP, Fnr, and ModE); and the PhoB, PurR, RpoH, and FhlA regulons in other archaebacterial species. We also used motif conservation to aid in finding new motifs by grouping upstream regions from closely related bacteria, thus increasing the number of instances of the motif in the sequence to be aligned. For example, by grouping upstream sequences from three archaebacterial species, we found a conserved motif that may regulate ferrous ion transport that was not found in individual genomes. Discovery of conserved motifs becomes easier as the number of closely related genome sequences increases.
Motif-finding algorithms can be used to discover alignments of sites, which correspond to transcriptional regulatory motifs in upstream regions of genes. These motifs are often binding sites for DNA-binding proteins. Several different algorithms have been used previously for motif finding, including Gibbs sampling (Lawrence et al. 1993; Liu et al.1995), MEME (Bailey and Elkan 1995; Grundy et al.1996), ClustalW (Thompson et al.1994), MACAW (Schuler et al.1991), and algorithms based on computational analysis of oligonucleotide frequencies (van Helden et al.1998).
AlignACE (Roth et al.1998) is a local alignment program based on the Gibbs-sampling algorithm, optimized for DNA sequence alignment. AlignACE chooses statistically significant alignments in the input sequence. However, regulatory signals often have relatively weak alignments when compared to other common genomic elements found by AlignACE, such as ribosome-binding sites and repetitive elements. Additional filters have been developed to select the motifs found by AlignACE that are the most likely to correspond to biologically relevant motifs (Roth et al. 1998; Hughes et al. 2000). Together with these filters, AlignACE has been used for motif-finding in Saccharomyces cerevisiae, using expression data from microarrays (Roth et al. 1998; Tavazoie et al. 1999) as well as metabolic functional group categories (Hughes et al. 2000). KnownS. cerevisiae motifs were computationally identified in these studies, as well as new motifs.
Computationally identifying upstream-regulatory motifs in bacterial genomes by AlignACE is complicated by the presence of operons. It is difficult to locate the regulatory region for a gene found within an operon, since the promoter for that operon can lie several genes upstream, and it is difficult to predict which gene is at the head of the operon. In addition, there are fewer instances of most regulatory motifs in a bacterial genome than in the S. cerevisiae genome, as there is usually only one instance of a regulatory motif per operon instead of one instance per gene. It is easier to discover a motif that is found in more copies in the genome. However, one can increase the number of instances of a conserved regulatory motif by pooling together upstream sequence from orthologous genes in closely related organisms, assuming the motif is conserved across these organisms.
A similar method was employed recently by Gelfand et al. (2000) to predict transcriptional regulatory sites in archaeal genomes by comparative genomics for four specific groups of genes. In phylogenetic footprinting analysis, noncoding sequences from several organisms are aligned to identify conserved regions (Tagle et al. 1988; Duret and Bucher 1997; Hardison et al. 1997; Wasserman and Fickett 1998). In addition, genomic comparisons of regulatory sites have been done between Escherichia coli and Haemophilus influenzaefor several known motifs by using search algorithms employing position weight matrices (Robison 1997; Mironov et al. 1999).
Microarray data, as well as at least five additional methods based on comparative genomics might be used to obtain functionally linked sets of genes that are good candidates for coregulation. Pellegrini et al. (1999) describe a method for predicting functional linkages between nonhomologous genes based on the assumption that proteins that function together in the cell are likely to evolve in a correlated fashion.Marcotte et al. (1999) describe a method based on protein fusions. We obtained potentially coregulated sets of bacterial genes for motif finding by three different methods. We used groups of genes that make up functional pathways, genes homologous to the members of regulons from E. coli, and groups of genes derived from conserved operons. Genes with similar expression profiles will be used for motif finding in bacteria as more microbial expression data becomes available.
By aligning the upstream regions of potentially coregulated sets of genes, we were able to find many known bacterial regulatory motifs, and to predict new regulatory motifs in 17 bacterial genomes. In addition, by pooling together upstream sequences from orthologous genes in closely related organisms, we were able to find conserved motifs with only a few instances in each genome. We also analyzed conservation between organisms of motifs in orthologous sets of upstream regions.
RESULTS
Potentially Coregulated Groups of Genes
We searched in each species for orthologs to the E. coligenes known to be regulated by 55 DNA-binding proteins (http://arep.med.harvard.edu/ecoli_matrices/; Robison et al.1998; see Methods). This results in 55 potentially coregulated groups in each of 17 genomes. These groups allow us to assess our motif-finding techniques in E. coli, as well as to look at conservation ofE. coli DNA motifs in other organisms and to identify potential new mechanisms for regulating the same cellular process in more distantly related organisms.
The second method we used to predict coregulated groups of genes is based on analysis of conserved operons. Functional couplings between genes can be inferred from conserved spatial proximity of gene pairs on a chromosome (Dandekar et al. 1998; Overbeek et al. 1998,1999). Two genes that are spatially separated on the chromosome in one organism, but have homologs which are spatially close in two or more other species, are good candidates for being coregulated. We can discover regulatory motifs by aligning the upstream regions of sets of genes that were predicted to be coregulated in this manner. We used 343 groups of genes from the WIT database (http://wit.mcs.anl.gov/WIT2/) in each of 17 complete bacterial genomes. These groups, containing between 0 and 54 genes in a particular organism, were constructed by searching for pairs of genes contained in conserved operons in >30 complete or partial genomes (Overbeek et al. 1999). In E. coli, most of these gene clusters contain parts of known metabolic systems, and at least 11 of these clusters are large groups that correspond to substantial pieces of known pathways (including the purine and arginine biosynthesis pathways).
We also used 68 different functional group categories in each of 17 bacterial species from the KEGG database (http://www.genome.ad.jp/kegg/). These groups are based on a compilation of experimental data on metabolic pathways (Ogata et al. 1999).
Motif Discovery Strategy
We used the AlignACE program (Roth et al. 1998) together with a set of filters based on known properties of binding sites for DNA regulatory proteins to find motifs in the upstream regions of potentially coregulated sets of genes. Our method for selecting upstream-regulatory regions to be searched in genomes containing operons is described in Methods. In addition to aligning groups of upstream regions from a single organism, we also combined sequence from orthologous genes in closely related organisms (see Methods for a listing of groups).
Running AlignACE on all of these groups of genes in each of 17 organisms, as well as in sets of closely related organisms, results in 104,282 motifs. To select significant motifs that are the most likely to be functional, regulatory motifs, we calculated several indices for each matrix: MAP score, site specificity score (S site), positional bias, AT content, and palindromicity. The MAP score is a measure used by the AlignACE program to judge alignments sampled during the course of the algorithm, based on the over-representation of the motif in the input sequence (Liu et al. 1995). S site is a measure of how specific a motif is for the sequence in which it was aligned, compared to the genome as a whole. The positional bias is a measure of the tendency of the top-scoring motif instances in the genome to be unevenly distributed relative to the start codon of the closest gene (Hughes et al. 2000). A large fraction (almost half) of the footprinted E. coli motifs are palindromic. To identify palindromic motifs, we used the CompareACE program (Hughes et al. 2000) to compare a motif with its reverse complement. These indices are described in more detail in Methods.
Table 1 shows how many motifs score above various cutoffs in the values for these indices. The AlignACE output files, as well as the values for these indices for all of the motifs, are available on our website (http://arep.med.harvard.edu/microbial_motifs).
Number of Controls Scoring Above Different Cutoffs
Controls
By searching the upstream regions of genes making up the known regulons in E. coli with AlignACE, we can determine what fraction of the known E. coli DNA-binding motifs can be found, and how these known motifs score in terms of the parameters that we use to rank motifs. We used the 32 E. coli footprinted regulons in our database with between 5 and 100 known binding sites (Robison et al. 1998) as controls. Twenty-six of these regulons have known regulatory motifs that can be found by AlignACE (81%). The six motifs that are not found by AlignACE have low or dispersed information content (Ihf, Hns, Lrp, Fis, OmpR, and SoxS;http://arep.med.harvard.edu/ecoli_matrices). Table 1 shows that by using our highest cutoffs we can eliminate the majority of false positives, but we also only find the true positives with the strongest motifs. Without using any additional cutoffs with AlignACE, the false positive rate is very high (95%). However, by restricting our analysis to the motifs with the lowest S site and those that are palindromic, we are able to make high-confidence predictions. Optimization of the choice of cutoffs for a certain false positive rate could also be performed using automatic classification schemes, such as decision trees and linear discriminant functions. Here we applied very stringent cutoffs based on simple criteria obtained from the biological properties of promoters. Table 2 lists the number of motifs from all of the AlignACE runs in all organisms that score above each cutoff.
Number of Motifs Scoring Above Different Cutoffs
Figure 1 shows the MAP scores andS site values for the positive controls (green triangles), as well as all of the motifs found by AlignACE with MAP scores greater than 5.0 (30,252 motifs from all AlignACE runs in three kinds of gene groupings in 17 organisms). Some of the controls are plotted more than once because the motif was found multiple times. Motifs in the upper right hand corner of the plot (nonspecific motifs with good alignments) tend to be either repetitive elements (i.e.,E. coli BIME elements) or common elements in the genome such as Shine–Dalgarno sequences. The fraction of motifs corresponding to known controls is highest in the upper left corner of the plot. Motifs with MAP score > 10.0 and S site < 10−25 are listed in Table 3a and3b. Pooling together upstream regions from orthologs in several closely related organisms in order to increase the number of instances of a motif improves the ability to discriminate known conserved motifs (magenta diamonds). These conserved motifs tend to have higher MAP scores and lower Ssite than the same motif found in sequence from E. coli alone (green triangles).
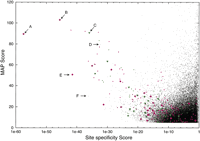
All of the motifs with MAP scores >5.0 (30,252 motifs) from the AlignACE runs in all 17 organisms are presented here (black points). The controls (E. coli motifs found in alignments of upstream regions of genes with >five footprints and which are known to constitute a regulon) are plotted with colored symbols: (green triangles) E. coli motifs found in alignments of upstream regions from genes making up the regulon in E. coli only; (magenta diamonds) conserved E. coli motifs found in alignment of sequence from E. coli together with sequence from B. subtilis, H. influenzae, or both. Several of the footprinted motifs are represented several times on this plot, as we perform two AlignACE runs on every group of genes, one for each of two different kinds of operon prediction. Also, a very strong motif can occasionally show up more than once in the same AlignACE run despite iterative masking in AlignACE. Therefore, for each control, we have represented the motif instance with the lowest value forSsite with a large colored symbol, and all other instances of the same motif with a small colored symbol. Several of the most specific motifs are labeled: (A) PurR conserved in E. coliand H. influenzae; (B) LexA conserved in E. coliand H. influenzae; (C) LexA in E. coli.; (D) T-box in B. subtilis; (E) ArgR conserved in E. coli., H. influenzae, and B. subtilis; (F) new motif found in the methane metabolism functional group in M. thermoautotrophicum. See text and Table 3 for details.
Top Motifs Found by AlignACE
The most useful parameter for discriminating the known motifs in theE. coli regulon controls from the rest of the motifs found by AlignACE is Ssite (see Fig. 1). Palindromicity is also a useful parameter. Almost half of the known motifs are palindromic, but only ∼5% of the 104,282 motifs found in our analysis are palindromic. Thus, selecting for palindromic motifs does increase greatly the fraction of biologically relevant motifs. The MAP score was not as useful as Ssite for distinguishing known motifs in the E. coli regulon controls from the rest of the motifs, since many nonspecific chromosomal features have high MAP scores (i.e., Shine–Dalgarno sequences and repetitive elements). The positional bias statistic was also not useful in distinguishing theE. coli regulon controls, because most known motifs do not have significant values for our positional bias parameter (data not shown). However, there are many other motifs found in this study that do have significant positional bias. Most of these likely correspond to locationally conserved features such as the ribosome binding site (Shine–Dalgarno sequence), which is always located close to the start codon (4–13-bp upstream of ATG).
Conservation of Known E. coli Motifs in Other Bacteria
In organisms other than E. coli, running AlignACE on upstream regions of orthologs of members of E. colifootprinted regulons allows for study of the conservation of E. coli regulatory motifs, as well as identification of other possible mechanisms for regulating the same cellular process. For each of the 34E. coli regulons with more than five members, Figure2 shows in which organisms the E. coli motif is conserved, and in which organisms there is a new and significant motif in the upstream regions of the homologous regulon.
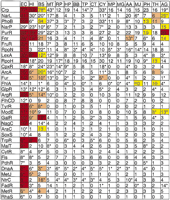
This figure summarizes the results in each organism from AlignACE runs on groups derived from E. coli footprinted regulons. The 34 known E. coli DNA-binding proteins that regulate at least five genes in E. coli are listed in the left column. The numbers indicate how many regulon members are present in that organism. (*) Ortholog to the E. coli DNA-binding protein was found. (Red or pink square) Motif similar to the known E. coli motif (CompareACE score >0.7) is conserved in this organism and was found by AlignACE with MAP score >5 and Ssite < 1e − 10. (Red square) Motif was found by aligning sequence from this organism only; (pink square) motif was found by aligning sequence from this organism together with sequence from E. coli (>30% of the aligned instances of the motif were from this organism). (Yellow square) New and significant motif found in sequence from this organism that is not similar to the E. coli motif. Yellow squares correspond to the motifs described in Tables 3a and 3c that were found in groups derived from E. coli regulons (i.e., these motifs score better than one of two cutoffs: (1) Ssite < 1e − 25, MAP score >10; (2) Ssite < 1e − 10, MAP score >10, palindromicity >0.7, % AT <80%. (White square) No new motif that scores better than these cutoffs, and the E. coli motif is not conserved in this organism.
A new motif can indicate either a different mechanism for regulating a similar cellular process, or divergence of binding site residues in a conserved DNA-binding protein. In Bacillus subtilis, there is no Crp protein; instead, CcpA regulates carbon metabolism by a different mechanism (Henkin 1996). We detect the CcpA binding motif in our analysis. An example of divergence of binding site residues is LexA in B. subtilis. H. influenzae and E. colihave very similar LexA binding motifs. However, gram-positive bacteria, including B. subtilis and Mycobacterium tuberculosis, have a completely different binding motif for LexA (Lovett et al. 1993;Cheo et al. 1991). The LexA protein is conserved but its binding-site residues are mutated. Thus B. subtilis LexA binds to a completely different motif, which is found using our strategy.
Five new motifs were identified in B. subtilis, and ten were identified in archaebacterial species (Methanococcus janaschii, Pyrococcus horokoshii, Methanobacterium thermoautotrophicum, and Archaeoglobus fulgidus). These motifs (Fig. 2, yellow) are listed in Tables 3a and 3c. In B. subtilis, the CcpA motif is found in the Crp, AraC, and PhoB categories. The motif found in the B. subtilis ArcA category is similar to the E. coli –Crp motif. Therefore this could simply be a variation on the Fnr motif, which is closely related to the Crp motif in E. coli. The Fnr and ArcA regulons overlap significantly.
In A. fulgidus, there are new motifs in the categories for NarL, NarP, Fnr, and ModE. In E. coli, all four of these DNA-binding proteins regulate overlapping regulons related to anaerobic metabolism. The motifs from the NarL, NarP, and Fnr categories are very similar to each other (pairwise CompareACE scores < 0.7). However, the motif that is found in the ModE category is different. Thus, we predict that these two new motifs control anaerobic metabolism inA. fulgidus (see Tables 3a and 3c).
Combining upstream sequences from orthologous genes in closely related organisms can help in the discovery of conserved motifs with few instances in each genome. A known E. coli motif (MetR) is not found when AlignACE is run on the upstream regions of the members of the MetR regulon in E. coli alone because there are too few instances of the motif in the E. coli genome. However, whenE. coli and B. subtilis upstream regions are pooled together, the MetR motif can be found because it is also present inB. subtilis. Eleven additional E. coli motifs can be identified in B. subtilis and/or H. influenzae using this method. Even for the motifs that occur frequently enough to be found in a single genome, pooling together sequence from closely related organisms increases the number of instances of the motif. This improves the values of the MAP and Ssite parameters in the alignments, making the motif easier to identify (see Fig. 1).
New Motifs
Table 3 shows the most specific motifs, as well as the top palindromic motifs, that result from AlignACE runs. Some of these motifs are known, and some of these correspond to potential new regulatory motifs. Only motifs scoring above stringent cutoffs are presented here; alignments and parameters for all motifs found in this analysis are available (http://arep.med.harvard.edu/microbial_motifs).
Specific Motifs Found by Aligning Sequence from Individual Organisms
Table 3a displays the most specific motifs found in AlignACE runs on sequence from single organisms. Out of 53,778 motifs, the 41 motifs with Ssite < 1e − 25 were clustered according to their similarity using pairwise CompareACE scores (see Methods), resulting in 18 motif clusters. The member from each cluster with the lowestSsite value is shown in the Table 3. Three of these clusters correspond to known E. coli motifs (the LexA, Crp, and PurR control sets), and one corresponds to a known B. subtilis motif (the T-box). The T-box is a known regulatory motif found in the functional group made up of aminoacyl-tRNA synthetases in gram-positive bacteria (Henkin et al. 1992).
The other 14 clusters correspond to new motifs. The motif with the lowest value for Ssite (Ssite = 5.6e − 37) for which we are not aware of any documentation in the literature is an AT-rich motif found in the methane metabolism functional group (00680) of the methane-producing archaebacteriaM. thermoautotrophicum. Another very specific member of this motif cluster (Ssite = 3.5e − 31) shows up in the closely related folate biosynthesis functional group (00790) in M. thermoautotrophicum. Therefore this motif could regulate metabolism of one-carbon units in this organism. A similar motif also shows up with a slightly higher S site in the glyoxylate and dicarboxylate metabolism functional group (00630) and the group corresponding to the orthologs of the RpoH (heat shock) regulon inM. thermoautotrophicum. The relationship between these two groups and metabolism of one-carbon units is not clear. This motif resembles the central AT-rich core (ATATAAAxxTT) of the known archaeal heat-shock promoter (Thompson and Daniels 1998; Gelfand et al. 2000). It has a CompareACE score of 0.72 when compared to this known heat shock motif (consensus TTCTATATAAAxxTTTCG). Another new motif in this list is the motif discussed earlier which we predict to regulate anaerobic metabolism in A. fulgidus (see Fig.2). It is not clear why this motif is also found in the tryptophan metabolism functional group (00380) in A. fulgidus. Another very specific motif is the AT-rich motif found in the PurR (purine biosynthesis) group in Pyrococcus horokoshii. This motif shows similarity to the purine biosynthesis motif found by Gelfand et al. (2000) in three Pyrococcus genomes, except that the highly conserved C and G positions are not present.
Many of these 14 new clusters contain AT-rich archaebacterial motifs that are very specific to the groups in which they are found. There are no known E. coli motifs with AT content greater than 80%. However, not much is known about regulatory mechanisms in the archaebacteria, so these could be functional regulatory elements. These are not AT-rich genomes (A. fulgidus and M. thermoautotrophicum both have 51% AT content).
Specific Motifs Found by Combining Sequence from Closely Related Organisms
Of the 50,504 motifs found by aligning sequence from closely related organisms together, there are 18 motifs with Ssite < 1e − 25, which fall into seven distinct clusters (Table 3b). Six of these clusters correspond to known E. coli motifs conserved in either H. influenzae or B. subtilis (PurR, LexA, TrpR, Crp, Fur, and ArgR). Many of these conserved motifs show up with lower values for Ssite and higher MAP scores than when sequence from only one organism containing the motif is aligned, because a stronger alignment can be obtained when more instances of the motif are present. Therefore, more known E. coli motifs are present in Table 3b than in Table 3a, which contains alignments of sequence from E. coli only.
The only motif in this list that has not been documented previously is found in a group derived from conserved operons (group 103) in A. fulgidus, M. thermoautotrophicum, and P. horokoshii. In A. fulgidus, the motif is found upstream of two genes with homology to ferrous ion transporters; in M. thermoautotrophicum, the motif is found upstream of a ferrous ion transporter and a gene with homology to the iron repressor; and inP. horokoshii, the motif is found upstream of a ferrous ion transporter (see Table 3d). This motif is highly palindromic (consensus TTAGG-x4-CCTAA). This pattern of two palindromic halfsites separated by a short linker sequence is common among the binding sites for known bacterial regulatory DNA-binding proteins. Our prediction is that this motif is a binding site for a protein regulating iron transport in these archaebacteria. Since the motif is found upstream of a putative iron repressor in M. thermoautotrophicum, it is possible that this putative repressor (MTH214) is the regulatory protein that binds to these sites, and that it is autoregulatory.
Top Palindromic Motifs Found by Aligning Sequence from Individual Organisms
Palindromicity is a parameter that is strongly correlated with regulatory function. Only about 5% of all of the motifs found by AlignACE in this study were palindromic, whereas almost half of all of the known motifs are palindromic. By considering palindromic motifs separately, we can increase the Ssite cutoff and retain a high rate of true positives (see Table 1). A selection of these motifs is shown in Table 3c. All 101 palindromic motifs with MAP >10, Ssite 1e − 10, and AT-content <80% were clustered into 30 clusters. One representative from each cluster is shown here. If we do not impose the AT-content cutoff, there are over twice as many motifs present (220 motifs). Ten of these 30 clusters correspond to known E. coli or H. influenzaemotifs, and two correspond to known B. subtilis motifs. The known E. coli motifs scoring above this cutoff are Crp, LexA, ArgR, Fnr, TyrR, FruR, TrpR, and GalR. The known B. subtilismotifs are the Cheo motif (the B. subtilis SOS box), and the CcpA motif. In B. subtilis there is also a variant of the Fnr motif that resembles the E. coli Crp motif. ArgR and PurR are also found above this cutoff in groups derived from conserved operons (groups 054 and 001, which contain parts of the purine and arginine biosynthesis pathways, respectively). Of the 22,134 motifs found by running AlignACE on the upstream regions of all 343 gene groups predicted from conserved operons, the ArgR and PurR motifs inE. coli are among the most specific palindromic motifs.
Eighteen of the 30 motif clusters in Table 3c show no similarity to known motifs. Thirty percent of these motifs are also AT rich (70%–80% AT content). Three of these motifs are found in groups derived from conserved operons. These are group 156 (Hyp operon genes) in P. horokoshii, group 255 (heat shock genes) in M. genitalium, and group 033 (pyruvate synthase genes) in M. janaschii. The presence of regulatory motifs upstream of the genes making up these groups lends additional support to the hypothesis that these are indeed functionally coupled groups of genes. Since this method for predicting regulons is based purely on the chromosomal gene order across genomes, the high-scoring motifs found in these groups are not biased toward well-studied organisms. In contrast, the other kinds of groups of potentially coregulated genes that we used in this study (groups from metabolic pathways and groups based on footprinted E. coli regulons) both originate from experimental information determined in the well-studied organisms, so the high-scoring motifs from these two methods are largely from E. coli and B. subtilis.
The motif found upstream of group 255 (heat shock genes) in M. genitalium does not resemble the CIRCE (Controlling Inverted Repeat of Chaperone Expression) motif, which is known to regulate several heat shock-related genes in a wide variety of organisms, including M. genitalium, through the binding of the repressor HrcA (Naberhaus 1999). Since HrcA is the only transcription factor known to be involved in regulating heat shock genes in M. genitalium, it is not clear what transcription factor could bind to our predicted motif. This motif could represent a false positive since it is also not found in the closely related Mycoplasma pneumoniae. A motif resembling the CIRCE element is found upstream of this group of heat shock genes in M. genitalium; however, it did not score below our stringent specificity score cutoff (Ssite = 2.9e − 7).
Top palindromic motifs found by combining sequence from closely related organisms
Using this same cutoff (Ssite < 1e − 10, MAP >10, AT content < 80%) on the motifs obtained from aligning upstream sequence from orthologous genes in two or more closely related organisms together, we obtain 65 motifs that reduce to 19 clusters (Table 3d). Of these 19 clusters, 12 correspond to known E. colimotifs conserved in either H. influenzae or B. subtilis (PurR, ArgR, LexA, TrpR, Crp, GalR, Fur, TyrR, ModE, HipB, ArcA, and Fnr). Again, these conserved motifs show up with lowerSsite and higher MAP scores when they are aligned in multiple organisms containing the same motif because there are more instances of the motif to align.
Of the seven clusters corresponding to new motifs, one is the ferrous ion-transport motif in A. fulgidus, M. thermoautotrophicum, and P. horokoshii described above. Four of the remaining six clusters correspond to conserved motifs found in groups derived from conserved operons. These are group 190 (ribonucleoside diphosphate) in E. coli and B. subtilis, group 199 (transcription) in M. janaschii andP. horokoshii, group 073 (fatty acid biosynthesis) in E. coli and H. influenzae, and group 009 (nucleotide biosynthesis) in E. coli and B. subtilis.
DISCUSSION
We found many significant new motifs in 17 bacterial genomes. Known regulons in E. coli were used as controls to calibrate the significance of these motifs. More motifs were found in larger genomes with more complex regulation. Some of the highest-scoring new motifs are found in archaebacteria, for which there is relatively little experimental data because of difficulties in performing experiments in these organisms. New, significant motifs include two motifs potentially regulating anaerobic metabolism in A. fulgidus, a motif potentially regulating methane metabolism in M. thermoautotrophicum, and a palindromic motif regulating ferrous ion transport that is conserved in M. thermoautotrophicum, A. fulgidus, and P. horokoshii.
We also identified a number of motifs that are conserved in several eubacterial species. At least 22% of the known E. coli DNA regulatory motifs are conserved in H. influenzae. In the more distantly related organism B. subtilis, at least 15% of the known E. coli motifs are conserved. In even more distantly related organisms, motifs can differ considerably. We found cases in which there are different but significant motifs upstream of genes homologous to known E. coli regulons, including Crp, LexA, and ArcA in B. subtilis; four anaerobic regulons in A. fulgidus (NarL, NarP, Fnr, and ModE); and PhoB, PurR, RpoH, and FhlA in other archaebacterial species. This can indicate that the organisms have evolved different methods for regulating the same cellular processes, or it can indicate parallel mutations in the DNA-binding protein and the motif that it recognizes.
The three methods of predicting regulons that we use here are based on different sources of biological information. The gene groups obtained from homologs to members of known E. coli regulons and the groups based on functional pathways in each organism are based on the prior body of knowledge from biological experiments. Therefore, many known motifs are found in these groups. In contrast, the groups derived from conserved operons in other organisms are not based on any biological information other than the positions of genes within the genomic sequence. Therefore, these groups are less biased towards motifs that were already known. However, some known motifs are found in these groups as some of the known regulons can be reconstructed in this manner (i.e., ArgR and PurR). By our criteria for ranking motifs, the PurR and ArgR motifs are two of the top 10 motifs to come out of the AlignACE runs on the groups derived from conserved operons, which lends credibility to the use of this method for predicting regulons and their regulatory motifs.
The usefulness of motif finding upstream of potential regulons depends strongly on how good the regulon predictions are. The groups of genes that were constructed based on conserved operons in other organisms were limited in several ways. The first limitation is that only top reciprocal FASTA hits were considered orthologs (Overbeek et al. 1999), which is a restrictive ortholog definition. When groups of orthologs are constructed across many organisms, and one or more organism contains close paralogs, actual orthologs will be missed or split into separate ortholog groups. This ortholog definition also causes problems in the case of multidomain protein fusions, which can be orthologous to multiple nonhomologous proteins in another species. Thus, using a method that treats multidomain protein fusions as multiple separate entities and employs a looser ortholog definition will increase the size of the functionally related gene clusters that can be predicted. Including multidomain protein fusions as separate entities will also extend the usefulness of this approach to include eukaryotic genomes. The second limitation in the way the groups were constructed is that only tandemly oriented genes were considered be potentially functionally related (Overbeek et al. 1999). However, divergently transcribed genes are also good candidates for coregulation in prokaryotes (Robison 1997), as well as in eukaryotes (Smith and Zhang 1998). Thus, a looser operon definition that also includes divergently transcribed genes should increase the usefulness of this approach for predicting coregulated sets of genes.
These methods will continue to become more powerful as the amount of available sequence data increases. Overbeek et al. (1998) suggest that the number of ortholog pairs contained in conserved operons increases roughly as the square of the number of organisms included in the analysis. This method of predicting regulons based on conserved operons will work best when using sequence from a variety of distantly related organisms, including representatives from all families of bacteria. A conserved operon is more likely to represent a functional coupling when it has been conserved across a large evolutionary distance. However, motif finding in bacteria using these predicted regulons will be most effective when there are many closely related genomic sequences available. As we have seen, regulatory motifs conserved more often in closely related genomes. With more bacterial genomes, upstream sequences from large groups of closely related bacteria can be pooled together, increasing the number of instances of conserved motifs.
The three methods that we use separately for predicting coregulated sets of genes can be combined to obtain larger and more complete groups. Groups obtained by these three methods can also be combined with groups of genes experimentally observed to have similar expression profiles, as well as groups obtained by methods such as that ofPellegrini et al. (1999), assuming that proteins that function together in the cell are likely to evolve in a correlated fashion. Combining different methods for predicting coregulated sets of genes, together with alignment of upstream regions of these groups using AlignACE, should be a very powerful method for predicting functionally related and coregulated sets of genes, discovering the upstream regulatory motifs controlling these groups, and generating hypotheses concerning gene regulation.
The biological significance of some of the motifs presented here should be verified experimentally, including determination of factors binding to these motifs. Predictions for which DNA-binding protein might be interacting with the motif can be obtained by computational methods, such as finding which predicted DNA-binding proteins have the motif in their upstream region (assuming autoregulation), and searching for a member of a known DNA-binding protein family that is linked to the regulon via a conserved operon in another organism. The regulon prediction and motif discovery methods described here should be an increasingly powerful addition to the current array of tools used to elucidate connectivities in bacterial regulatory networks.
METHODS
Organisms
AlignACE runs were performed on upstream regions from the following 17 bacterial organisms (abbreviations used in the tables and figures are given in parentheses): A. fulgidus (AG), Aquifex aeolicus (AA), Borrelia burgdorferei (BB), B. subtilis (BS), Chlamydia trachomatis (CT), E. coliK12 (EC), H. influenzae (HI), Helicobacter pylori(HP), Mycoplasma genitalium (MG), M. janaschii(MJ), Mycoplasma pneumoniae (MP), M. thermoautotrophicum (TH), M. tuberculosis (MT), P. horokoshii (PH), Ricksettia prowazekii (RP),Synechocystis sp. (CY), and Trepenoma pallidum (TP). For AlignACE runs on groups of genes derived from conserved operons, sequence from the following 12 groupings of closely related organisms were pooled together: EC and HI; EC and BS; EC, HI, and BS; BS and MT; EC and MT; AG and TH; AG, TH, and MJ; AG, TH, MJi, and PH; MJ and PH; MG and MP; CY and CT; and TP and BB. For AlignACE runs on upstream regions of groups of genes derived from E. coli-footprinted regulons, 16 groups are constructed by pooling E. colisequence together with sequence from each organism separately, including distantly related organisms. Two additional groups were also used: EC, HI, and BS; and BS and MT.
Identification of Orthologs
In each organism, we searched for orthologs to the members of the footprinted-E. coli regulons (http://arep.med.harvard.edu/ecoli_matrices), as well as for orthologs to the E. coli DNA-binding proteins controlling these regulons. To identify potential orthologs, we performed reciprocal BLAST searches between E. coli and each of 16 other completely sequenced organisms. To not discount closely related paralogs from our analysis, we allowed up to five potential orthologs for each gene to be included. It is desirable to include upstream regions from several potential orthologs in the alignments because AlignACE can tolerate some superfluous sequence, and we want to make sure that the correct ortholog is included; however, if too much extra sequence is added, the real motif will not be found.
To find potential orthologs in genome Gb for a genexa in genome Ga , we performed a BLAST search over all genes in genome Gb usingxa as a query. We identified the raw BLAST score of the top hit in genome Gb , and selected up to five hits in genome Gb with raw score >70% of this value. For each of these genes xbi in genomeGb , we performed a BLAST search over all genes in genome Ga . If the original genexa turned up with a raw score >70% of the value of the top BLAST hit in genome Ga , thenxa and xbi are potential orthologs.
Identification of Upstream Regulatory Regions
If a gene lies within an operon, its promoter and regulatory region could lie several genes upstream. It is difficult to predict the first gene in an operon, especially in less well-studied organisms. To ensure that the sequence we align contains the regulatory intergenic region, we must include several possible intergenic regions. However, if we add too many extra intergenic sequences, the regulatory motif will not be found by AlignACE because there will be too much noise.
Our definition of an operon is two or more tandem genes separated by less than a certain cutoff distance. We used two different operon cutoff distances (100 and 300 bp). For each operon defined in this manner, we recorded the entire sequence of all of the intergenic segments of length greater than 20 bp between the gene of interest and the operon head, as well as 300 bp upstream of the operon head (see Fig. 3). We ran AlignACE twice for each group of potentially coregulated genes: once using a loose distance cutoff in the operon definition (300 bp) to ensure inclusion of the correct upstream region, and once using a more conservative distance cutoff (100 bp) to reduce inclusion of extraneous intergenic regions.

Example of upstream region prediction. This shows the predicted upstream region for gene E taking up to 300-bp of sequence upstream of the predicted operon head, and using two different cutoffs for operon prediction (300 and 100 bp). First the algorithm checks the length of the upstream region for the gene in question. If an intergenic region is shorter than the distance cutoff, then the entire intergenic region is stored for motif finding and the next intergenic region further upstream is considered as well. This continues until an intergenic region is encountered that is either divergently transcribed, or longer than the distance cutoff.
To increase the signal to noise ratio, we took no more than 300 bp of sequence upstream of the operon head because the overwhelming majority of the binding sites for DNA-binding proteins in bacteria are found within the first 300 bp upstream of the start codon. In cases where there is a site further upstream, there is usually also a site close to the promoter (Gralla and Collado-Vides 1996). We looked only at noncoding regions, because most binding sites for DNA-binding proteins are found within noncoding regions. Thus, we excluded regulatory sites downstream of start codons, as well as rare sites far upstream of the start codon.
AlignACE Runs
The AlignACE (Roth et al. 1998), ScanACE, and CompareACE (Hughes et al. 2000) programs are available athttp://arep.med.harvard.edu/mrnadata/mrnasoft.htm. Default AlignACE parameters were used, except that the expected number of sites was set to five, and the fractional GC content was set to the proper value for each genome. A total of 14,863 AlignACE runs took two months of CPU time on eight 300–400 MHz Pentium II processors.
Each motif found in this analysis was compared to the 55 footprintedE. coli motifs (Robison et al. 1998) using the CompareACE program (Hughes et al. 2000). CompareACE finds the best alignment between two motifs and calculates the correlation coefficient between the two position-specific scoring matrices. Two motifs with a CompareACE score >0.7 were considered to be similar. This cutoff was determined by looking at sequence logos and alignments for motifs similar to the known E. coli motifs, but containing slightly different sets of aligned sites. Most variants of the same motif had CompareACE scores > 0.7 when compared to the original motif alignment. Only 0.8% of 100,000 randomly selected pairs of motifs have CompareACE similarity scores > 0.7.
The CompareACE program was also used to compare and cluster new motifs coming out of the analysis. A matrix of all pairwise CompareACE scores was calculated, and then motifs were clustered using a simple joining algorithm (Hartigan 1975). The member of each cluster with the best value for Ssite is shown in Table 3.
Parameters Used in Motif Analysis
Site Specificity Score (Ssite )
Ssite is a measure of how specific a motif is for the sequence in which it was aligned. This statistic is similar to the specificity score described by Hughes et al. (2000) , but modified to better accommodate bacterial motifs, which are located upstream of operons rather than individual genes and are often found in multiple copies within a single regulatory region.
For each motif, we constructed a position-specific weight matrix and searched for additional instances of the motif in the
whole genome. We used the Berg and von Hippel weight matrix (Berg and von Hippel 1987,1988; Berg 1988a, 1988b), implemented in the ScanACE program (Hughes et al. 2000) to perform this search, and saved the best 200 sites. Of these top 200 sites, we calculated the number of sites that are
located within the upstream regions used to align the motif. Then we calculated the probability of obtaining this number of
sites or more within this particular subset of the genomic sequence by chance using the hypergeometric distribution: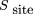 N is the total number of possible sites (the total number of base pairs in the genome(s) considered), s
1 is the number of ScanACE hits considered (here we are using the 200 highest-scoring hits in the genome), s
2 is the total number of possible sites in the set of upstream regions used to align the motif (equal to the number of base
pairs in the sequence input to AlignACE), and x is number of the top 200 ScanACE hits that fall within the upstream regions input to AlignACE.
N is the total number of possible sites (the total number of base pairs in the genome(s) considered), s
1 is the number of ScanACE hits considered (here we are using the 200 highest-scoring hits in the genome), s
2 is the total number of possible sites in the set of upstream regions used to align the motif (equal to the number of base
pairs in the sequence input to AlignACE), and x is number of the top 200 ScanACE hits that fall within the upstream regions input to AlignACE.
AT Content
Many of the motifs that were found by AlignACE, including motifs with low values of Ssite , are AT rich (>90% AT content). However, no known matrices for E. coli DNA-binding proteins have AT content greater than 80% (Robison et al. 1998). We are also not aware of any known motifs in other organisms, including archaebacterial genomes (Gelfand et al. 2000), with AT content > 80%. Thus, we explored the use of this measure to exclude AT-rich motifs that do not resemble known regulatory motifs. However, many high-scoring motifs in less well-studied organisms, including archaebacteria, are AT rich. Since we know very little about binding sites for DNA regulatory proteins in these bacteria, we did not exclude the most specific AT-rich motifs.
Palindromicity
To select palindromic motifs for further analysis, we used the CompareACE program to compare a motif with its reverse complement. We used the same CompareACE cutoff score for comparing motifs to one another (0.7).
Additional Cutoffs used in Selecting Interesting Motifs
To exclude repetitive elements from our analysis, we excluded motifs in which more than half of the aligned sites came from a single upstream region. In the AlignACE runs combining sequence from several closely related organisms, we only looked at those motifs where < 70% of the aligned instances of the motif came from a single organism, in order to limit our analysis to conserved motifs.
Acknowledgments
We thank Jason Johnson, John Aach, Jong Park, Martha Bulyk, and Ann Nichols for help and discussions. A.M.M. is a Howard Hughes Medical Institute predoctoral fellow. This work was supported by the office of Naval Research (grant N00014–97–1–0865), the Department of Energy (grant DE-FG02–87-ER60565), and a grant from Hoechst Marion Roussel.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL church{at}rascal.med.harvard.ed; FAX (617) 432-7266.
-
- Received October 28, 1999.
- Accepted March 28, 2000.
- Cold Spring Harbor Laboratory Press








