
Serpins are a widely distributed family of structurally very closely related medium molecular weight proteins (mostly 40–50 kDa) that are primarily known as inhibitors of serine proteinases (hence the origin of the name serpin;Gettins et al. 1996). They are found in most organisms, with notable exceptions being bacteria and fungi. There are both extracellular and intracellular serpins. The proteinase targets are often involved in complex physiological processes that must be carefully regulated both temporally and spatially, such as blood coagulation, fibrinolysis, and inflammation. Some of the best known human serpins are antithrombin, the principal inhibitor of the blood clotting cascade proteinases thrombin and factor Xa, α1-antitrypsin, the principal inhibitor of elastase secreted by neutrophils at sites of inflammation, and plasminogen activator inhibitor-1 (PAI-1), an inhibitor of the plasminogen activators t-PA and u-PA. Because serpins do not inhibit proteinases by a simple noncovalent lock and key mechanism but, instead, use a conformational change-based trapping mechanism that depends on their structural and thermodynamic properties, understanding the common elements of serpin structure, folding and conformational change is at the heart of understanding how serpins function in both normal and pathological states. In this issue, Irving and colleagues report serpin sequence comparisons that provide insight not only into the evolutionary relationships between serpins but also into those residues that must play critical roles for serpin structure and function (Irving et al. 2000).
Metastability of Serpins
A corollary of Anfinsen's proposal that all the information necessary for the correct folding of a protein is present in its primary structure (Anfinsen 1973) is that the native structure of a protein is expected to represent the most stable state. The large and growing family of serpins, whose primary structures are analyzed and compared by Irving and colleagues (Irving et al. 2000), represents one of a handful of exceptions to this expectation. Serpins instead fold into a metastable state that represents the active state. Subsequent conformational change, involving major reorganization of a large, exposed proteinase-recognition loop and one of the three β sheets, is fundamental to the normal functioning of many serpins as suicide inhibitors of serine and cysteine proteinases. Other properties of serpins, such as antiangiogenic activity (O'Reilly et al. 1999), chemotactic response (Potempa et al. 1991; Banda et al. 1988; Hoffman et al. 1989), or ability to regulate cell migration (Stefansson and Lawrence 1996) also appear to depend on the serpin's ability to undergo conformational change. The primary structure of a serpin must therefore ensure not only that a folding pathway leading to the metastable conformation be followed but that, under appropriate circumstances, structural rearrangement to the most stable state can occur rapidly and smoothly enough to carry out the physiological function of the serpin. It is likely that many of the conserved residues identified by Irving and colleagues are essential for correct metastable folding and for subsequent smooth conformational change.
Use of Conformational Change to Inhibit Proteinases
By far the largest group of serpins are inhibitors of serine proteinases. With only a few exceptions, where reversible inhibition may occur, serpins inhibit proteinases irreversibly as suicide substrate inhibitors in which the acyl enzyme intermediate of the normal substrate cleavage pathway becomes kinetically trapped as a result of a major conformational change in the serpin (Gettins et al. 1996). The inhibition pathway is thus a branched one, with one branch leading to the kinetically trapped covalent serpin:proteinase complex and the other to the normal cleavage products of the reaction (Fig.1), which in the case of a serpin involves cleavage within the reactive center loop, but without loss of any polypeptide. There is, thus, a competition between the two branches, with the outcome determined by the relative values of the rate constants k3 and k4 (Fig. 1; Patston et al. 1991).
Branched pathway mechanism of serpin (I) inhibition of proteinase (E), illustrated using α1PI as the serpin and trypsin as the proteinase. Serpin and proteinase bind to form an initial noncovalent Michaelis-like complex (EI) in which the proteinase docks with the exposed reactive center loop (red) with no distortion of the proteinase or the body of the serpin (Peterson et al. 2000). Normal substrate-like cleavage occurs with rate constant k2 to form the acyl enzyme intermediate EI#. A competition ensues between the inhibitory pathway (rate constant k4) and the substrate pathway (rate constant k3). Completion of the substrate reaction by hydrolysis of the acyl enzyme intermediate results in release of cleaved serpin (I*) and free proteinase. The inhibition pathway involves insertion of the cleaved reactive center loop into β-sheet A of the serpin and concomitant dragging of the covalently bound proteinase from the top to the bottom of the serpin to give the kinetically trapped covalent serpin–proteinase complex (EI+; Stratikos and Gettins 1999;Huntington et al. 2000). In the covalent complex, the proteinase (shown in red in the EI+ structure) is distorted through compression against the bottom of the serpin and, hence, rendered catalytically incompetent (Huntington et al. 2000). Because the catalytic competence of the proteinase has been severely compromised by the distortions within the active site, completion of the hydrolysis of the acyl intermediate from the trapped complex (rate constant k5) is extremely slow. In vivo complexes are cleared before appreciable dissociation has time to occur.
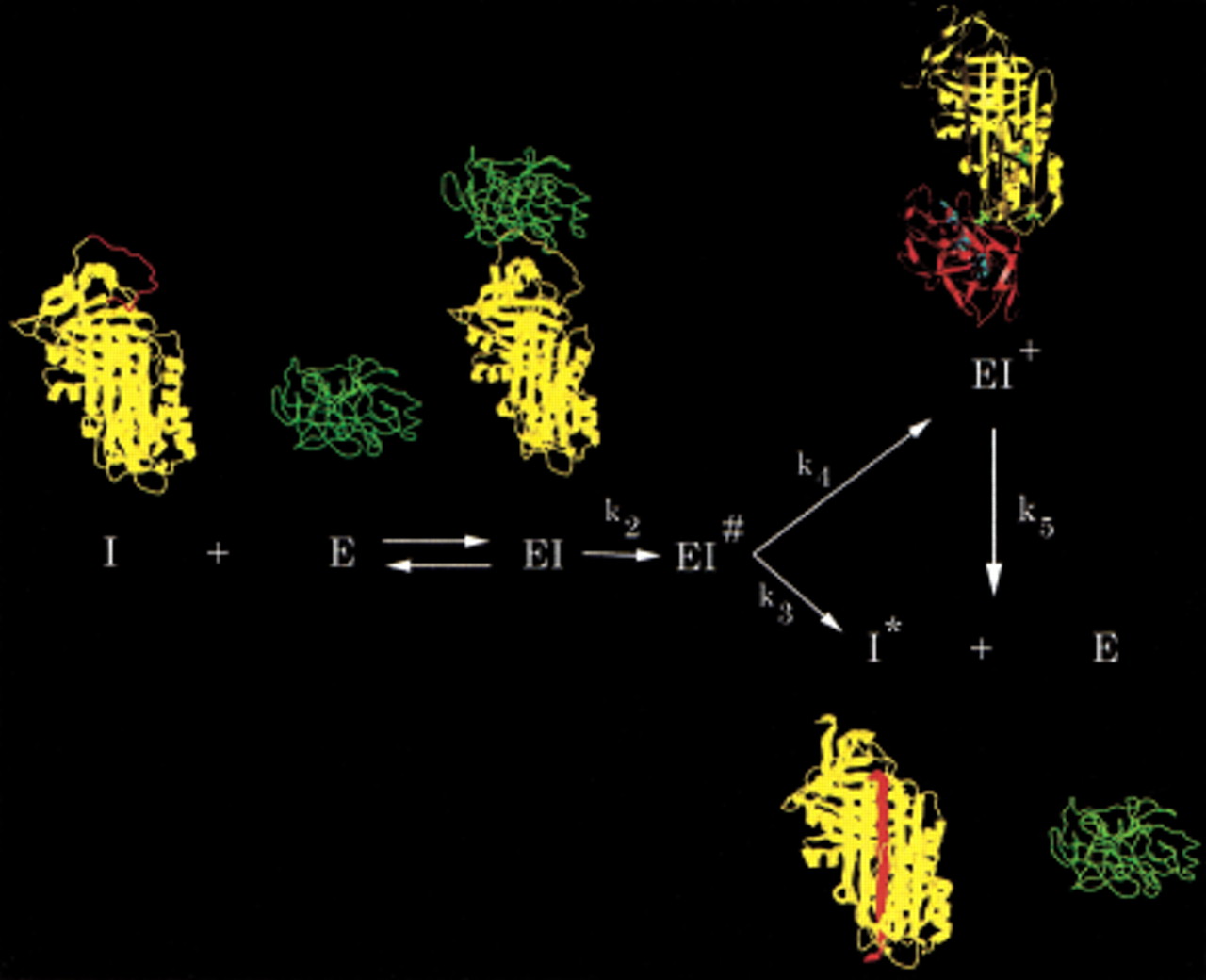
X-ray structures of several cleaved serpins had already shown that the previously exposed reactive center loop of the native serpin underwent a remarkable change in conformation on cleavage, with integration into the principal β sheet of the protein (sheet A) as a central strand and with the consequent need for this β sheet to be broken open to permit insertion of the cleaved reactive center loop. Recent structural studies by fluorescence resonance energy transfer (Stratikos and Gettins 1999) and by X-ray crystallography (Huntington et al. 2000) have shown that the serpin moiety in the kinetically trapped covalent serpin:proteinase complex very closely resembles the loop-inserted structure present in cleaved serpins and that the covalently linked proteinase has moved over 70 Å from its initial docking site with the reactive center loop (Fig. 1). The X-ray structure of the α1-proteinase inhibitor (serpin):trypsin (proteinase) complex further showed that the structural basis for kinetic trapping, and hence inhibition of the proteinase, is a gross distortion of the active site of the proteinase and repositioning of the acyl ester intermediate (Huntington et al. 2000), resulting from steric compression of the proteinase against the base of the serpin. Critical to generating this compression is that the reactive center loop that inserts into β sheet A, and to which the proteinase active site serine (or cysteine in the case of cysteine proteinases) is covalently linked, be just long enough to allow the proteinase to be squeezed in at the bottom of the serpin. If the loop is too long, there is too much slack to generate the compression; if it is too short, the proteinase cannot reach the bottom of the serpin.
For the serpin inhibition “machine” to function properly, there are thus many structural requirements that must be met. First, the serpin must fold into the correct metastable state; second, this state must be stable long enough for reaction with proteinase; third, the reactive center loop must retain residues at the scissile bond that are recognized by the cognate proteinase and at the hinge point for insertion into β-sheet A that are compatible with burial into a β sheet; fourth, the composition of β-sheet A must allow splitting of the sheet to accommodate the new strand; fifth, other secondary structure elements that contact β-sheet A above or below must allow the necessary movement of the sheet during loop insertion at a fast enough rate to compete with the substrate branch of the pathway; and sixth, other structural features that might impede movement of the proteinase to the bottom of the serpin, such as helix F, must be able to move out of the way during proteinase passage. Irving and colleagues have shed considerable light on which residues may be involved in meeting these requirements from their sequence comparisons of 219 serpins (Irving et al. 2000). It is striking how many of the ∼50 conserved residues are buried in locations related to loop insertion into β-sheet A; either within the sheet flanking the site of insertion or in regions underlying the sheet. This insight has provided excellent starting points for future mutagenesis studies that will allow more precise identification of which of the requirements enumerated above are met by which conserved residues.
Why Use Conformational Change?
The above description of proteinase inhibition by serpins, involving a well-coordinated series of machine-like movements, invites the question of why such a complicated inhibition mechanism has evolved when there are numerous examples of alternative lock-and-key type inhibitors of proteinases available in each of the different mechanistic classes, including serine and cysteine proteinases. Probably the most important reason is the opportunities for regulation that are so afforded, which are absent from reversible, rigid noncovalent inhibition. In this regard, it is significant that, as Irving and colleagues point out (Irving et al. 2000), the distribution of serpins and the processes in which they are involved appear to be those that have arisen as a result of the organism being multicellular. These are processes where inhibition per se is not sufficient: In addition, there must be regulation of inhibition.
As proteinase inhibition by a serpin is a biochemical reaction, there is an associated rate of inhibition that can, in principle, be increased or decreased by allosteric effectors. The dramatic modulation by heparin of the rate of inhibition of blood coagulation proteinases by the serpin antithrombin is the best-studied example of such rate regulation in a serpin (Olson et al. 1992), but it is unlikely to be the only one. The branched nature of the serpin inhibitory pathway also ensures that there are two possible products of the reaction between serpin and proteinase: kinetically trapped complex and cleaved, conformationally altered, serpin. Examples of biological function associated with the cleaved serpin, such as antiangiogenic activity for cleaved antithrombin (O'Reilly et al. 1999) and chemotactic activity for cleaved α1-antichymotrypsin (Potempa et al. 1991) or heparin cofactor II (Church et al. 1991) suggest why operation of this branch might not be inefficient or wasteful but actually be desirable. Cofactor proteins that bind to the serpin and promote or inhibit loop insertion could also alter the relative proportions of cleaved versus complexed serpin and so regulate the biological function associated with the cleaved form. Because serpin effectors such as heparin can also be physically localized, for example, by being on the vascular surface, they allow control of the rate of serpin inhibition in a spatially regulated manner. Finally, the ability of the metastable serpin conformation to convert to the more stable, latent (inactive) conformation provides the opportunity to control the active lifetime of a serpin by promoting or inhibiting such conversion. Here the best-documented example is the stabilization of the active conformation of plasminogen activator inhibitor-1 (PAI-1) by bound vitronectin (Wiman et al. 1988). Dissociation of vitronectin allows rapid inactivation of the PAI-1 by spontaneous conversion to its most stable, latent, state.
Breakdown of the Machine
As with any machine that relies on moving parts, inhibitory serpins that use the mechanism in Figure 1 are prone to malfunction when mutations interfere with the necessary motions. There are thus many examples of natural serpin variants that result in diseases such as thrombosis, emphysema, and hemorrhage (Stein and Carrell 1995). The mutations have involved changes not only in the reactive center loop but also in those conserved regions identified by Irving and colleagues (Irving et al. 2000) as being critical to the functioning of the conformational changes needed for inhibition. In addition, the use by serpins of a transformation of an exposed loop into a strand of β sheet has permitted opportunities for inappropriate insertion of the loop of one molecule into the sheet of another, leading to polymerization. This is now well documented in the case of α1PI polymerization leading to emphysema (Elliott et al. 1998) and of neuroserpin leading to dementia (Davis et al. 1999).
Given the many ways in which serpins can malfunction as a result of their complex inhibition mechanism, it is therefore all the more striking that they are so widely used as proteinase inhibitors. The principal human plasma proteinase inhibitors, including those of blood coagulation, fibrinolysis, and complement activation, are nearly all serpins. It is clear, therefore, that the advantages of regulation alluded to above must far outweigh the disadvantages that arise when the mechanism goes wrong. It is also striking that our understanding of the specific details of regulation for most serpins is still very limited.
Phylogenetic Analysis as a Structural Tool
The phylogenetic analysis of serpin sequences by Irving and colleagues (Irving et al. 2000) has revealed remarkable patterns of residue and structural conservation within the whole family that is fundamental to operation of the serpin machine. As more sequences from different species become available for a given serpin, it should be possible to extend such analyses to also reveal common surface elements that might be involved in hitherto unsuspected, but functionally important, interactions with other cofactor proteins. More generally, the use of phylogenetic analysis on other protein families should provide great insight into functionally important regions, whether needed for enzymatic activity or binding to other macromolecules. Examples of such successful analyses already exist for several abundant protein families such as zinc finger DNA-binding domains and SH2 and SH3 modular signaling domains (Lichtarge et al. 1996a) as well as G proteins (Lichtarge et al. 1996b).
I thank Steven Olson for helpful comments on the text and Efstratios Stratikos for the figure of the serpin pathway.
Notes
[1] Corresponding author.
Notes
[2] E-MAIL [email protected]; FAX (312) 413-0364.
[3] Article and publication are at www.genome.org/cgi/doi/10.1101/gr.168900.
REFERENCES
- ↵C. Anfinsen(1973) Science 181:223–230.
- ↵M.J. BandaA.G. RiceG.L. GriffinR.M. Senior(1988) J. Biol. Chem. 263:4481–4484.
- ↵F.C. ChurchC.W. PrattM. Hoffman(1991) J. Biol. Chem. 266:704–709.
- ↵R.L. DavisA.E. ShrimptonP.D. HolohanC. BradshawD. FeiglinG.H. CollinsP. SondereggerJ. KinterL.M. BeckerF. Lacbawan(1999) Nature 401:376–379.
- ↵P.R. ElliottD. BiltonD.A. Lomas(1998) Am. J. Respir. Cell Mol. Biol. 18:670–674.
- ↵P.G.W. GettinsP.A. PatstonS.T. Olson(1996) Serpins: Structure, function and biology. (Landes, Austin, TX).
- ↵M. HoffmanC.W. PrattR.L. BrownF.C. Church(1989) Blood 73:1682–1685.
- ↵J.A. HuntingtonR.J. ReadR.W. Carrell(2000) Nature 407:923–926.
- ↵J.A. IrvingR.N. PikeA.M. LeskJ.C. Whisstock(2000) Genome Res. 10:1845–1864.
- ↵O. LichtargeH.R. BourneF.E. Cohen(1996a) J. Mol. Biol. 257:342–358.
- ↵O. LichtargeH.R. BourneF.E. Cohen(1996b) Proc. Natl. Acad. Sci. 93:7507–7511.
- ↵S.T. OlsonI. BjörkR. ShefferP.A. CraigJ.D. ShoreJ. Choay(1992) J. Biol. Chem. 267:12528–12538.
- ↵M.S. O'ReillyS. Pirie-ShepherdW.S. LaneJ. Folkman(1999) Science 285:1926–1928.
- ↵P.A. PatstonP. GettinsJ. BeechemM. Schapira(1991) Biochemistry 30:8876–8882.
- ↵F.C. PetersonN.C. GordonP.G.W. Gettins(2000) Biochemistry 39:11884–11892.
- ↵J. PotempaD. FedakA. DubinJ. Travis(1991) J. Biol. Chem. 266:21482–21487.
- ↵S. StefanssonD.A. Lawrence(1996) Nature 383:441–443.
- ↵P.E. SteinR.W. Carrell(1995) Nature Struct. Biol. 2:96–113.
- ↵E. StratikosP.G.W. Gettins(1999) Proc. Natl. Acad. Sci. 96:4808–4813.
- ↵B. WimanÅ. AlmquistO. SigurdardottirT. Lindahl(1988) FEBS Lett. 242:125–128.