
A Preliminary Gene Map for the Van der Woude Syndrome Critical Region Derived from 900 kb of Genomic Sequence at 1q32–q41
- Brian C. Schutte1,7,
- Bryan C. Bjork1,4,7,
- Kevin B. Coppage1,7,
- Margaret I. Malik1,
- Simon G. Gregory5,
- Deborah J. Scott5,
- Luci M. Brentzell6,
- Yoriko Watanabe1,
- Michael J. Dixon6, and
- Jeffrey C. Murray1,2,3,4,8
- Departments of 1Pediatrics, 2Biological Sciences, and 3Preventive Medicine and Environmental Health, and 4Program in Genetics, University of Iowa, Iowa City, Iowa 52242 USA; 5Sanger Centre, Hinxton, Cambridgeshire B10 1SA, UK; and 6Departments of Dental Medicine and Surgery, University of Manchester, Manchester M13 9PT, UK
Abstract
Van der Woude syndrome (VWS) is a common form of syndromic cleft lip and palate and accounts for ∼2% of all cleft lip and palate cases. Distinguishing characteristics include cleft lip with or without cleft palate, isolated cleft palate, bilateral lip pits, hypodontia, normal intelligence, and an autosomal-dominant mode of transmission with a high degree of penetrance. Previously, the VWS locus was mapped to a 1.6-cM region in 1q32–q41 between D1S491 and D1S205, and a 4.4-Mb contig of YAC clones of this region was constructed. In the current investigation, gene-based and anonymous STSs were developed from the existing physical map and were then used to construct a contig of sequence-ready bacterial clones across the entire VWS critical region. All STSs and BAC clones were shared with the Sanger Centre, which developed a contig of PAC clones over the same region. A subset of 11 clones from both contigs was selected for high-throughput sequence analysis across the ∼1.1-Mb region; all but two of these clones have been sequenced completely. Over 900 kb of genomic sequence, including the 350-kb VWS critical region, were analyzed and revealed novel polymorphisms, including an 8-kb deletion/insertion, and revealed 4 known genes, 11 novel genes, 9 putative genes, and 3 psuedogenes. The positional candidates LAMB3, G0S2, HIRF6, and HSD11 were excluded as the VWS gene by mutation analysis. A preliminary gene map for the VWS critical region is as follows: CEN-VWS33-VWS34-D1S491-VWS1-VWS19-LAMB3G0S2-VWS26-VWS25-HSD11-ADORA2BP-VWS17-VWS14-HIRF6-VWS2-VWS18-D1S205-VWS23-VWS20-VWS30-VWS31-VWS35-VWS37VWS38-HIPP-RNASEH1P-VWS40-VWS42-VWS41-TEL. The data provided here will help lead to the identification of the VWS gene, and this study provides a model for how laboratories that have a regional interest in the human genome can contribute to the sequencing efforts of the entire human genome.
There are >300 described syndromes that have cleft lip and palate as an associated characteristic (OMIM, http://www.ncbi.nlm.nih.gov/Omim/). Van der Woude syndrome (VWS) is the most common form of syndromic cleft lip and palate and accounts for ∼2% of all cleft lip and palate cases (OMIM no. 119300). VWS has been recognized for more than a century (Murray 1860) and was assigned its eponym following Anne Van der Woude's description of the disorder in 1954. Distinguishing characteristics include cleft lip with or without cleft palate, isolated cleft palate, bilateral lip pits, hypodontia, normal intelligence, and an autosomal-dominant mode of transmission with a high degree of penetrance (Burdick et al. 1985). VWS is distinguished from nonsyndromic cleft lip and palate by the presence of lower lip pits, which are found in most affected individuals with the disorder (Janku et al. 1980; Shprintzen et al. 1980; Burdick et al. 1985). The unusual lip pits seen in VWS are believed to be an embryonic remnant from an early stage of development (Onofre et al. 1997), and are rarely seen in other settings. There are no other systemic, cognitive, or craniofacial anomalies to differentiate VWS from nonsyndromic forms of clefting.
Isolated clefts of the palate (CPO; secondary palate defects) are genetically and embryologically distinct from clefts that include the lip or the lip and palate together (CL/P; primary palate defects;Fraser 1955). VWS is the only single-gene form of clefting in which affected individuals within the same family commonly have either isolated cleft palate only or clefts of the lip and palate. This unique feature suggests that VWS may arise from an abnormality in a gene that disrupts a very early stage of palate development when a common factor is involved in the formation of both the primary and secondary palates.
Positional cloning of the VWS gene has progressed through genetic and physical mapping. Initially, the locus for VWS was suggested through the reporting of a patient with a large cytogenetic anomaly at 1q32–q41 by Bocian and Walker (1987) and by a suggestion of linkage to the Duffy blood group by Wienker et al. (1987). Murray et al. (1990)confirmed linkage of Van der Woude syndrome to 1q32, and subsequently, two microdeletions (Sander et al. 1994; Schutte et al. 1999) as well as individual recombinants (Schutte et al. 1996) further narrowed the region to a 1.6-cM region between the flanking markers D1S491 and D1S205. The identification of deletion mutations in three independent cases of VWS (Bocian and Walker 1987; Sander et al. 1994; Schutte et al. 1999), suggest that VWS is caused by haploinsufficiency of a gene at the VWS locus (Schutte et al. 1999). Haploinsufficiency is a common theme in autosomal-dominant clefting syndromes that include Waardenburg syndrome (OMIM no. 193500), Basal Cell Nevus syndrome (OMIM no. 109400), Rieger syndrome (OMIM no. 180500), Treacher Collins syndrome (OMIM no. 154500), and Stickler syndrome (OMIM no. 108300, 184840). In these syndromes, haploinsufficiency is evidenced by deletions and/or loss-of-function mutations (Lu-Kuo et al. 1993; Wu et al. 1993; Semina et al. 1996; Edwards et al. 1997; Wicking et al. 1997; DeStefano et al. 1998; Snead and Yates 1999). Thus, from a VWS mutation search, we expect to find a range of loss-of-function mutations in one of the positional candidates in addition to the three previously identified deletions.
The autosomal-dominant clefting syndromes described above also suggest the types of genes that would make ideal candidates for the VWS locus. Those genes encode for either transcription factors (Tassabehji et al. 1992; Semina et al. 1996), extracellular matrix proteins (Ahmad et al. 1991) or proteins involved in signal transduction (Johnson et al. 1996). Additional candidate functions for the VWS gene can be deduced from transgenic mice whose phenotype includes an orofacial cleft. To date, ∼30 knockout strains of mice exhibit some form of orofacial clefts (http://tbase.jax.org), and the product of those genes includegrowth factors in addition to transcription and signaling factors (for review, see Schutte and Murray 1999). Although genes with these functions are excellent candidates for the VWS locus, we note that not every gene involved in an autosomal-dominant clefting syndrome has such obvious developmental functions (Dixon et al. 1997).
To identify the gene responsible for VWS, we constructed a contig of bacterial clones that spans the VWS locus. STS content analysis and large-scale sequencing of this entire contig resulted in the identification of 4 known genes, 11 novel genes, 9 putative genes, and 3 psuedogenes in the 1.1-Mb region surrounding the 350-kb VWS critical region. In addition, mutation analysis excluded several positional candidates for the VWS locus.
RESULTS
Identification of Bacterial Clones from the VWS Critical Region at 1q32–q41
In a previous study (Schutte et al. 1996), we constructed a physical map of the VWS critical region composed of a 4.4-Mb contig of yeast artificial chromosomes (YACs). From that contig, we demonstrated that the VWS critical region, as defined by genetic recombinants at D1S491 and D1S205, was contained within a single 850-kb CEPH YAC clone, 785B2. To facilitate the identification of transcribed sequences in the VWS critical region, a contig of bacterial artificial chromosome (BAC) clones was constructed.
BAC clones were identified by PCR screening of the California Institute of Technology (CITB) human BAC DNA pools (B and C libraries) (Kim et al. 1996). Initially, the sequence-tagged sites (STSs) used for this screen (Table 1) were developed from three sources. The first set of STSs was obtained directly from the previous YAC physical map, which included five genetic markers—D1S245, D1S471, D1S491, D1S70, and D1S205—and five YAC end-clones—yAS9L, yAS9R, yAS10L, yAS2R, and yAS8L. When the BAC library was screened with these STSs, only a subset yielded full BAC addresses (Table 1). The STSs D1S205 and yAS8L identified the same BAC address, suggesting that they were located near each other.
STSs Used to Construct Contigs of BAC Clones
The second set of STSs was developed from transcripts that were located at chromosome bands 1q32–q41. From a screen of 36 genes from 1q32–1q41 (Table 2), 4 of the genes mapped to the VWS critical region—HSD11, G0S2, Hs.179758 (VWS31), and LAMB3. One other gene, TRAF5, was contained on several YACs near the VWS critical region but did not amplify a product from YAC 785B2, suggesting that it was located just outside of the VWS critical region (data not shown). When the STSs fromHSD11, G0S2, Hs.179758, and LAMB3 were screened against the BAC library, all but the STS for LAMB3 identified a full BAC address.
Genes from 1q32–q41 Tested as Positional Candidates for VWS
Finally, the third set of STSs was developed from sequences derived from the YAC clone 785B2. As described in Methods, the STSs were derived from sequences from cosmid subclones of the YAC (c53), island rescue PCR (IR2, IR6, IR7), and Alu-splice PCR (AS1.30, AS3.23). All of these STSs identified BAC addresses, except AS1.30 and IR2.
In total, 16 BAC clones were identified with these three sets of STSs.
Assembly of BAC Clones
Initially, the order and orientation of the BAC clones were investigated by STS content analysis with all of the STSs against all of the BAC clones (Fig. 1). The analysis indicated that the BAC clones fell into two nonoverlapping contigs. The larger contig included BAC clones 55i10–11c7, whereas the smaller contig included clones 432o17–189L14. At this point, all of the BAC clones and STSs used to construct these contigs were shared with the Sanger Centre. With these resources, the Sanger Centre constructed two independent contigs, ctg320 and ctg348 (http://webace.sanger.ac.uk/cgi-bin/display?db=acedb1). Importantly, the clones contained in these contigs were derived from a different library, the human Roswell Park Cancer Institute (RPCI) PAC library (Ioannou et al. 1994). A subset of the clones from these contigs is shown in Figure 1.
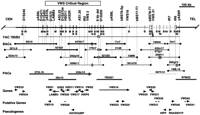
Physical and gene map of the VWS critical region. The VWS critical region is defined by genetic recombinants at D1S491 and D1S205 (open box at top). The CEPH YAC clone 785B2 contains both flanking markers in the original YAC contig (Schutte et al. 1996). STS content for this YAC and each of the BAC clones is indicated by long vertical lines. Restriction sites are indicated by short vertical bars and includeBssHII (B), MluI (M), NotI (N), andNruI (Nr). Parentheses indicate restriction sites that were absent on the indicated clone. The specific address for each BAC and PAC clone is shown. Sequencing of these clones is complete (thick line), partial (thick, shaded line), or only at their ends (thin lines). The sequence at the T7 (■) and Sp6 (●) ends for each BAC clone was determined. The presence and location of the 8-kb deletion/insertion polymorphism is indicated by an open rectangle. The order and orientation of genes, putative genes, and psuedogenes are indicated with arrowheads. For the putative genes VWS26,VWS37, VWS38, and VWS42, the direction of transcripton could not be deduced (flat bar). See text for definitions of genes, putative genes, and psuedogenes.
To complete the contig across this region, the CITB BAC library was rescreened with two additional STSs—VWS18 and bBS77-T7. The STS for VWS18 was derived from the 3′ end of the EST 222591. The 5′ end of this EST was identical to the STS yAS8L. Two new BAC clones were identified by the STS VWS18—21d8 and 438f6. The STS bBS77-T7 was derived from the sequence of the T7 end of the BAC clone 438f6 and identified the BAC clone 501d19. STSs were produced from the sequence of each end of clone 501d19. When they were tested against the BAC clones from the two contigs, the appropriate product was amplified from BAC clones from each contig (Fig. 1), suggesting that 501d19 spanned the gap between the two contigs.
As an independent test of the order and orientation of the BAC clones, a restriction map was generated for each BAC clone and for the YAC clone 785B2 (Fig. 1). The restriction map of the BAC clones was very similar to the restriction map of the YAC clone. The few sites that were different may represent sequence variants. One such site is theNotI site predicted to be about 330 kb from the centromeric end of the YAC clone 785B2. To test this hypothesis, the sequence from a subclone from this YAC clone that contained this NotI site was compared with the sequence from the clone 447d17 (see next section), which lacks this NotI site (Fig. 1). As predicted, the sequence of the YAC subclone contained the NotI site, GCGGCCGC, but the corresponding sequence at position 137083 of the BAC clone was GCGGCCAC. Similarly, a MluI polymophism was observed in the PAC clone 434o14 at position 126196. In addition, the restriction map provided an independent test for the overlap between the BAC clone 501d19 with the distal clones 189h6, 432o17, 672p1, and 259f11. Each of these distal clones contained a NotI site near their centromeric ends (Fig. 1). The BAC clone 501d19 contained a corresponding NotI site at its distal end, providing further evidence that these clones overlap. As final confirmation of the order, orientation, and overlap of the clones in this contig, the sequence from all the BAC ends was generated (GenBank accession nos.AQ853159–AQ853198) and aligned with the sequence contigs described in the next section. Together, these results confirm that these 19 BAC clones represent a complete contig that spans ∼1.1 Mb at chromosome bands 1q32–q41.
Analysis of High-Throughput Genomic Sequence
In preparation for high-throughput DNA sequence analysis at the Sanger Centre, all clones from both the BAC contig and the PAC contigs were mapped by FISH to verify their position at 1q32–q41 and fingerprinted to determine the degree of overlap (http://webace.sanger.ac.uk/cgi-bin/display?db=acedb1). On the basis of the fingerprint analysis, 11 clones from both libraries were chosen for high-throughput sequencing. The Sanger Centre has completed sequence analysis for all but two of the clones (Fig. 1; Table3). The sequence in GenBank for the clone 55i10 (GenBank accession no. AL035408) consisted of four large noncontiguous sequence islands. To complete the sequence for this clone, PCR primers were designed at the end of the sequence islands. PCR experiments were performed with every possible combination of primer pairs to join the sequence islands. Three of the primer pairs amplified products that were 734, 308, and 771 bp in length. Because these products were relatively small, the PCR primers were used to directly sequence the BAC clone (GenBank accession nos. AQ853199–AQ853201), thereby connecting these short sequence gaps. The complete sequence for 55i10 is 280944 bp.
Summary of Genomic Sequence of BAC/PAC Clones from the VDWS Critical Region
To date, >900 kb of sequence has been generated over the 1.1-Mb region. The sequences for the individual clones were assembled into two sequence contigs. The larger sequence contig is 721875 bp in length and includes the entire VWS critical region. The VWS critical region was defined as the sequence between the recombinant markers D1S491 and D1S205 (Schutte et al. 1996). To determine the length of this region, the sequences for these two markers were aligned with the 720-kb contig. The nucleotide distance between these markers is 350 kb, thus defining the physical interval for the VWS critical region.
Identification of Genes
As a first step toward constructing a complete gene map of this region, we analyzed all available sequences from this region by sequence similarity searches against the nonredundant nucleotide and peptide sequence database and nucleotide sequence EST database in GenBank. From this analysis, known genes, novel genes, putative genes, and psuedogenes were identified. In this study, known genes are defined as genomic sequences that are nearly 100% similar to a cDNA sequence derived from a gene of known function; the only sequence differences being the presence of introns and possible sequence variants. Novel genes are defined as genomic sequences that are nearly 100% similar to a cDNA or EST sequence and contain additional evidence of being a true transcript such as consensus splice sites that flank the regions of identity and/or a polyadenylation site and polyadenylation sequence in the cDNA. Putative genes also show nearly 100% similarity to at least one EST sequence, but lack the additional evidence of being a true transcript. Pseudogenes are defined as genomic sequences that have near identity with a known gene, but generally lack introns and contain at least one mutation that disrupts the ORF of the published cDNA sequence. A total of 4 known genes, 11 novel genes, 9 putative genes, and 3 pseudogenes were identified. For each of these, we listed the genomic clone that contains the homologous sequence (Table 3). We also listed the name of the cDNA or EST sequence with the most significant similarity score, and whether any contain polyadenylation sites and sequences (Table 4). Table 4 also includes the gene clusters from human, mouse, and rat that display near sequence identity and the minimum number of exons and genomic size. The location and direction of transcription for each potential transcript is diagrammed in Figure 1.
Genes, Putative Genes, and Psuedogenes from the VWS Critical Region
The sequence similarity search identified four known genes—LAMB3, G0S2, HSD11, and HIRF6. Mutations in LAMB3 cause the autosomal-recessive blistering disease Herlitz junctional epidermolysis bullosa (Pulkkinen et al. 1994). G0S2 encodes a small protein that is involved in the G0/G1 switch (Russell and Forsdyke 1991).HSD11 encodes for an enzyme that catalyzes the interconversion of cortisol with its inactive form cortisone. There are two isozymes of this protein, and mice lacking the homolog for this isozyme displayed decreased glucocorticoid-inducible responses and were resistant to hyperglycemia caused by obesity or stress (Kotelevtsev et al. 1997).HIRF6 is the human homolog of the mouse interferon regulatory factor 6 and belongs to a family of transcription factors that regulate the expression of interferon and interferon-stimulated genes (Grossman et al. 1996).
In addition, we identified genomic sequences with high similarity (83%–93% identity) to three other known genes, the human adenosine B2 receptor (ADORA2B), the HSC70-interacting protein (HIP), and the ribonuclease H1 (RNASEH1). The genomic sequence similar to the ADORA2B gene was identical to the previously identified pseudogene ADORA2BP (Jacobson et al. 1995). The ADORA2BP pseudogene lacked introns and contained numerous sequence differences that disrupted the ORF. Similarly, the sequences similar to the HIP and RNASEH1P genes also lack an ORF and lack introns indicating that they are also pseudogenes. The HIP pseudogene was also disrupted by two tandemly arrayed AluY repetitive elements that were flanked by a perfect 18-bp duplicated sequence.
The novel gene VWS1 appears to be the human homolog of the rat Ca2+/calmodulin-dependent protein kinase 1γ (CaMK1γ; B.C. Bjork and J.C. Murray, in prep.). The rat gene was isolated from brain and belongs to a family of serine/threonine protein kinases (Yokokura et al. 1997). Also of note, the VWS2 gene is very similar to a yeast ORF of unknown function (GenPept P40498). The remaining 18 genes or putative genes display high similarity with EST sequences, including those derived from other species. However, no functional information is available for the human gene or its homologs.
Mutational Analysis of LAMB3, G0S2, HIRF6, and HSD11
To date, three independent deletion mutations were demonstrated to cause VWS (Bocian and Walker 1987; Sander et al. 1994; Schutte et al. 1999), indicating that VWS is probably caused by haploinsufficiency of the VWS gene. We performed mutation analysis for LAMB3,G0S2, HIRF6, and HSD11, because their complete genomic structure (Table 4) was readily determined by aligning the full-length cDNA sequence with the genomic sequence. Although these genes are poor functional candidates, our approach is to screen all positional candidates, regardless of function, as different mutations in the same gene may cause different genetic disorders (Smith et al. 1994; Biesecker 1997). From our mutation screen, we observed three sequence variants in the LAMB3 gene, a single variant in theHSD11 gene, and no variants in the G0S2 norHIRF6 genes (Table 5). All four of these variants were observed in unaffected control samples, demonstrating that they were not etiologic mutations. These results, therefore, exclude LAMB3, G0S2, HIRF6, andHSD11 as candidates for the VWS gene.
Sequence Variants Identified from Known Genes in the VWS Critical Region
Comparison of Gene Recognition Programs
Because no etiologic mutations were found in the LAMB3,G0S2, HIRF6, and HSD11 genes, additional exons must be identified and mutation analysis performed. In addition to identifying exons from full-length cDNA sequence, we performed a preliminary analysis of the genomic sequence with gene recognition programs. However, the ability of gene recognition programs to detect specific exons varies significantly (Burge and Karlin 1997). To develop an efficient strategy for identifying putative exons, the gene recognition programs contained in Genotator (Harris 1997) were compared for their ability to detect the exons in the 270-kb region that contains the LAMB3, G0S2, HIRF6, andHSD11 genes (see Methods). The program Genotator performs sequence similarity searches of the GenPept and EST databases as well as gene recognition programs with Genscan (Burge and Karlin 1997), GeneFinder (Solovyev et al. 1994), GRAIL II (Xu et al. 1994), and Genie (Kulp 1996). Because the complete cDNA sequence for each of these genes tested is stored in GenBank, the GenPept search was very efficient at identifying the exons for these genes, only those exons that lacked an ORF failed to be identified with this tool (Fig. 2). Similarly, the dbEST search was also excellent for identifying exons for these genes. The exceptions were the exons located toward the 5′ end of the LAMB3 gene. This is expected, asLAMB3 is a large gene and sequences in dbEST are biased toward the 3′ ends of genes because the first-strand synthesis of the cDNA clones are generally made by priming the reverse transcription reaction with an oligo-dT primer. Because the number of genes and exons in this analysis was relatively small, specific comparisons between the programs are not valid. However, consistent trends were observed. Each of these programs failed to detect the first exon of these genes and exon 7 of LAMB3. One of the most important criteria for each of these gene prediction programs is the presence of a coding sequence. Because the first exon of each of these genes is not translated, the failure of these programs to predict the first exon is not unexpected. Exon 7 is short (64 bp) and can be skipped without disrupting the reading frame because the subsequent exon contains an AG dinucleotide adjacent to the real splice donor site. This second potential donor site was predicted to be the splice donor site in the gene prediction programs and places the predicted gene back in frame. In addition, within the 270-kb sequence that was analyzed, whenever two or more programs predicted an exon, that sequence was an exon (Murakami and Takagi 1998). From this analysis, top priority will be given to putative exons that are predicted by multiple programs. Sequence analysis with the programs in Genotator was subsequently performed on the entire 720-kb sequence that contains the VWS critical region. The graphical output from this analysis is available at our website (http://genetics.uiowa.edu/∼bschutte/genotator_results/). In addition, a complementary set of sequence analyses was performed by the Sanger Centre and the results are available at their website (http://webace.sanger.ac.uk/cgibin/webace?db=acedb1&class=Genome_Sequence).

Comparison of gene recognition programs. Genomic sequences containing the indicated genes were analyzed for exon content with the suite of programs contained in Genotator. Exons for each gene are numbered. Those that contain part of the translated ORFs are shaded. All other shaded boxes indicate a hit with the indicated analysis program.
Identification and Verification of Novel Polymorphisms
As demonstrated with the restriction site polymorphisms, it is possible to identify sequence variants by aligning the sequences from overlapping clones that are derived from the two different libraries. Because short tandem repeats are often polymorphic, we searched the genomic sequence from this region for short tandem repeats whose copy number was different in different clones. To verify that these short tandem repeats were polymorphic, primers were designed that flanked the repeats and were then used to genotype a normal control population. As expected, all of the short tandem repeats whose sequence lengths were different in the two clones were polymorphic (Table6). As new single nucleotide and short tandem repeat polymorphisms are discovered from this region, they are added to our web site (http://genetics.uiowa.edu/∼bschutte/polymorphisms/).
Novel STRP Markers from VWS Critical Region
In addition, when the sequences for the BAC clone 321i20 and PAC clone 782d21 were aligned, two regions of sequence variation were observed. Specifically, a 7922-bp sequence is absent at position 31766 of BAC clone 321i20 relative to the sequence present in PAC clone 782d21 at positions 1668–9591. To prove that this deletion in BAC 321i20 was not a sequencing or cloning artifact, all clones that span this region were tested with a set of primers that flanked the deleted sequences. As shown in Figure 1, the 7922-bp sequence was present in clones BAC 508k11 and PAC 782d21 but was absent in clones YAC 785B2, BACs 179n7, and 321i20. The absence of this sequence in three independent clones derived from two different libraries demonstrates that this nearly 8-kb region represents a deletion/insertion polymorphism. Additional genotyping was performed in a normal control population and showed that the 8-kb deletion/insertion polymorphism is quite common (Y. Watanabe and B. Schutte, in prep.).
A second region of sequence variation between these two clones was observed at position 25422 in BAC clone 321i20 and position 35012 in PAC clone 782d21. The region of sequence variation was 136 bp, and the obvious difference was that the PAC clone sequence was extremely G/C rich, whereas the BAC clone sequence was not. To determine whether the variation was a sequencing artifact, we designed sequencing primers to flank this region and resequenced both clones. The new sequence was virtually identical to the PAC clone sequence, indicating that the original sequence variation was due to a sequencing or sequence assembly error in BAC 321i20.
Mouse Syntenic Region for the VWS locus
Human genes that map to 1q32 have been localized to mouse chromosome 1F and 1H (Seldin 1994), suggesting that the mouse homolog for the VWS gene could map to either 1F or 1H (Fig. 3). In this study, both LAMB3 and TRAF5 were mapped to the 4.4-Mb YAC contig (Schutte et al. 1996) that contains the VWS critical region.LAMB3 is located near the proximal end of the critical region, whereas TRAF5 is located distal to the critical region. Thus, it is probable that the VWS gene is located between or very close to these two genes.
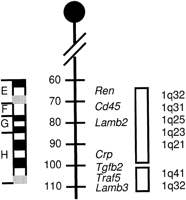
Mouse synteny map for human chromosome 1q32–q41. Ideogram (left) and genetic map (middle) for the distal end of mouse chromosome 1 are shown. Vertical rectangles at rightrepresent syntenic region at the indicated human chromosome band.
Because both the mouse Lamb3 (Aberdam et al. 1994) andTraf5 (Nakano et al. 1997) genes map to chromosome 1H, it is likely that the mouse homolog of the VWS gene also maps to mouse chromosome 1H. This information will allow us to more effectively identify positional and functional candidates from mouse. This is important because many new genes that are involved in early development are being identified from ESTs derived from embryonic tissues in mouse. No mouse phenotypes that include clefting map to this region.
DISCUSSION
The VWS locus was originally mapped by linkage analysis and microdeletions to a 1.6-cM region in 1q32–q41 between D1S491 and D1S205. In our efforts to positionally clone the VWS gene, gene-based and anonymous STSs were developed from the existing physical map and were then used to construct a contig of sequence-ready bacterial clones across the entire VWS critical region. All STSs and BAC clones were shared with the Sanger Centre, which developed a contig of PAC clones over this same region. A subset of 11 clones, derived from both the PAC and BAC contigs, were selected to represent the minimum tiling path across an ∼1.1-Mb region and were the template for large-scale genomic sequencing. All but two of the clones have been sequenced completely.
We analyzed over 900 kb of genomic sequence, including the entire 350-kb VWS critical region. Our efforts revealed that novel polymorphisms and sequence similarity searches led to the construction of a preliminary transcript map encompassing the entire VWS critical region that included 4 known genes, 11 novel genes, 9 putative genes, and 3 pseudogenes. This map permitted the precise localization and transcriptional orientation of LAMB3, G0S2, andHSD11. These known genes were mapped previously to this region, as well as the unmapped but previously described geneHIRF6. Our analyses also identified a collection of potential novel genes in the VWS critical region as evidenced by the identification of genomic sequences that were nearly 100% similar to EST sequences; many of these also contained consensus splice sites and polyadenylation sites, whereas the corresponding EST sequence contained polyadenylation sequences. Additional analyses were performed with gene recognition programs on 270 kb of contiguous sequence that contains the four known genes. No single analysis was 100% specific or sensitive at identifying exons. However, by setting criteria whereby an exon must be predicted by at least two of the programs, this suite of programs successfully identified 32 of 33 exons in these four genes that contain coding sequence. These results confirm the utility of these programs to predict exons and lay the foundation for identifying new exons from the region. The current gene map for the VWS critical region is as follows: CEN-VWS33-VWS34-D1S491-VWS1-VWS19-LAMB3-G0S2-VWS26-VWS25-HSD11-ADORA2BP-VWS17-VWS14-HIRF6-VWS2-VWS18-D1S205-VWS23-VWS20-VWS30-VWS31-VWS35-VWS37-VWS38-HIPP-RNASEH1P-VWS40-VWS42-VWS41-TEL. The VWS critical region was genetically defined by recombinant and deletion events at the markers D1S491 and D1S205 (Schutte et al. 1996). Thus, the genes located between these markers are positional candidates. From mutation analysis, four of these genes,LAMB3, G0S2, HIRF6, and HSD11, were excluded as candidates for the VWS gene. The contig of BAC clones presented here should greatly facilitate the identification of the gene associated with VWS.
The identification of a single gene involved in the etiology of Van der Woude syndrome will likely provide major insights into the more complex etiology of nonsyndromic cleft lip and palate. The extraordinary phenotypic overlap between VWS and nonsyndromic clefting, with only the lip pits being a differentiating factor, strongly suggests a common set of embryologic events. Because nonsyndromic clefting serves as a model for other complex traits thought to be secondary to both genetic and environmental factors, having this unique inroad into its etiology will prove beneficial in not only the specific case of clefting, but for other complex birth defects as well. Furthermore, it is possible that some of the unexplained cases of nonsyndromic clefting may, in fact, be allelic to VWS, but lack the lip pit component of the phenotype. Thus, substantial advances in the altering of recurrence risks from the 50% gene passage in the case of VWS to the lower 3%–5% recurrence risks usually given for nonsyndromic cleft lip and palate will also be of substantial benefit to families having a first affected child.
In summary, analysis of high-throughput genomic sequence is a powerful approach for developing a preliminary gene map and was greatly facilitated by the close collaboration with the Sanger Centre. Our commitment to developing a well-defined physical map at the outset and their commitment to making a sequence immediately available to the public provided us with the opportunity to investigate and analyze sequence in a real-time fashion. In addition, the interaction between our two groups was important to resolve sequence anomalies (the 8-kb deletion polymorphism and potential alignment errors) and to complete analysis of unfinished sequences. The latter will be a greater issue as the priority for the Human Genome Project shifts from finished sequence to a working-draft sequence (http://www.nhgri.nih.gov/NEWS/news.html). This collaborative effort provides a model for how laboratories that have a regional interest in the human genome can contribute to the sequencing efforts of the entire human genome. The resulting sequence information, along with the descriptive approaches, including recombinant and microdeletion mapping, and close clinical characterization of the syndrome itself, provide a targeted region most suitable for gene identification.
METHODS
Patient Identification
A total of 107 individuals diagnosed with VWS and 14 individuals with popliteal pterygia syndrome (PPS) were examined at various locations, including the University of Iowa, the Philippines, and Germany. VWS and PPS families were ascertained and examined by one or more geneticists or clinical collaborators, as described previously (Schutte et al. 1999). Individuals were considered to be affected with VWS if they had one or more of the following clinical phenotypes: cleft lip, cleft palate, hypodontia, or lower-lip pits. They were considered to have PPS if they had pterygia along with any of the classical VWS phenotypic characteristics. Nearly all pedigrees had at least one individual with lip pits. We included a few pedigrees in which no family member exhibited lip pits but at least one individual had a cleft lip with or without cleft palate and at least one individual with cleft palate only. We also included families with a single affected individual that showed lip pits along with one or more features of VWS. Development and mental performance appeared to be normal in all affected individuals in this study. Ten-milliliter samples of whole blood per kilogram of body weight were obtained from the adults and 1 ml of whole blood per kilogram of body weight was obtained from each of the children in the families studied.
PCR and DNA Sequencing
DNA from these individuals was purified from whole blood (Miller et al. 1988) or blood spots (Qiagen, Germany). Approximately 40 ng of template genomic DNA were analyzed by PCR by standard conditions (GDB no. 9798291). BAC DNA was prepared as described in Identification and Characterization of BAC clones (below). Direct sequencing of BAC clones was performed by end sequencing and primer walking and carried out on an Applied Biosystems DNA Sequencing System Model 373 with fluorescently labeled dye terminators as implemented in the Taq Dye Deoxy Terminator Cycle Sequencing Kit from Applied Biosystems (Foster City, CA). Sequencing reactions included 1–2 μg of BAC DNA and 20 pmoles of primer. For end sequencing of BAC clones, standard T7 (TAATACGACTCACTATAGGG) and Sp6 (ATTTAGGTGACACTATAG) primers were used.
Development of Novel STSs from the VWS Critical Region
Novel STSs from the VWS critical region were developed by three approaches. First, STS primer pairs were designed from the 3′ untranslated regions of 36 genes that were mapped previously to 1q32–q41 (Table 2). To identify genes that map to the VWS critical region, content analysis was performed with these STSs against the following DNA templates: human genomic DNA, a hamster somatic cell line containing human chromosome 1, and YAC clone 785B2.
Second, STSs were derived from the sequence of cosmid subclones from YAC clone, yCEPH785B2, which spans the entire VWS critical region. A total of 72 cosmid clones were obtained in the sCos-1 vector by established techniques (Sambrook et al. 1989). The sequence of the T3 and T7 ends of each cosmid was determined and was then analyzed by BLAST (Altschul et al. 1990) to identify sequences with similarity to known gene sequences and/or repeats. Oligonucleotide primer pairs were designed within unique, nonrepetitive end sequence. These STSs were also tested as above to verify that they map to the VWS critical region.
Third, STSs were also obtained from the sequence of clones generated by two previously described PCR-based methods, Island Rescue PCR (Valdes et al. 1994) and Alu–splice PCR (Morgan et al. 1992). PCR products yielded by these methods were cloned into pBSKII, after restriction enzyme digestion, and sequenced. As above, STSs were designed to unique sequence and mapped back to the VWS critical region.
Identification and Characterization of BAC Clones
BAC clones were identified by PCR screening of the Research Genetics (Huntsville, AL) human CITB BAC library. BAC DNA was purified as recommended with either a Genome Systems KB-100 Magnum or a Qiagen QIAquick-Maxi DNA purification kit. The resulting BAC DNA was quantified via UV spectrophotometry. Following NotI restriction enzyme digestion, BAC clones were sized by pulsed-field gel electrophoresis (PFGE) with a CHEF DR-II apparatus (BioRad) through a 1% agarose gel in 0.5× TBE (14°C, 200 V, 5–30-sec ramp time, 14 hr). Additional rare-cutting restriction enzymes were used to generate a restriction map of the BACs. DNA from each BAC was digested singly with BssHII, MluI, and NruI and doubly withMluI–NotI and NruI–NotI and then analyzed by PFGE.
Assembly of Unfinished Clone Contig Sequences
The sequence for BAC clone 55i10 is presently contained in four large unfinished sequence contigs—55i10.03033, 55i10.03548, 55i10.02402, and 55i10.1090. The sequence gaps between each of these contigs was filled by direct sequence analysis of the BAC clone 55i10 with the primers derived from the ends of the unfinished sequence contigs. Prior to sequence analysis, the orientation of the unfinished contigs and the size of the gaps was determined by PCR. Each primer from a single contig was paired with all possible combinations of primers from the other contigs and used in PCR reactions with 2 ng of 55i10 as template. For these experiments, standard PCR conditions were modified to include a 3-min, 72°C extension per cycle. Product sizes and primer pairs were as follows: 734 bp for oBS1158F, 5′-GCAGCCTTACTCAATCTGAGG-3′, and oBS1152R, 5′-CAGTCAAGAAGAAATGGGCT-3′; 308 bp for oBS1153F, 5′-GGCTACAAACCTGTACTGCA-3′, and oBS1155R, 5′-GCTATGTGCAGTGGATCACAC-3′; and 778 bp for oBS1154F, 5′-GTCTGTGCCTTCTCCATTAGC-3′, and oBS1159R, 5′-GGTCAGTTGTGTTGTGATTGTC-3′. These successfully linked contigs 55i10.03033–55i10.03548, 55i10.03548–55i10.02402, and 55i10.02402–55i10.1090, respectively. The resulting sequences were aligned and sequence ambiguities resolved with the computer program Sequencher (GeneCodes, Ann Arbor, MI).
Analysis of High-Throughput Genomic DNA Sequence
Genomic sequence from the Sanger Centre was downloaded as multiple, random Unfinished sequence contigs generated from specific clones in the sequencing pipeline as soon as they became publicly available on the Sanger Centre ftp site (ftp://ftp.sanger.ac.uk/pub/human/sequences/Chr_1/unfinished_sequence/). As the initial step in analyzing genomic DNA sequence, the repetitive elements within the genomic sequence were identified and masked with RepeatMasker 2 (http://ftp.genome.washington.edu/cgi-bin/RepeatMasker). Subsequently, BLAST (Altschul et al. 1990) analysis of these masked sequences was performed to find similarities to known gene and/or cDNA sequences in the nonredundant peptide sequence database and nucleotide sequence EST database in GenBank. Sequence similarity searches were repeated as each unfinished sequence contig was updated toward a complete Finished clone sequence contig. The finished sequences were aligned into two contigs, 721,875 and 137,679 bp (Figure 1). In addition, a 270-kb genomic sequence that contains the genesLAMB3, G0S1, HSD11, and HIRF6(nucleotides 90,000–360,000 of the 721-kb contig) was searched for putative exons with the suite of programs contained in Genotator (http://www-hgc.lbl.gov/inf/annotation.html). The graphical output from this analysis can be viewed at our website (http://genetics.uiowa.edu/∼bschutte/genotator_results).
Mutation Analysis of Candidate Genes by SSCP
To screen for sequence variants, oligonucleotide primers were designed in the introns flanking each exon containing coding sequence for the genes LAMB3, G0S2, HIRF6, andHSD11. Each primer pair was designed to amplify a 150–200-bp PCR product for increased sensitivity in detecting SSCPs (Sheffield et al. 1993). Consequently, larger exons required multiple, overlapping primer pairs.
The exon sequences were amplified from genomic DNA from a panel of 107 unrelated individuals diagnosed with VWS and 15 unrelated individuals with PPS by standard conditions. In parallel experiments, we used the same primers to amplify PCR products from a control panel of genomic DNA from 96 CEPH grandparents and parents (Dausset et al. 1990) to determine whether detected SSCP variants were potential etiologic mutations or normal sequence polymorphisms. In addition, the inheritance of any rare variants detected in the affected panel was studied in individual affected families to determine whether they segregated with the VWS phenotype. The amplified PCR products were denatured at 95°C for 5 min and electrophoresed for 5 hr at 20 W through 0.5× MDE (FMC, Rockland, MD) acrylamide gel that was cooled with a fan. DNA bands were visualized by silver staining and inspected for potential variants. DNA fragments representing potential variants were then excised from the gel and boiled in 50 μl of ddH2O. Ten microliters of the DNA suspension was used as template in 50-μl PCR reactions with the original primers. These PCR products were purified by extraction from 2% agarose gel with the Qiagen Gel Purification Kit (Chatsworth, CA) and sequenced in both directions. Parallel PCR products were amplified from genomic DNA of corresponding individuals possessing each potential sequence variant, purified from agarose gel, and sequenced. Sequences obtained from the variant bands were then compared with the common, normal sequence by the computer program Sequencher.
Genotyping of STRPs
Potentially polymorphic short tandem repeats were identified by aligning sequences from BAC and PAC genomic sequence by the program Sequencher (GeneCodes Corporation). PCR primers that flanked the repeats were designed with the program Primer 3.0 (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3.cgi). Standard PCR conditions were used to amplify these loci from 96 CEPH grandparents and parents (Dausset et al. 1990). The products were separated on 6% acrylamide denaturing gels for 2 hr at 60 W and visualized by silver staining.
Acknowledgments
We thank our collaborators at the Sanger Centre, especially the mapping, sequencing, and annotation teams, specifically Mark Vaudin and Richard Wooster for establishing the collaboration, Louise McDonald for assistance with mapping and Susan Rhodes for analysis and annotation. We acknowledge Bonnie Ludwig, Dave Spencer, Ann M. Basart, Nancy Leysens, Sheri Sekenske, Beth Pruessner, and Tom Forsha for contributory technical assistance, and Sandy Daack-Hirsch, Nancy Newkirk, and Buck Huppman for administrative support. We thank our colleagues Sue Kenwrick, Martin Tymms, T.J. Yen, Walter Becker, and Margit Burmeister for sharing cDNA sequence prior to publication. We thank our many clinical colleagues (listed in Schutte et al. 1999) and their patients for contributing samples for this study. This work was supported by National Institutes of Health grants P50-DE09170 and P60-DE13076 (J.C.M. and B.C.S.), R01-DE08559 (J.C.M.), P30-HD27748 (Frank Morriss and B.C.S.), Action Research grant S/P/3261 and Colgate-Palmolive (M.J.D.). Grant support was also provided to K.B.C. by the Pediatric Scientist Development Program of the National Institute of Child Health and Human Development administered by the Association of Medical School Pediatric Department Chairman.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.








