Abstract
A-to-I RNA editing, catalyzed by the adenosine deaminase acting on RNA (ADAR) enzymes, is a posttranscriptional process that modifies RNA sequences and diversifies the transcriptome. ADARs bind to double-stranded RNA (dsRNA), and their specificity and efficiency are affected by the structural properties of these substructures. In most cases, the dsRNA structure arises from homology between two segments of the same RNA molecule that fold into RNA stem structures. Another possible source of dsRNA is cotranscription of sense and antisense strands of the same genomic region. Binding of these complementary, naturally occurring, antisense transcripts (NATs) results in a perfect RNA duplex, which may be targeted by ADARs. To explore the scope of ADAR editing of NAT-derived dsRNA, we examine editing levels at genome locations where both strands are transcribed. Our findings indicate that editing is rare in regions for which both strands cotranscribe. Moreover, even when RNA editing does occur in NAT regions, it is typically associated with secondary structures on a single strand, suggesting that editing depends on intramolecular structures rather than binding of NATs.
Double-stranded RNA (dsRNA) structures play a vital role in the intricate balance of the immune system. Many viruses are made of dsRNA or produce dsRNA. Sensing of viral dsRNA by pattern recognition receptors, such as melanoma differentiation–associated protein 5 (MDA5), triggers the innate immune response through the mitochondrial antiviral signaling protein (MAVS) pathway, leading to the production of interferons and proinflammatory cytokines (Feng et al. 2012; Brisse and Ly 2019; Hur 2019). Although this antiviral defense mechanism is essential for combating infections, inappropriate activation by self-derived dsRNA can result in unnecessary immune activation, potentially leading to autoinflammatory or autoimmune diseases (Peisley et al. 2011, 2012; Wu et al. 2013; Chen and Hur 2022; de Reuver and Maelfait 2024). In fact, long and nearly perfect dsRNA structures undergo strong negative selection and are depleted from the genome (Barak et al. 2020). Adenosine deaminase acting on RNA (ADAR) enzymes are essential in preventing the immune system from mistakenly identifying self-derived dsRNA as a viral threat, thereby suppressing unwanted immune responses (Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015; Hu et al. 2023). ADARs deaminate adenosine (A) within dsRNA substrates into inosine (I), thus disrupting base-pairing of immunogenic dsRNAs, or marking them otherwise, to prevent their recognition as viral by MDA5 and suppress inappropriate activation of the innate immune response (Liddicoat et al. 2015; Chung et al. 2018; Hu et al. 2023). Recent studies have highlighted the intricate interplay between ADAR enzymes and the MAVS pathway. For instance, it was shown that ADAR1 dampens MAVS signaling not only by editing the dsRNA but also through competitive binding of these substrates (Hu et al. 2023), underscoring the multifaceted regulatory role of ADARs in dsRNA metabolism.
Endogenous dsRNAs often arise from base-pairing between complementary regions within the same RNA molecule, resulting in folding of single-stranded RNA molecules into intramolecular hairpin or stem-loop structures. These structures are often targeted by ADARs and edited. A-to-I editing occurs frequently in repetitive elements that fold into stable secondary structures (Morse et al. 2002; Porath et al. 2017). For example, the vast majority of mRNA editing in humans occurs within Alu repetitive elements. These ∼300 bp elements are abundant and make up ∼10% of the genome. Binding of neighboring, reversely aligned, Alu elements results in stable intramolecular secondary structures, which are targeted by ADARs (Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004; Ramaswami and Li 2014). Indeed, the editing level of Alu elements depends on the distance between the element and its neighboring reversely oriented Alu element (Bazak et al. 2014).
However, an additional potential source for endogenous dsRNAs is intermolecular pairing of natural antisense transcripts (NATs). cis-NATs are endogenous RNA molecules transcribed from two opposing strands of the same genomic locus (Vanhée-Brossollet and Vaquero 1998; Lavorgna et al. 2004; Katayama et al. 2005; Conley and Jordan 2012; Wight and Werner 2013). Advancements in RNA sequencing (RNA-seq) technology have facilitated identification and characterization of widespread cis-NAT expression (Ozsolak and Milos 2011). Several independent studies have shown that NAT expression is common in many species. In humans, between 5% and 20% of all genes were found to have a cis antisense counterpart (Shendure and Church 2002; Yelin et al. 2003; Chen 2004; Katayama et al. 2005; Conley et al. 2008; He et al. 2008), and similar results were reported for the mouse (Kiyosawa et al. 2003), Drosophila (Misra et al. 2002), Arabidopsis (Meyers et al. 2004; Wang et al. 2014), and rice genomes (Osato et al. 2003). In addition, many long intergenic noncoding RNAs (lincRNAs) have been identified as antisense transcripts, further contributing to the prevalence of antisense transcription (Derrien et al. 2012; Pelechano and Steinmetz 2013; Werner et al. 2024). Because of their perfect complementarity, binding of two NATs can, theoretically, generate long stretches of uninterrupted dsRNA. Studies have shown that perfectly paired dsRNA longer than ∼50 bp undergoes extensive ADAR-mediated editing, as demonstrated in synthetic dsRNA substrates (Bass and Weintraub 1988; Wagner et al. 1989; Nishikura et al. 1991; Polson and Bass 1994; Bass 1997), viral dsRNA (Pfaller et al. 2018), and engineered guide RNA systems (Katrekar et al. 2022). If they exist, these long perfect dsRNAs may pose a risk of triggering the immune system and also serve as perfect editing substrates for ADAR enzymes (Feng et al. 2012). A recent report has provided evidence supporting A-to-I editing in certain cis-NAT formations (Li et al. 2022). However, compelling evidence for widespread in vivo ADAR-mediated editing of endogenous NATs remains elusive (Neeman et al. 2005).
Although nearly all editing sites result from the folding of single-stranded RNA, there is considerable interest in understanding the full scope of intramolecular cis-NAT editing. It has been argued that most ADAR targets do not pose an immunogenic risk, and the search for the few critical targets is ongoing (Li et al. 2022; Levanon et al. 2024). cis-NAT editing targets, which are long and perfect dsRNAs, are prime candidates for critical ADAR targets. Identifying these may help to better understand ADAR's role in preventing inappropriate immune activation.
In this study, we systematically investigate whether intermolecular pairing of cis-NATs is a prevalent source of ADAR editing substrates in the human transcriptome. Specifically, we aim to determine how common editing is in regions where both strands are transcribed, assess whether such editing originates from intermolecular dsRNA structures, and evaluate evidence for coordinated editing activity on both strands.
Results
We employed a genomic-based approach to identify candidate regions in the human genome in which both strands are expressed and edited concurrently (Fig. 1).
Exploring the nature of ADAR substrates in regions where both strands are expressed. A double-stranded RNA (dsRNA) structure, essential for ADAR activity, may arise from either intramolecular folding of the transcript or pairing with a naturally occurring antisense transcript. (Top) We identify genomic regions exhibiting expression of both the plus (green) and minus (pink) strands. (Middle) dsRNAs may form due to intermolecular pairing of antisense transcripts (left) or intramolecular secondary structures (right). In both cases, ADAR may bind and edit the dsRNA structures. In this work, we aim to map systematically the scope of cis-NAT dependent editing.
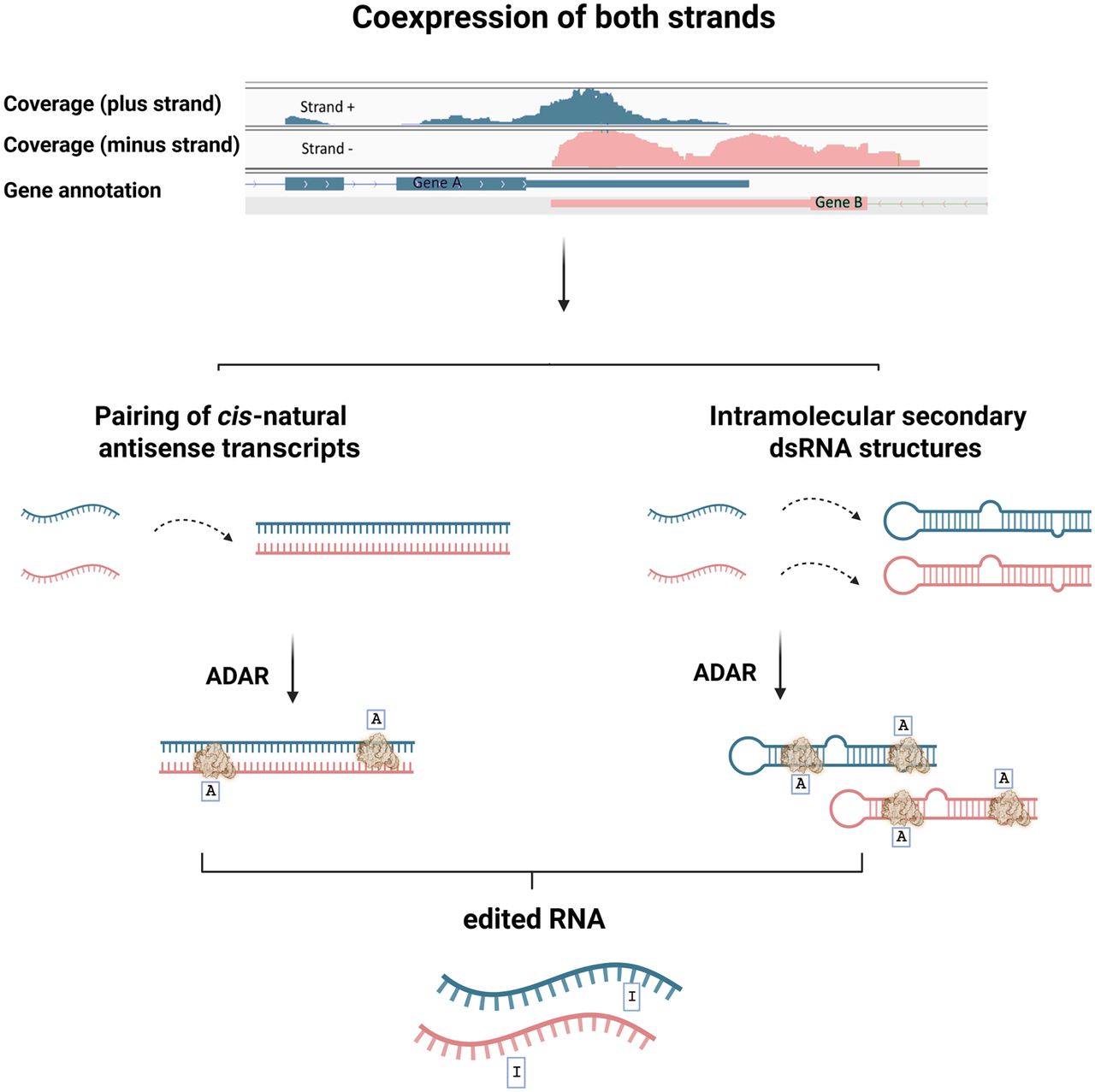
Define putative cis-NATs regions
To investigate the possibility of dsRNA structures forming due to pairing of cis-NAT transcripts, we first searched for genomic regions that potentially can give rise to cis-NAT editing. Our strategy involved two complementary approaches. First, we focused on identifying regions exhibiting extensive editing on at least one strand and tested whether the editing may be associated with cis-NAT transcription. Complementarily, we focused on regions exhibiting expression of both strands and tested whether they undergo editing.
To detect highly edited regions, we employed the hyperediting approach (Methods) (Porath et al. 2014), looking for extensively edited reads. This approach looks for clusters of A-to-G mismatches, which are a clear and specific signal of A-to-I editing. cis-NAT duplexes are perfectly paired, typically >50 bp. Thus, they are expected to be extensively edited (Bass and Weintraub 1988; Wagner et al. 1989; Nishikura et al. 1991; Polson and Bass 1994; Bass 1997; Pfaller et al. 2018; Katrekar et al. 2022). Accordingly, we anticipated these regions would be detectable with the hyperediting approach. Large clusters of mismatches of other types are rare, and thus, this method allows determining the edited strand even for unstranded RNA-seq data: A-to-G clusters attest for editing on the positive strand, whereas T-to-C clusters (as observed when mapping the reads to the positive strand, i.e., the reference genome sequence) attest for editing of an RNA molecule that was expressed from the negative strand. We analyzed RNA-seq data from the Genotype-Tissue Expression (GTEx) database (Lonsdale et al. 2013), encompassing 8603 samples across 47 tissues from 548 donors, and identified 899,142 regions, ≥50 bp long, in which A-to-I hyperediting was observed on at least one strand (for at least one sample). For comparison, the number of hyperedited regions for all other mismatch types combined is 278,728 (Supplemental Fig. S1).
Although the search itself is not inherently biased toward Alu elements, we found that 77.8% of hyperedited regions overlapped Alu repeats (Levanon et al. 2004). Alu elements tend to cluster in the genome, and two oppositely oriented Alu’s that appear in proximity are likely to create a stable dsRNA that is a substrate for ADAR-mediated editing (Bazak et al. 2014). Thus, editing in these repeats is probably due to intramolecular binding. We discarded these Alu regions and kept only 199,689 regions (22.2%) non-Alu regions for further analysis (Fig. 2).
Defining putative cis-NAT regions using three approaches. Putative cis-NATs were identified in two ways: (1) searching for genomic regions expressed from both strands using stranded RNA-seq expression data (left) and gene annotation data (middle) and (2) searching GTEx data for regions that are hyperedited on at least one strand (right). Alu regions were excluded as they are likely to form dsRNA structures due to two oppositely oriented elements on the same strand. Regions exhibiting expression of both strands and editing levels exceeding the background were considered putative cis-NAT regions.
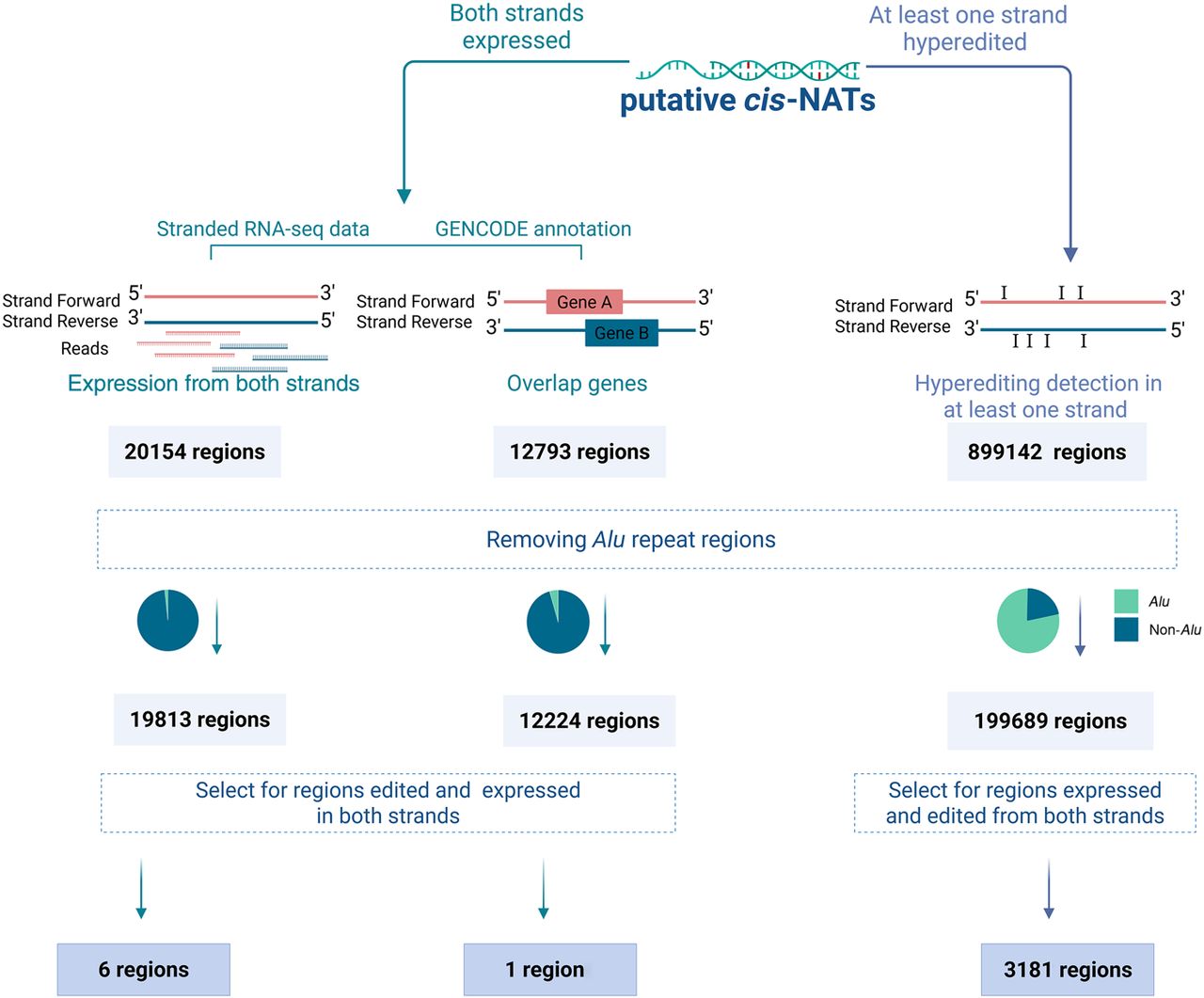
For each of these remaining regions, we calculated, for each sample and for each strand, the editing index (Roth et al. 2019), defined as the weighted average over the mismatch levels observed across a region. Here, too, we assumed T-to-C mismatches represent A-to-G editing events on the opposite strand. Thus, A-to-G index measures editing in transcripts expressed from the positive strand, whereas T-to-C index is a measure of editing of RNA molecules transcribed from the reverse strand. We defined regions as expressed and edited in a given sample if coverage exceeded 100 A nucleotides and 100 T nucleotides and the editing index for each of the two strands was >0.5%. To minimize the effect of isolated genomic polymorphisms and suppress detection noise, we required each editing index to be supported by at least three mismatches sites (i.e., at least three sites showing A-to-G mismatches). Additionally, to avoid false positives due to alignment noise, we excluded index values that were not at least 10-fold higher than the indices calculated for each of the other 10 mismatch types. To assess the robustness of these thresholds, we performed sensitivity analyses using various parameter combinations (Supplemental Fig. S2A). These filters resulted in the identification of 3181 regions with expression and editing on both strands, which serve as candidates for cis-NAT-mediated editing. We verified that the results are not very sensitive to the detection parameters (Supplemental Fig. S2).
As a complementary approach, we first defined regions as NAT regions based on their expression profile and then looked into the editing levels therein. Strand-specific expression and RNA editing levels were calculated using a second RNA-seq data set, including 216 stranded RNA-seq samples from 45 different tissues (Lorenzi et al. 2021). We consider a region of ≥50 bp to be expressed on both strands if its aggregated coverage (over all samples) was greater than 10 reads for each of the two strands. We found 20,154 such regions and kept only 19,813 of them that did not overlap with Alu repeats (Fig. 2). We then calculated the editing index for these regions using the abovementioned criteria. This approach yielded only six NAT candidate regions exhibiting expression and editing on both strands.
In addition, the GENCODE database, a well-regarded repository of gene annotations, was utilized to search for cis-NAT regions. We identified 12,793 genomic regions annotated as being expressed from both the positive and negative DNA strands (regardless of whether they are expressed concurrently). Exclusion of regions located within Alu repeats and calculation of editing index per region using the abovementioned criteria resulted in only one additional candidate region, located in the MDM2 gene. The main reason for the low number of regions detected using the complementary approach, as compared with the hyperediting one, is the need for sufficient reads coverage.
Four of the seven regions found in this complementary approach have overlapped regions found in the first method. We merged the candidate regions from the two approaches, resulting in 8184 suspected regions.
Putative intramolecular RNA structures in NAT-candidate regions
Editing occurs in the candidate regions which may indicate cis-NAT editing but could also result from intramolecular folding of the molecules transcribed from both strands, independently. Note that the secondary structures expected for two reverse-complemented RNA sequences are highly similar, as the interaction between RNA bases is nearly symmetrical (an exception is the difference between G:U and A:C pairing energies). Thus, strong dsRNA structure for one strand is highly correlated with the existence of such a structure for molecules transcribed from the reverse strand. To assess the possibility of intramolecular folding, we employed the Vienna's RNAplfold code (Lorenz et al. 2011) to calculate the locally stable secondary structures and the pairing probabilities for each of the single-strand RNAs transcribed from the candidate regions. The probability of a given nucleotide to base pair with another nucleotide within the same molecule was computed along a 2000 bp window around the center of each candidate region (RNAplfold failed for 98 of the 3184 regions, and they were excluded) and averaged over sliding windows of 100 bp and across all candidate regions (Fig. 3A). A clear peak is seen around the center of the regions, indicating a marked overall tendency toward intramolecular pairing.
Most cis-NAT-candidate regions are characterized by putative intramolecular RNA structures. (A) The averaged base pair probability in a window surrounding 3184 NAT-candidate regions. The average was calculated using a 100 bp sliding window across a 2000 bp sequence surrounding the identified region, centered at the center of the NAT-candidate region. Results are presented for the plus (top) and minus (bottom) strands, separately. The gray area indicates the standard deviation. (B) Base pair probability and editing levels for both strands for a specific candidate region. (C) A secondary RNA structure predicted for the region shown in B. (D) Distribution of correlation coefficients between editing indices and pairing probabilities, calculated for regions exhibiting high pairing probability values (>0.8, n = 866; red) compared with the regions with the lowest base-pairing probability (n = 866; blue). Results are presented for the plus (left) and minus (right) strands, separately. The analysis was performed using samples from the brain cerebellum tissue, and a similar analysis for five tissues is shown in Supplemental Figure S3. (E) Venn diagram showing the number of regions with putative dsRNA structure based on either BLAST or RNAplfold and their overlap.
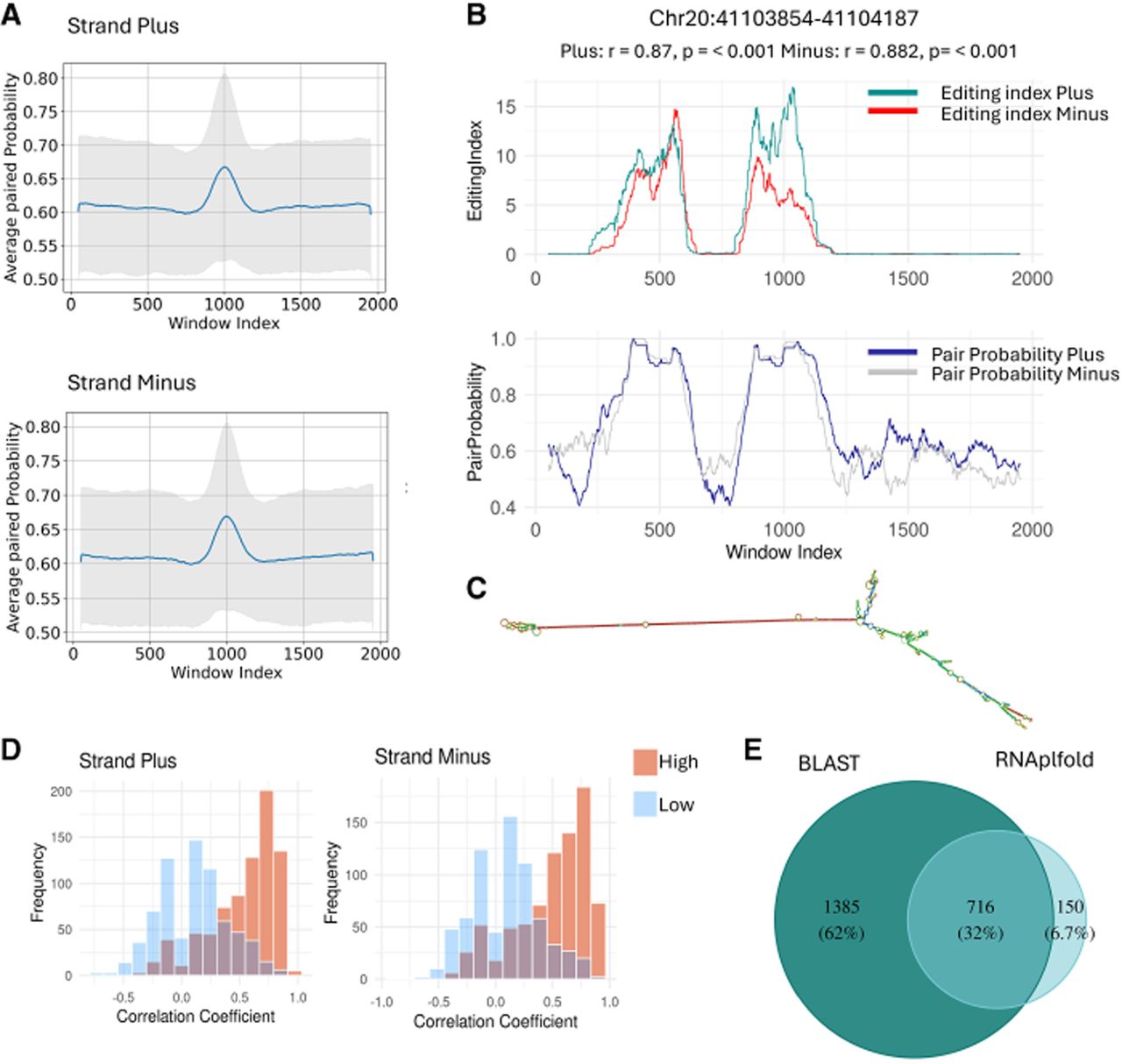
Figure 3B shows a specific illustration of this scenario. For one candidate region (Chr 20: 41,103,854–41,104,187), the two editing indices (corresponding to editing on both strands) and the pairing probability (averaged over 100 bp running windows) are presented. The structure predicted for this region (using RNAplfold) is presented in Figure 3C. If editing were associated with antisense pairing, one would have expected the editing signal to appear uniformly across the region coexpressed from both strands, and there would be no correlation between regions of high pairing probability and high editing levels. However, the data show editing, on both strands, in specific segments of the region, and these are closely correlated to the ones predicted to be paired due to intramolecular folding of each strand, independently.
To quantify the extent of this phenomenon across our regions, we first defined regions exhibiting intramolecular pairing with high probability as those in which at least one window within the region exhibited an average pairing probability exceeding 0.8 on both the positive and negative strands, resulting in a total of 866 regions. For these regions, we calculated the A-to-G and T-to-C editing indices (corresponding to adenosines on the positive and negative strands, respectively) for 100 bp running windows along the 2000 bp region and looked at the correlation of this spatially dependent index with the pairing probability. Figure 3D illustrates the distribution of correlation coefficients for these 866 regions (both strands) compared with the 866 regions that presented the lowest base-pairing probability. Almost all regions that are expected to pair intramolecularly do exhibit high positive correlation coefficients, in contrast with the control regions. This further supports the connection between editing and secondary structure.
The computing power required for folding analyses increases substantially with sequence length, and we thus predicted the folding for sequences up to 2000 bp. To explore the possibility of intramolecular pairing for longer sequences, we complemented our approach with a BLAST (Altschul et al. 1990) search to identify potential complementary sequences within a 10,000 bp window surrounding the candidate region.
Our analyses revealed compelling evidence of intramolecular dsRNA structures within candidate regions. Among the 3184 examined regions, BLAST-based dsRNA structures were predicted for 2101 regions (65%) compared with only 336 (10.5%) same-strand matches with the same parameters. Most of the RNAplfold structures were also picked up in this search, and altogether, we found 2251/3184 regions (70%) for which we have evidence for intramolecular dsRNA structures (Fig. 3E). Notably, sensitivity analyses using different threshold parameters consistently show that, combining both methods, most regions contain predicted intramolecular structures (Supplemental Fig. S2B,C). For the remaining 933 regions, it is possible that secondary structures were missed due to the limitations of our prediction methods, detection thresholds, or window size. However, some of these may exhibit editing due to intermolecular binding of NATs.
Co-occurrence of editing on both strands is rare
Another indication for cis-NAT-dependent editing is co-occurrence of editing in the same sample(s). We hypothesized that if editing depends on the pairing of molecules transcribed from the two strands, it should appear concomitantly on both strands. We thus quantified for each region the fraction of individual samples (among well-covered samples, and only for regions with editing detected in at least one strand in five or more samples) for which editing is seen (index > 0.5%) in both strands, only one strand, or no editing is seen at all. As illustrated in Figure 4A, for nearly all candidate regions, editing of both strands occurs much less frequently than editing of one strand only. Detailed per-sample data are presented in Figure 4B for two sample regions. This observation further supports the notion that even within our candidate regions editing occurs in each strand independently, indicating that most of the editing activity does not stem from sense–antisense interactions. Supplemental Figure S4A presents the same information only for the 872 regions (with editing detected in at least one strand in five or more samples) in which no secondary structure was found, and shows a similar trend. Among these, only a single region (Chr 9: 40,904,767–40,904,914) showed concurrent editing on both strands for all samples for which editing was seen (six samples, out of 23 samples with adequate coverage). Detailed genomic annotation analysis of these regions (Supplemental Fig. S4B) reveals that potential cis-NAT candidates are predominantly located in noncoding contexts.
Concurrent editing of both strands. (A) Each candidate region was analyzed to determine the percentage of samples exhibiting editing (index > 0.5%) in both strands, only one strand, or no editing. Each row presents data for one candidate region, showing the percentage of samples in which editing is observed in both strands (pink) or only one strand (blue). Other samples have exhibited no detectable editing in the region. The red and blue arrows point to specific regions whose data are presented in panels B. (B) Focused views of specific regions, showing the editing index over this specific region on both strands (A to G for the positive strand and T to C for the negative strand) across individual samples. Overall, in all regions analyzed, many samples exhibit editing on one strand only. If NAT pairing were the primary mechanism driving editing, we would expect to see editing occur simultaneously on both strands. (C) Distribution of Pearson's correlation coefficients between A-to-G editing indices (positive strand) and T-to-C editing indices (negative strand). Gray bars represent all candidate regions (n = 2503), and green bars highlight the subset with statistically significant correlation (FDR < 0.05, n = 906). The vertical dashed line marks r = 0. (D) Scatter plots showing the correlation between positive-strand (A-to-G) and negative-strand (T-to-C) editing indices for the two specific regions highlighted in panel B. Each point represents one sample. Pearson correlation coefficients (r) and P-values are shown.
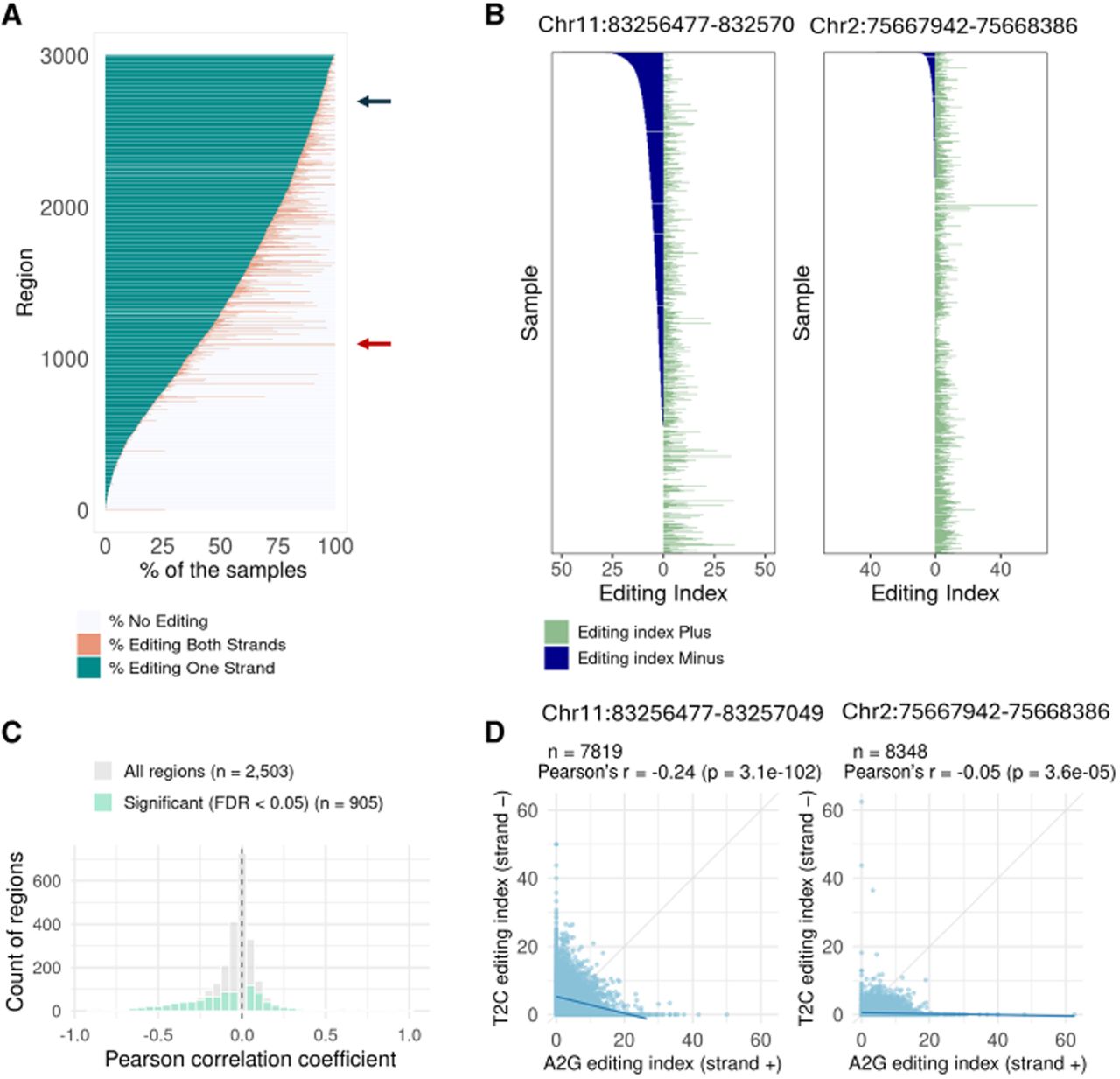
To further investigate whether editing events on opposite strands are coordinated, we performed correlation analyses between editing indices of the two strands. For regions with sufficient editing events (30 or more samples showing editing in at least one strand), we filtered samples with adequate coverage (100 or more A nucleotides for the positive strand and 100 or more T nucleotides for the negative strand) and calculated Pearson's correlation coefficients between A-to-G editing indices (positive strand) and T-to-C editing indices (negative strand). Figure 4C shows the distribution of correlation coefficients across all qualifying regions. The resulting correlation coefficients are distributed around zero, and positive coefficients are less frequent than negative ones (Fig. 4C). This finding further supports independent editing on each strand rather than coordinated NAT-dependent editing. Figure 4D presents detailed correlation analyses for the two specific regions highlighted in Figure 4B. Both regions exhibit negative correlation coefficients, demonstrating the lack of coordinated editing between strands.
Discussion
In this study, we examined the possible role of cis-NATs in mediating editing, seeking to understand their prevalence, characteristics, and potential implications for RNA editing. NAT regions were considered a prime candidate for RNA editing due to their naturally occurring perfect double-stranded structure. For example, RNA editing in the TNFRSF14/TNFRSF14-AS1 locus was suggested to prevent an immune response to dsRNA structure created by two-strand expression, demonstrating the importance of editing in NAT regions (Li et al. 2022). However, our structural analysis of this locus reveals that the detected editing sites overlap regions that are likely to produce strong intramolecular dsRNA structures, whereas the gene overlap region itself shows no significant editing (Supplemental Fig. S5). Consistent with this finding, our exploration of the human genome revealed that although cis-NAT expression is not rare, instances of editing within these regions are relatively uncommon. Even when editing does occur in these regions, it is most likely due to intramolecular dsRNA structures in most cases. Folding of an RNA transcript to create a stable secondary structure may be favored thermodynamically over base-pairing with a second transcript, which may not be in close physical proximity within the cell at that moment. Therefore, it is plausible that the probability of forming a dsRNA structure that will lead to editing by ADAR is higher when the transcript folds upon itself.
Note, however, that ADARs can certainly be recruited by intermolecular dsRNA structures. In recent years, the prospects of utilizing endogenous ADAR for RNA engineering have attracted much interest. These technologies rely on engineered guide RNAs that bind to RNA molecules of interest to form dsRNAs that recruit and activate ADAR editing (Azad et al. 2017; Fukuda et al. 2017; Merkle et al. 2019; Diaz Quiroz et al. 2023; Schneider et al. 2024; Reautschnig et al. 2025). The success of these methods clearly demonstrates that intermolecular pairing may drive ADAR editing. It is therefore unclear why NATs appear to play a limited role in facilitating editing. Possibly, even for regions in which both strands are expressed, coexpression and colocalization of both transcripts do not happen often, and thus, the creation of dsRNA structures due to NATs is a rare event. In addition, splicing can disrupt pairing of sense and antisense transcripts, preventing stable dsRNA formation, and RNA-binding proteins may wrap RNA molecules, interfering with their ability to pair with complementary transcripts.
Several limitations of our analysis warrant consideration. First, we conservatively excluded candidate cis-NAT editing regions whenever a plausible alternative explanation (e.g., intramolecular secondary structure) existed. Although this does not definitively rule out antisense pairing, we prioritized simpler, well-characterized mechanisms in which multiple explanations were possible. Second, our reliance on poly(A)-selected RNA-seq means we cannot assess whether edited cis-NAT duplexes form in nonpolyadenylated nuclear transcripts. As the primary interest in these duplexes stems from their potential cytoplasmic immunogenicity, this limitation may be less critical; however, applying our approach to matched total RNA and poly(A)-selected libraries would be an informative extension. A third constraint is the use of unstranded GTEx data, which precludes direct quantification of strand-specific expression. Although supplemented with a strand-specific data set, its lower coverage limited our sensitivity. Finally, our model assumes that cis-NAT pairing leads to extensive bidirectional editing, consistent with prior studies of synthetic (Bass and Weintraub 1988; Wagner et al. 1989; Nishikura et al. 1991; Polson and Bass 1994; Bass 1997), viral (Pfaller et al. 2018), and engineered dsRNA (Katrekar et al. 2022). However, significant asymmetry in strand expression could mean only a small fraction of the abundant transcript is paired, resulting in detectable editing being largely confined to the low-abundance strand, which may itself fall below detection thresholds and lead to an underestimate of true intermolecular editing.
Additional biological processes may further complicate the detection of endogenous RNA editing of cis-NAT pairs. One such mechanism is RNA interference (RNAi), which could, in principle, process sense–antisense duplexes into small RNA species, thereby reducing their abundance and availability for ADAR-mediated editing and also the detectability of editing at these loci. Although RNAi activity is generally considered limited in human somatic cells and although endogenous small interfering RNAs (siRNAs) are thought to be expressed at very low levels, their potential contribution cannot be completely excluded. However, even if siRNA-like molecules are generated, they lack the extended dsRNA features associated with innate immune activation. Consequently, such processed RNA species are unlikely to represent major physiological substrates for ADAR-mediated editing or to play a central role in dsRNA-driven immune regulation.
Finally, although our findings suggest a less prominent role for cis-NATs in widespread RNA editing, it is important to acknowledge that our analysis of candidate regions revealed a small number of cases for which we found no evidence of intramolecular dsRNA structures. The quest to identify the primary targets of ADAR enzymes is still ongoing, but our findings strongly suggest that the secondary structures responsible for ADAR's functional phenotype predominantly arise within the same transcript (Levanon et al. 2024).
Methods
RNA-seq data sets and annotations
RNA-seq data were obtained from the GTEx project (v7; unstranded, 8603 samples across 47 tissues) (Lonsdale et al. 2013) and from the RNA Atlas (strand-specific, poly(A)-selected, 216 samples across 45 tissues; obtained from the NCBI BioProject database [https://www.ncbi.nlm.nih.gov/bioproject/] under accession number PRJNA576920) (Lorenzi et al. 2021). Reads were aligned to the GRCh38 human reference genome. Genomic repeat annotations were obtained from the RepeatMasker database, and regions overlapping Alu elements were excluded.
Hyperediting detection
Hyperedited regions were identified using RNA-seq reads from the GTEx project with the cluster scan method of Porath et al. (2014), with their recommended default parameters: high-quality (Phred ≥ 30) A→G (T→C) mismatches had to comprise ≥5% of the read length and ≥60% of all mismatches. The cluster, spanning from the first to last A→G/T→C site, had to cover ≥10% of the read and must not lie wholly within the first or last 20% of the read. Clusters were excluded if >60% of bases within the cluster were a single nucleotide type. For paired-end reads, both mates had to map in opposite orientation within 500 kb of each other. To assess the robustness of our detection approach, we performed a sensitivity analysis using more lenient hyperediting detection parameters: mismatch density ≥4% (reduced from ≥5%) and mismatch specificity ≥50% (reduced from ≥60%). After detection, overlapping clusters were merged, and intervals ≥50 bp supported by hyperediting on at least one strand were retained. This minimum length threshold ensures that each region contains at least three mismatch sites when applying a 5% editing density criterion. Thus, editing contributions come from multiple sites rather than isolated events that likely represent SNPs rather than genuine editing. Regions overlapping with Alu elements were excluded as they are likely to form dsRNA structures due to two oppositely oriented elements on the same strand (Katrekar et al. 2022).
Editing index calculation
The editing index was calculated per region, strand, and sample as the weighted average of mismatch levels across the region, as previously described (Roth et al. 2019). A-to-G mismatches were used for the positive strand, and T-to-C mismatches were interpreted as A-to-G events on the reverse strand. Regions with total read coverage (fewer than 100 A nucleotides for positive strand or fewer than 100 T nucleotides for negative strand) were excluded. This threshold ensures sufficient statistical power to reliably distinguish editing events from technical artifacts. Lower coverage would increase false-positive rates due to sequencing errors. Regions showing an editing index exceeding 0.5%, at least 10-fold higher than the highest index value calculated for all other mismatch types (excluding A to G and T to C), and at least three mismatch events of the relevant mismatch type were retained for further analysis. The editing index threshold must be appreciably larger than the noise level (0.1%). Sensitivity analyses using thresholds ranging from 0.1% to 0.5% showed no substantial change in the number of candidate regions, indicating that the conclusions are robust across this parameter range (Supplemental Fig. S2A).
Correlation analysis between strands
Pearson correlation coefficients were calculated between A-to-G editing indices (positive strand) and T-to-C editing indices (negative strand) across samples from all GTEx tissues. Analysis was performed for regions with 30 or more samples showing editing in at least one strand. From these selected regions, only samples with adequate coverage (100 or more A nucleotides for positive strand, 100 or more T nucleotides for negative strand) were included in the correlation calculation. Correlation was calculated only if, for each of the two strands, the variance of editing level is positive.
Secondary structure prediction
We used RNAplfold from the ViennaRNA Package v2.0 (Lorenz et al. 2011) to compute base-pairing probabilities over 2000 bp windows centered at the center of each candidate region, with the parameters -u 1 -S 1.17. The probabilities per base pair were averaged over 100 bp sliding windows. To detect longer-range intramolecular pairing, we used BLASTN 2.7.1+ (Altschul et al. 1990) with default parameters to search for reverse-complement matches within ±10 kb. In this approach, a region was considered structured if a match was found whose length was at least 60% of the length of the candidate region. The 60% BLAST match threshold was chosen to prevent spurious matches in which antisense pairing occurs in one region, whereas editing occurs in a distant, unrelated region. The 0.8 RNAplfold pairing probability threshold was selected as approximately one standard deviation above the mean pairing probability. Sensitivity analyses using alternative thresholds for both approaches are presented in Supplemental Figure S2, B and C.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Jin Billy Li and Qin Li for fruitful discussions. We also acknowledge helpful comments from Haim Krupkin. This research was supported by the Israel Science Foundation (grant 2637/23 to E.Y.L. and 867/25 to E.E.) and by the Foundation Fighting Blindness (E.Y.L.)
Author contributions: Z.R. performed the data analyses. R.C.-F. assisted in data collection and preprocessing. O.S. contributed to the early stages of the project. E.Y.L. and E.E. conceived the study, as well as designed and supervised the analyses. Z.R., E.Y.L., and E.E. wrote the paper.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280820.125.
[3] Freely available online through the Genome Research Open Access option.
References
- ↵Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. 10.1016/S0022-2836(05)80360-2
- ↵Athanasiadis A, Rich A, Maas S. 2004. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol 2: e391. 10.1371/journal.pbio.0020391
- ↵Azad MTA, Bhakta S, Tsukahara T. 2017. Site-directed RNA editing by adenosine deaminase acting on RNA for correction of the genetic code in gene therapy. Gene Ther 24: 779–786. 10.1038/gt.2017.90
- ↵Barak M, Porath HT, Finkelstein G, Knisbacher BA, Buchumenski I, Roth SH, Levanon EY, Eisenberg E. 2020. Purifying selection of long dsRNA is the first line of defense against false activation of innate immunity. Genome Biol 21: 26. 10.1186/s13059-020-1937-3
- ↵Bass B. 1997. RNA editing and hypermutation by adenosine deamination. Trends Biochem Sci 22: 157–162. 10.1016/S0968-0004(97)01035-9
- ↵Bass BL, Weintraub H. 1988. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55: 1089–1098. 10.1016/0092-8674(88)90253-X
- ↵Bazak L, Levanon EY, Eisenberg E. 2014. Genome-wide analysis of Alu editability. Nucleic Acids Res 42: 6876–6884. 10.1093/nar/gku414
- ↵Blow M, Futreal PA, Wooster R, Stratton MR. 2004. A survey of RNA editing in human brain. Genome Res 14: 2379–2387. 10.1101/gr.2951204
- ↵Brisse M, Ly H. 2019. Comparative structure and function analysis of the RIG-I-like receptors: RIG-I and MDA5. Front Immunol 10: 1586. 10.3389/fimmu.2019.01586
- ↵Chen J. 2004. Over 20% of human transcripts might form sense-antisense pairs. Nucleic Acids Res 32: 4812–4820. 10.1093/nar/gkh818
- ↵Chen YG, Hur S. 2022. Cellular origins of dsRNA, their recognition and consequences. Nat Rev Mol Cell Biol 23: 286–301. 10.1038/s41580-021-00430-1
- ↵Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann H-H, Rosenberg BR, 2018. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172: 811–824.e14. 10.1016/j.cell.2017.12.038
- ↵Conley AB, Jordan KI. 2012. Epigenetic regulation of human cis-natural antisense transcripts. Nucleic Acids Res 40: 1438–1445. 10.1093/nar/gkr1010
- ↵Conley AB, Miller WJ, Jordan IK. 2008. Human cis natural antisense transcripts initiated by transposable elements. Trends Genet 24: 53–56. 10.1016/j.tig.2007.11.008
- ↵de Reuver R, Maelfait J. 2024. Novel insights into double-stranded RNA-mediated immunopathology. Nat Rev Immunol 24: 235–249. 10.1038/s41577-023-00940-3
- ↵Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, 2012. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22: 1775–1789. 10.1101/gr.132159.111
- ↵Diaz Quiroz JF, Ojha N, Shayhidin EE, De Silva D, Dabney J, Lancaster A, Coull J, Milstein S, Fraley AW, Brown CR, 2023. Development of a selection assay for small guide RNAs that drive efficient site-directed RNA editing. Nucleic Acids Res 51: e41. 10.1093/nar/gkad098
- ↵Feng Q, Hato SV, Langereis MA, Zoll J, Virgen-Slane R, Peisley A, Hur S, Semler BL, van Rij RP, van Kuppeveld FJM. 2012. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep 2: 1187–1196. 10.1016/j.celrep.2012.10.005
- ↵Fukuda M, Umeno H, Nose K, Nishitarumizu A, Noguchi R, Nakagawa H. 2017. Construction of a guide-RNA for site-directed RNA mutagenesis utilising intracellular A-to-I RNA editing. Sci Rep 7: 41478. 10.1038/srep41478
- ↵He Y, Vogelstein B, Velculescu VE, Papadopoulos N, Kinzler KW. 2008. The antisense transcriptomes of human cells. Science 322: 1855–1857. 10.1126/science.1163853
- ↵Hu S-B, Heraud-Farlow J, Sun T, Liang Z, Goradia A, Taylor S, Walkley CR, Li JB. 2023. ADAR1p150 prevents MDA5 and PKR activation via distinct mechanisms to avert fatal autoinflammation. Mol Cell 83: 3869–3884.e7. 10.1016/j.molcel.2023.09.018
- ↵Hur S. 2019. Double-stranded RNA sensors and modulators in innate immunity. Annu Rev Immunol 37: 349–375. 10.1146/annurev-immunol-042718-041356
- ↵Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, 2005. Antisense transcription in the mammalian transcriptome. Science (1979) 309: 1564–1566. 10.1126/science.1112009
- ↵Katrekar D, Yen J, Xiang Y, Saha A, Meluzzi D, Savva Y, Mali P. 2022. Efficient in vitro and in vivo RNA editing via recruitment of endogenous ADARs using circular guide RNAs. Nat Biotechnol 40: 938–945. 10.1038/s41587-021-01171-4
- ↵Kim DDY, Kim TTY, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A. 2004. Widespread RNA editing of embedded Alu elements in the human transcriptome. Genome Res 14: 1719–1725. 10.1101/gr.2855504
- ↵Kiyosawa H, Yamanaka I, Osato N, Kondo S, Hayashizaki Y, RIKEN GER Group GSL Members. 2003. Antisense transcripts with FANTOM2 clone set and their implications for gene regulation. Genome Res 13: 1324–1334. 10.1101/gr.982903
- ↵Lavorgna G, Dahary D, Lehner B, Sorek R, Sanderson CM, Casari G. 2004. In search of antisense. Trends Biochem Sci 29: 88–94. 10.1016/j.tibs.2003.12.002
- ↵Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 22: 1001–1005. 10.1038/nbt996
- ↵Levanon EY, Cohen-Fultheim R, Eisenberg E. 2024. In search of critical dsRNA targets of ADAR1. Trends Genet 40: 250–259. 10.1016/j.tig.2023.12.002
- ↵Li Q, Gloudemans MJ, Geisinger JM, Fan B, Aguet F, Sun T, Ramaswami G, Li YI, Ma JB, Pritchard JK, 2022. RNA editing underlies genetic risk of common inflammatory diseases. Nature 608: 569–577. 10.1038/s41586-022-05052-x
- ↵Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120. 10.1126/science.aac7049
- ↵Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, 2013. The Genotype-Tissue Expression (GTEx) Project. Nat Genet 45: 580–585. 10.1038/ng.2653
- ↵Lorenz R, Bernhart SH, Höner zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. 2011. ViennaRNA package 2.0. Algorithms Mol Biol 6: 26. 10.1186/1748-7188-6-26
- ↵Lorenzi L, Chiu H-S, Avila Cobos F, Gross S, Volders P-J, Cannoodt R, Nuytens J, Vanderheyden K, Anckaert J, Lefever S, 2021. The RNA atlas expands the catalog of human non-coding RNAs. Nat Biotechnol 39: 1453–1465. 10.1038/s41587-021-00936-1
- ↵Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. 10.1016/j.celrep.2014.10.041
- ↵Merkle T, Merz S, Reautschnig P, Blaha A, Li Q, Vogel P, Wettengel J, Li JB, Stafforst T. 2019. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat Biotechnol 37: 133–138. 10.1038/s41587-019-0013-6
- ↵Meyers BC, Vu TH, Tej SS, Ghazal H, Matvienko M, Agrawal V, Ning J, Haudenschild CD. 2004. Analysis of the transcriptional complexity of Arabidopsis thaliana by massively parallel signature sequencing. Nat Biotechnol 22: 1006–1011. 10.1038/nbt992
- ↵Misra S, Crosby MA, Mungall CJ, Matthews BB, Campbell KS, Hradecky P, Huang Y, Kaminker JS, Millburn GH, Prochnik SE, 2002. Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol 3: RESEARCH0083. 10.1186/gb-2002-3-12-research0083
- ↵Morse DP, Aruscavage PJ, Bass BL. 2002. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc Natl Acad Sci 99: 7906–7911. 10.1073/pnas.112704299
- ↵Neeman Y, Dahary D, Levanon EY, Sorek R, Eisenberg E. 2005. Is there any sense in antisense editing? Trends Genet 21: 544. 10.1016/j.tig.2005.08.005
- ↵Nishikura K, Yoo C, Kim U, Murray JM, Estes PA, Cash FE, Liebhaber SA. 1991. Substrate specificity of the dsRNA unwinding/modifying activity. EMBO J 10: 3523–3532. 10.1002/j.1460-2075.1991.tb04916.x
- ↵Osato N, Yamada H, Satoh K, Ooka H, Yamamoto M, Suzuki K, Kawai J, Carninci P, Ohtomo Y, Murakami K, 2003. Antisense transcripts with rice full-length cDNAs. Genome Biol 5: R5. 10.1186/gb-2003-5-1-r5
- ↵Ozsolak F, Milos PM. 2011. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 12: 87–98. 10.1038/nrg2934
- ↵Peisley A, Lin C, Wu B, Orme-Johnson M, Liu M, Walz T, Hur S. 2011. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci 108: 21010–21015. 10.1073/pnas.1113651108
- ↵Peisley A, Jo MH, Lin C, Wu B, Orme-Johnson M, Walz T, Hohng S, Hur S. 2012. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc Natl Acad Sci 109: E3340–E3349. 10.1073/pnas.1208618109
- ↵Pelechano V, Steinmetz LM. 2013. Gene regulation by antisense transcription. Nat Rev Genet 14: 880–893. 10.1038/nrg3594
- ↵Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. 2015. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43: 933–944. 10.1016/j.immuni.2015.11.001
- ↵Pfaller CK, Donohue RC, Nersisyan S, Brodsky L, Cattaneo R. 2018. Extensive editing of cellular and viral double-stranded RNA structures accounts for innate immunity suppression and the proviral activity of ADAR1p150. PLoS Biol 16: e2006577. 10.1371/journal.pbio.2006577
- ↵Polson AG, Bass BL. 1994. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J 13: 5701–5711. 10.1002/j.1460-2075.1994.tb06908.x
- ↵Porath HT, Carmi S, Levanon EY. 2014. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat Commun 5: 4726. 10.1038/ncomms5726
- ↵Porath HT, Knisbacher BA, Eisenberg E, Levanon EY. 2017. Massive A-to-I RNA editing is common across the metazoa and correlates with dsRNA abundance. Genome Biol 18: 185. 10.1186/s13059-017-1315-y
- ↵Ramaswami G, Li JB. 2014. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res 42: D109–D113. 10.1093/nar/gkt996
- ↵Reautschnig P, Fruhner C, Wahn N, Wiegand CP, Kragness S, Yung JF, Hofacker DT, Fisk J, Eidelman M, Waffenschmidt N, 2025. Precise in vivo RNA base editing with a wobble-enhanced circular CLUSTER guide RNA. Nat Biotechnol 43: 545–557. 10.1038/s41587-024-02313-0
- ↵Roth SH, Levanon EY, Eisenberg E. 2019. Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity. Nat Methods 16: 1131–1138. 10.1038/s41592-019-0610-9
- ↵Schneider N, Steinberg R, Ben-David A, Valensi J, David-Kadoch G, Rosenwasser Z, Banin E, Levanon EY, Sharon D, Ben-Aroya S. 2024. A pipeline for identifying guide RNA sequences that promote RNA editing of nonsense mutations that cause inherited retinal diseases. Mol Ther Nucleic Acids 35: 102130. 10.1016/j.omtn.2024.102130
- ↵Shendure J, Church GM. 2002. Computational discovery of sense-antisense transcription in the human and mouse genomes. Genome Biol 3: RESEARCH0044. 10.1186/gb-2002-3-9-research0044
- ↵Vanhée-Brossollet C, Vaquero C. 1998. Do natural antisense transcripts make sense in eukaryotes? Gene 211: 1–9. 10.1016/S0378-1119(98)00093-6
- ↵Wagner RW, Smith JE, Cooperman BS, Nishikura K. 1989. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc Natl Acad Sci 86: 2647–2651. 10.1073/pnas.86.8.2647
- ↵Wang H, Chung PJ, Liu J, Jang I-C, Kean MJ, Xu J, Chua N-H. 2014. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res 24: 444–453. 10.1101/gr.165555.113
- ↵Werner A, Kanhere A, Wahlestedt C, Mattick JS. 2024. Natural antisense transcripts as versatile regulators of gene expression. Nat Rev Genet 25: 730–744. 10.1038/s41576-024-00723-z
- ↵Wight M, Werner A. 2013. The functions of natural antisense transcripts. Essays Biochem 54: 91–101. 10.1042/bse0540091
- ↵Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, Chu F, Walz T, Hur S. 2013. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152: 276–289. 10.1016/j.cell.2012.11.048
- ↵Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A, Diber A, Biton S, Tamir Y, Khosravi R, 2003. Widespread occurrence of antisense transcription in the human genome. Nat Biotechnol 21: 379–386. 10.1038/nbt808