
A 7.5 Mb Sequence-Ready PAC Contig and Gene Expression Map of Human Chromosome 11p13–p14.1
Abstract
The region p13 of the short arm of human chromosome 11 has been studied intensely during the search for genes involved in the etiology of the Wilms' tumor, aniridia,genitourinary abnormalities, mental retardation (WAGR) syndrome, and related conditions. The gene map for this region is far from being complete, however, strengthening the need for additional gene identification efforts. We describe the extension of an existing contig map with P1-derived artificial chromosomes (PACs) to cover 7.5 Mb of 11p13–14.1. The extended sequence-ready contig was established by end probe walking and fingerprinting and consists of 201 PAC clones. Utilizing bins defined by overlapping PACs, we generated a detailed gene map containing 20 genes as well as 22 anonymous ESTs which have been identified by searching the RH databases. RH maps and our established gene map show global correlation, but the limits of resolution of the current RH panels are evident at this scale. Initial expression studies on the novel genes have been performed by Northern blot analyses. To extend these expression profiles, corresponding mouse cDNA clones were identified by database search and employed for Northern blot analyses and RNA in situ hybridizations to mouse embryo sections. Genomic sequencing of clones along a minimal tiling path through the contig is currently under way and will facilitate these expression studies by in silico gene identification approaches.
One step towards the functional understanding of the human genome is large-scale sequencing followed by sequence-based gene identification. Sequencing is performed in a directed strategy to reduce redundancy to a minimum, as applied to theCaenorhabditis elegans genome ( C. elegans consortium 1998). This strategy relies on prior mapping information and sequence-ready contigs as resources for the generation of a minimal tiling path to be followed during sequencing. More recently, a random sampling strategy, as applied in bacterial sequencing, has been suggested to also be practicable for the human genome. To assess the potential of this strategy, the Drosophila melanogaster genome is being used as a model (Pennisi 1999b). Sequencing of cDNAs from a large variety of different libraries (i.e., IMAGE consortium; Lennon et al. 1996) has been employed as a shortcut for gene identification. However, the neglect of mapping information, as well as information hidden in intronic sequences or promoters with their regulatory elements, is a drawback of this method. Moreover, genes with very low or highly restricted expression still pose a severe problem as they are underrepresented in the commonly used cDNA libraries. Nevertheless, partial information on a significant fraction of genes and their corresponding gene products can be obtained in less time and at lower cost compared to genomic sequencing (Adams et al. 1991; Hillier et al. 1996). With the recent proposal to speed up genome sequencing (Pennisi 1999a; Wadman 1999), these ESTs can soon be integrated into the emerging human genome sequences.
We have focused on the genetic content of the region p13 on human chromosome 11. Intense analysis of the deletion region of patients with WAGR syndrome (Wilms' tumor,aniridia, genitourinary abnormalities and mental retardation) has led to the isolation of theWT1 and PAX6 genes (Call et al. 1990; Gessler et al. 1990; Ton et al. 1991). However, the molecular mechanisms underlying their regulation are still unclear. For PAX6, position effects detected are caused by rearrangements well downstream of the known exons (Fantes et al. 1995; Schedl et al. 1996). The tissue-restricted expression of WT1 appears to be driven by far awaycis-regulatory elements that have yet to be identified (Scholz et al. 1997). The knowledge of the complete genomic sequence flanking these two genes and subsequent interspecies comparison with corresponding mouse or fugu data will be an essential step towards the elucidation of these mechanisms (Miles et al. 1998). Mental retardation and growth retardation, which is seen in ∼30% of WAGR patients (Turleau et al. 1984), represent two other features of this syndrome but no candidate genes have yet been identified. Frequent allele loss in distinct regions of 11p13 suggests the presence of one or more tumor suppressor genes other than WT1 that may be involved in a large variety of tumors, including lung, bladder, breast, and ovarian cancers (Bepler and Garcia-Blanco 1994; Kiechle-Schwarz et al. 1994;Shipman et al. 1998; Shipman et al. 1993; Vandamme et al. 1992).
In order to create the tools for sequence based gene identification, we extended a 3 Mb sequence-ready PAC contig described previously byNiederführ et al. (1998) to cover a region of 7.5 Mb of 11p13 reaching into p14.1. We also report the characterization of 22 novel 11p13 specific EST clones, their integration into the physical map, and their initial expression analyses.
RESULTS
Contig Construction
Niederführ et al. (1998) have constructed a 3 Mb sequence-ready PAC contig that covers the inner part of 11p13. We have extended this contig in the telomeric (t;PAX6–D11S155) and centromeric (c; D11S935–CAT) direction by hybridizing high density grid filters of the RPCI-1, -3, -4, and -5 PAC libraries with 11p13 specific markers and probes derived from a YAC contig covering 11p13 (Gawin et al. 1995). 125 PAC clones for the centromeric region c and 94 for the telomeric region t were identified and used as starting material for further end probe walking.
The mapping strategy followed Niederführ et al. (1998). End probes of each PAC were generated by linker mediated PCR and hybridized to colony filters of the arrayed sublibrary to identify overlapping clones. Clone insert sizes were determined by NotI restriction digests of the respective PAC clones. Additionally, we used markers that were already integrated into a detailed physical map of 11p13 as hybridization probes (Gawin et al. 1995). The combination of these data sets allowed an accurate alignment of the analyzed PAC clones along the physical map of 11p13.
With the first set of 219 PAC clones we could establish 10 individual, preliminary contigs (c:4; t:6). To close intercontig gaps between the new clone assemblies and to link them to the CAT–PAX6 contig, the RPCI PAC libraries were rescreened with end probes of clones located at the outer edges of these contigs. Two more cycles of clone ordering and rescreening led to the identification of 199 additional clones (c:43;t:156). Out of a total of 418 clones, we employed 125 (c:36; t:89) to build the two final contigs. Clones with either repetitive sequences at the insert ends that inhibited successful hybridizations, or with poor growth on colony filters were rejected from further analysis. Similarly clones that turned out to be located outside our region of interest were disregarded. Another 61 PAC clones that were part of the working draft contigs, are not included in the final version due to abound coverage of several regions or failure to determine insert sizes by PFGE, likely due to large inserts prone to instability. The ordered clone contig is shown in Figure1.
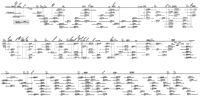
PAC contig covering 7.5 Mb of 11p13–14.1. 201 PAC clones are displayed, represented by horizontal bars with clone names (original well location) written above and insert sizes in kb below. PACs drawn as light grey horizontal bars between CAT and PAX6are from the PAC contig described previously in Niederführ et al. (1998). Square boxes at the ends of most clones allude to the SP6 (empty box) and the T7 ends (filled box) that have been used for hybridization analysis. Every PAC clone crossing a presumed vertical line through these boxes had shown a positive hybridization signal with the respective end probe and was thus proven to overlap. At thePAX6 locus several cosmid clones are shown that have been sequenced previously by the Sanger Centre (GenBank accession nos.Z83301, Z83306-83309, Z86001, and Z95332). The horizontal line on top of the contig represents the integrated map of 11p13–14.1 with known markers and NotI restriction sites that are indicated by tick marks. Genes are highlighted by shaded boxes and the newly integrated EST clones are written at a 45° angle. The positions ofRAG1/2 and TRAF6 as well as of the three EST clones 60587, 125727, and 41188, have been determined by hybridization to YAC clones located at the centromeric border of 11p13 (Gawin et al. 1995).
A complementary approach for contig construction was followed by performing fingerprint analyses on all PAC clones that had been identified during the screening process. The comparison ofHindIII/Sau3AI digest patterns of all clones strongly corroborates the overlaps suggested by the hybridization data without apparent inconsistencies. However, in the few regions with single clone coverage or short overlaps, fingerprinting could not reach statistical significance (a random probability of < 10-4, data not shown). In these cases, overlap is supported by a minimum of three shared hybridization markers. Only two reciprocal hybridizations were detected with two overlapping PACs (815A16/562D20) centromeric to theFSHB locus, pointing to an underrepresentation of this genomic DNA fragment in the libraries used. All fingerprinting data are stored in the chromosome 11 database 11DB (http://chr11.bc.ic.ac.uk).
Combined with the previously described 3 Mb CAT–PAX6 contig of Niederführ et al. (1998), we have now constructed a sequence-ready PAC contig covering approximately 7.5 Mb of 11p13–14.1. This contig extends from the marker D11S935 at the centromeric end to 250 kb beyond the marker D11S155 located in band p14.1. Our newly established contig extensions comprise 36 and 89 PAC clones for the centromeric and telomeric regions respectively, with an average 3.75-fold coverage.
Identification of 11p13 Specific Transcripts-Gene Map
The complete PAC contig represents an excellent tool for the positioning of genes. The nested nature of the PAC clones and their exact anchoring along the physical map, allows the localization of genes within an interval of generally <100 kb with respect to known markers.
All available versions of radiation hybrid (RH) maps were searched for ESTs assigned to 11p13 (especially http://ncbi.nlm.nih.gov/genemap98;http://shgc.stanford.edu/Mapping/rh/MapsV2/index.html;http://carbon.wi.mit.edu:8000/cgi-bin/contig/phys_map). We concentrated mainly on three GeneMap'98 RH intervals defined by the markers D11S1324 (t) and D11S4102 (c) that flank the PAC contig. To compensate for possible differences between the statistically based RH maps and our physical data, we also included the region D11S1324–S904 telomeric to FSHB, and a subset of clones from RH bins located centromeric to RAG1/2 in 11p12 (Table 1).
Listing of Putative 11p13–14.1-Specific Transcripts
With the identified RH map entries we performed extensive similarity searches and UniGene cluster comparison to eliminate redundancy arising through multiple entries of sequences that are likely derived from the same transcript. Several ESTs could be assigned to 14 genes mapped previously to our region of interest (cen-RAG1/2, GLT1/SLC1A2, CD44, CAT, p137GPI /M11S1, LMO2, CD59, G2, WT1, RCN1, PAX6, FSHB, KCNA4-tel). We identified an additional 47 nonredundant EST clusters or single ESTs and selected one representative cDNA clone from each for further analyses (Table 1). Three of the 47 ESTs correspond to known genes, namely the t umor necrosis factorreceptor-associated factor 6 (TRAF6), the X component of the pyruvate dehydrogenase complex (DLAT/PDX1/E3BP) and the 77 kD subunit of the c leavage stimulationfactor (CSTF3). Whereas CSTF3 andTRAF6 had not yet been assigned to 11p13, PDX1 has been mapped previously to 11p13 by FISH analysis (Aral et al. 1997;Ling et al. 1998). We also included four cDNAs from sources other than the RH maps: The cDNA sequence of the GA17 protein (GenBank accession no. AF064603) shows similarity to the Fugu cosmid clone 38e12, neighboring the Fugu WT1, RCN1 and PAX6 gene loci, suggesting a possible synteny (Miles et al. 1998; C. Miles and G. Elgar, pers. comm.). The fact that GA17 maps to chromosome X (DXS983–995) according to GeneMap'98 likely results from the presence of a truncated GA17 pseudogene that can be detected in the PAC 849L7 located on Xq21 (GenBank accession no. AL008987). The similarity of the 1.25 kb cDNA sequence of GA17 (GenBank accession no.AF060643) to this PAC clone is restricted to the last 300 bp of the 3′ end without any indication for splice sites. By using the STS A002N23 located within these 300 nucleotides for mapping GA17on the RH map, the intronless X-derived sequence is being amplified preferentially, explaining the observed mapping inconsistency. TheTR2 and 3K cDNAs (GenBank accession nos. AJ245600 andAJ245599, respectively), the latter has been identified as the human homolog FJX1 of the D. melanogaster fj gene (Ashery-Padan et al. 1999), have previously been identified by CpG island analyses on 11p13 and subsequent cDNA selection (Thäte et al. 1995). The IMAGE clone 814116 shows sequence similarity to another CpG island clone NE70 (D11S3899) located on 11p13.
To determine the location of all novel cDNA clones within the PAC contig, we hybridized them to colony filters of the 11p13-specific PAC sublibrary. Out of 52 independent clones tested, 28 could finally be assigned to our region of interest (Fig. 1). Four of these 28 clones were located outside of the PAC contig to the centromeric border of 11p13, as they hybridized to YAC clones from a contig that extends further centromeric (Gawin et al. 1995).
The integration of all hybridization data into the existing physical map resulted in the generation of a detailed gene map for the region 11p13–14.1 containing 20 genes, as well as 22 anonymous transcripts (Fig. 1).
Comparison Between RH and PAC Contig Mapping Data
Current RH maps represent a rich source of detailed mapping information that is readily available to the research community. To assess the concordance of the data obtained by this statistical method and our physical approach, we graphically correlated the localization of GeneMap'98 entries within the D11S4102–D11S1324 interval with the positions of corresponding markers on the contig map as shown in Figure2.

Comparison of GeneMap'98 (left) and PAC contig mapping data (right). Corresponding loci are connected by lines. GeneMap'98 entries that could not be mapped to our PAC contig are enclosed by brackets; gene names are underlined. Dotted lines and arrows mark RH framework markers. Shaded triangles describe the positional variation of genes or markers resulting from the use of multiple STSs (compare Table 2).
In cases where one transcript is characterized by multiple independent EST-derived entries in GeneMap'98, only one representative cDNA clone was employed for hybridization to the PAC contig (Table2). In one instance, three seemingly independent markers may actually be derived from a single transcription unit as suggested by the alignment of cDNA sequences shown in Figure3.
Redundancy of GeneMap'98 Entries
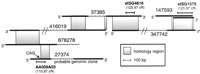
cDNA contig 37385 as established by BLAST search. Insert sizes of 27374, 37385, and 147493 have been determined experimentally; no information about insert sizes is available for clones 878278, 416019, and 347742. Shaded areas represent sequence similarity. Clone 27374 seems to be derived from an unspliced genomic sequence which is supported by two findings, namely lack of sequence similarity to the clones 416019 and 37385 and the presence of a CAG splice donor site at position 184 of the 5′ end sequence of 27374. The orientation of the transcript cannot be deduced because directions are not consistent for overlapping clones.
Generally good overall correspondence between the two data sets can be observed with local variations for several transcripts of up to 6 cR generated by multiple RH entries. In three cases (CD44,CSTF3, and cDNA contig 37385) these mapping differences were much larger, extending to 10–15 cR (compare Table 2). The estimate that 1 cR3000 is equivalent to ∼270 kb on chromosome 11 (Gyapay et al. 1996) is in agreement with our own findings for 11p13, where 1 cR3000 corresponds to ∼300 kb. However, out of 34 clones listed in the RH interval D11S1324–D11S4102 that is completely covered by our PAC contig, only 24 (71%) do map to the corresponding contig region. This fact, together with observed mapping uncertainties regarding order and distance between single entries, points to the difficulties when translating statistically derived coordinates into basepairs.
Human Northern Blot Analyses
To generate expression profiles for all new transcripts assigned to our PAC contig, we hybridized the corresponding human IMAGE clone inserts to Northern blots containing RNA from multiple human tissues as summarized in Figure 4.
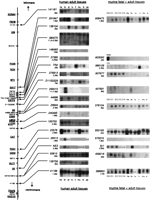
Northern blot analyses of the new transcripts. The results obtained by hybridization of cDNA clones to Northern blots with RNA from human (middle) or murine tissues (right) are displayed according to the genomic order of the probes. The names of the cDNA probes employed as well as the transcript sizes are indicated next to the autoradiograms. Quantitative comparison between human and murine expression is limited as the human Northern blots carry poly(A) + RNAs with amounts normalized to β-actin expression, whereas an invariable amount of 20 μg of total RNA of each tissue had been applied to the murine Northern blots. RNA samples used are either derived from whole embryos of embryonic stages E10 through E14 or from adult tissues (heart, he; brain, br; placenta, pl; lung, lu; liver, li; skeletal muscle, mu; kidney, ki; pancreas, pa). For analysis of clone 603000, a testis-derived RNA sample has been added.
Out of a total of 25 cDNA clones employed, we could detect signals on the Northern blots with 20 clones. Failure to detect transcripts for the other cDNA clones might be due to low expression or expression in tissues other than those employed on the Northern blots. The fact that some of the EST clones may be derived from contaminating genomic DNA may also play a role.
In general, rather ubiquitous expression of the analyzed transcripts was detected. However, some are obviously restricted to certain tissue types like liver (TR2) or brain (148493, 290015, and 26513). In two cases (380575 and 290015), two bands on the autoradiogram point to alternative splice or polyadenylation variants of the respective transcripts. The similarity in size and expression pattern of the transcript detected by 290015, as well as the genomic localization, provide strong evidence that this cDNA may be part of the G2transcript (Glaser et al. 1989). However, it does not match the published 5 kb partial cDNA sequence (GenBank accession no. U10991).
In three cases, cDNA clones from different EST entries colocalized on the PAC contig (50813/136759, 44686/29944, 148493/37385/TR2), but extensive BLAST search with additional overlapping sequences did not reveal any connection between them. The similarity of expression patterns and transcript sizes suggests that 50813 and 136759 are nevertheless derived from the same gene, while 44686/29944 and 148493/37385/TR2 do indeed represent independent transcription units each (Fig. 4).
Expression Analyses with Mouse cDNA Clones
For those human cDNA clones that mapped to our PAC contig, we searched the mouse EST database for homologous clones. To render the similarity search more successful, we not only used the 3′ and 5′ end sequences of the respective IMAGE clones, but also where available, the annotated cDNA sequences of corresponding genes (3K/FJX1, PDX1, G2, TR2, and GA17). The murine pBs13 cDNA was also used to search for EST clones, as human clone 37385 is annotated as “highly similar to testis-specific mouse protein pBs13” in GeneMap'98, showing up to 42 % identity and 62 % similarity for this protein (summary Table3).
Mouse Gene Probes Used for Expression Analysis
Northern blots containing RNA of whole embryos and of various tissues of adult mice were probed with these mouse cDNA clones and results are summarized in Figure 4. The mRNAs of most transcripts are distributed ubiquitously in all tissues analyzed. Exceptions are 407691, 658218,fjx1, where a brain-specific expression in adult tissues is detected, and 989128, which showed strong expression in liver and relatively weak expression in kidney. The IMAGE clone 603000 did detect a transcript in adult testis tissue only. However, size differences between transcripts of 37385 (human)/603000 (mouse) or 26513 (human)/676616 (mouse) provide strong evidence that the mouse cDNA clones identified do not represent the orthologous transcripts. For two of 14 independent clones analyzed, no corresponding mRNAs could be detected in the tissues used for the Northern blots (962716 and 670550).
Expression analyses by Northern blotting is restricted to whole mouse embryos or tissues of adult mice that can be obtained in sufficient quantity. Therefore, we performed RNA in situ hybridization on paraffin sections of E14.5 to E17.5 mouse embryos to detail the fetal expression data as shown in Figure 5.
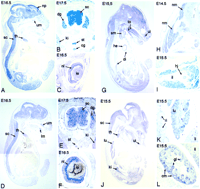
RNA in situ hybridizations to paraffin sections of mouse embryos, stages E14.5–17.5. (A-C) The global expression of G2in the nervous system is demonstrated. An even staining of spinal cord, dorsal root ganglia and brain with emphasis to the neopallial cortex can be observed as well as expression in the sympathetic trunk and the coeliac ganglion. Additionally, G2 transcripts are seen in the inner neuroblastic area of the retina, thymus, and the primordia of the teeth. (D-F) The anonymous cDNA clone 658218 detects an expression pattern in the nervous system that is very similar to that of G2. (G-I) The mRNA of the X component of the PDH complex is expressed mainly in skeletal muscle, diaphragm, and the heart, but also at lower levels in brain, spinal cord, and teeth primordia. (J-L) The GA17 mRNA is present in various tissues, mainly kidney (primitive glomeruli), lung, thymus, skeletal muscle, and teeth precursors, as well as in brain and spinal cord. Abbreviations: cg, coeliac ganglion; cm, condensing mesenchyme; dg, dorsal root ganglion; di, diaphragm; he, heart; hj, hip joint; il, iliac bone; ki, kidney; le, lens; li, liver; lm, primordium of lower molar teeth; lu, lung; nm, neck muscle; np, neopallial cortex; nr, neuroblastic layer of retina; gl, primitive glomeruli; ri, ribs; sc, spinal cord; sm, sternohyaloid muscle; st, sympathetic trunk; th, thymus gland; to, ex- and intrinsic muscles of the tongue; ui, primordium of upper incisor tooth; um, primordium of upper molar teeth.
The expression of the G2 transcript detected in human, as well as in murine adult neuronal tissues, is already present in the embryonic stage. The mouse embryo sections analyzed show a global but specific staining of neuronal tissues with an emphasis on the neopallial cortex along with expression in the retina, the developing teeth, and the thymus gland (Fig. 5A–C).
A very similar pattern but at lower levels can be observed for the anonymous IMAGE clone 658218 with the exception of a less explicit staining of the neopallial cortex region (Fig. 5D–F).
For PDX1 we detected strong expression in skeletal muscle and heart which agrees with data obtained from the literature (Aral et al. 1997; Ling et al. 1998) as well as the preceding Northern blot analyses. Weaker expression of PDX1 mRNA is also present in neuronal tissues as well as in the primordia of the teeth (Fig. 5G–I).
A high concentration of the GA17 mRNA is found in the mouse embryo in various organs, mainly lung, thymus, or the developing kidney, but also at lower levels in muscle and neuronal tissues. The expression in the kidney is restricted to the condensing mesenchyme and their derivatives, the primitive glomerular precursors that will later form nephrons (Fig. 5J–L).
No expression of the TR2 mouse homolog could be detected in the mouse embryo, either by Northern blot analysis, or by the in situ experiments. The strong expression in liver and the weaker expression in kidney seems to occur in adult tissues only.
A detailed expression analysis of the 3K/fjx1 gene has already been published (Ashery-Padan et al. 1999; A. Vortkamp in prep.).
In situ hybridization with the remaining murine cDNA clones as probes (388893, 962716, 670550, 907677) did not reveal any specific expression patterns.
DISCUSSION
Contig Construction
By end probe walking we have extended a PAC contig that now consists of 201 PAC clones and covers 7.5 Mb of 11p13–14.1. Determination of the marker content and insert sizes of overlapping PACs from a highly redundant set formed the basis for exact positioning of the clones. During contig construction we found a remarkable nonrandom distribution of the PAC clones. Whereas some markers detect up to ten PACs (data not shown) we could achieve only single clone coverage in five places of the newly established clone assemblies. However, single coverage never exceeded 50 kb and was smaller than 20 kb in two cases. A fragment of genomic DNA centromeric of the FSHB locus (PACs 815A16/1134O24) seems to be especially difficult to clone, not only in the PAC cloning system. We could not find any additional informative clone by screening three chromosome 11 specific cosmid libraries, LLNL (Evans and Lewis 1989), EJNAC (Heding et al. 1992), and ICRF (Nizetic et al. 1994) providing an approximate 12-fold genomic coverage with PAC ends of clones neighboring this critical area.
The newly created data fit the already existing physical maps of this region very well, reflecting the high accuracy achieved. However, an exact definition of the extent of clone overlaps is precluded at the outer edges of our region due to the paucity of NotI restriction sites and other mapped markers. Importantly, data obtained by fingerprinting all PAC clones strongly corroborate the hybridization results and did not provide any evidence for interstitial deletions. However, the statistical significance of results obtained with this method is necessarily limited to regions with large clone overlaps.
Gene Map of 11p13–14.1
Bins defined by overlapping PAC clones are an excellent tool to accurately place ESTs and genes along the physical map of 11p13. We were able to position 15 already known genes within our PAC contig, as well as five genes that had not yet been precisely mapped (TRAF6, 3K/fjx1, PDX1, GA17, andCSTF3) and 22 anonymous ESTs. According to the established gene map, there is a striking difference in concentration of genes in certain regions on 11p13. The 4 Mb region flanked by RAG1/2and GA17 harbors 13 genes and 20 ESTs with two particular gene dense regions close to the CSTF3 and CD59/G2 loci. In contrast, a region of similar size telomeric of GA17 is of low gene density and contains only seven genes (GA17, WT1, RCN1, PAX6, 239FB, FSHB, and KCNA4) and two ESTs (Fig. 1,2). However, the established gene map may still be incomplete and in particular, genes with restricted spatial or temporal expression may be missing. Thus, gene density may change in the course of further investigations. Nevertheless, the current distribution fits the paradigm of higher gene density in R-band versus G-band regions.
RH maps are currently the most detailed source available for whole genome gene maps. It is estimated that about half of the human genes are already annotated (Deloukas et al. 1998), most of them as ESTs derived from anonymous cDNA clones. Although most of the known 11p13 genes are already included in GeneMap'98, we located three genes,FJX1, GA17, and 239FB to our PAC contig that are not accounted for by the latest release. FJX1 and239FB had been detected by cDNA selection (Thäte et al. 1995; Schwartz et al. 1994) and GA17 by comparative mapping with pufferfish sequences (C. Miles and G. Elgar pers. comm.). Thus, alternative gene identification approaches are still necessary to discover the entire gene content of a region of interest.
Of 34 EST clones derived from the GeneMap'98 interval D11S4102–D11S1324 that is completely covered by our PAC contig, only 24 (71%) map to the contig clones. Especially on the level of single RH intervals, some major discrepancies occur regarding the order and distances of the markers entered (Figure 2). These inaccuracies originate from the use of the GB4 RH panel providing an average resolution of 1 Mb that is too large for fine mapping. Nevertheless, an overall concordance can be observed between the RH mapping and physical mapping approaches. Therefore, the current RH maps represent an excellent source to identify candidate genes in a distinct genomic region provided that the distortion of RH coordinates and the still preliminary gene content are taken into account. Unfortunately, the presentation of GeneMap'98 with small increments in cR values between most markers, suggests the presence of a much better resolution that cannot be gained with the panel used. The less widely used G3 panel, generated with a higher radiation dose, will certainly improve resolution.
Expression Analyses
The decision whether neighboring EST entries are part of the same gene can not always be made by mapping studies or sequence similarity searches, as cDNAs can span large genomic regions. In this respect, the comparison of their expression patterns provides valuable additional information.
Knowledge about transcript size and tissue specific expression of the newly mapped transcripts and their mouse homologs could be achieved rapidly by probing Northern blots with the corresponding cDNA clones. In some cases, RNA in situ hybridizations to sections of mouse embryos extend these expression profiles. The G2 transcript and the transcript detected by the anonymous cDNA clone 658218, are both expressed specifically and almost exclusively in neuronal tissues, pointing to their potential relevance for the development and maintenance of the nervous system. The fact that PDX1 is prominently expressed in muscle reflects the role of the PDH complex during glycolysis, namely the conversion of pyruvate to acetyl-CoA that is entered into the TCA cycle providing energy for muscle contractions. In the case of GA17, the widespread expression does not yield any clues as to its function in the organism. Similarly, the analysis of the GA17 protein for sequence motifs only revealed the presence of a PINT domain, which itself has no ascribed function yet.
The search for homologous mouse cDNA clones was often impeded by the nature of IMAGE cDNA clones that are usually derived from oligo-dT primed libraries. This creates a strong bias towards clones representing the 3′ ends of the transcripts which are often untranslated and much less conserved between species. For human and mouse cDNA probes that showed a rather weak similarity (BLAST E-values ≥1e-30), Northern blot analyses revealed considerably differing transcript sizes in some instances (37385/603000 and 26513/676616). This strongly suggests that the mouse cDNA clone identified is related, but not orthologous. Where the annotated coding sequences of genes were available for the similarity search, the identified probes generally resulted in hybridization signals consistent among all three approaches (human, as well as mouse Northern blot and in situ analyses), thus generating highly informative expression patterns.
Genomic sequencing of the present PAC contig is already under way at the Sanger Centre UK (ftp.sanger.ac.uk/pub/human/sequences/Chr_11). The knowledge of the genomic sequence will enable us in part to circumvent the lack of cDNA sequence information and to put the anonymous 3′-derived ESTs into their genomic context. The identification of murine counterparts, as well as the subsequent detailed expression analyses in the mouse, will thus become more reliable.
METHODS
Clone Libraries
PAC clones used for contig construction are derived from the RPCI-1, -3, -4, and -5 PAC libraries (Ioannou et al. 1994) and cDNA clones from the IMAGE consortium clone libraries (Lennon et al. 1996). PAC and IMAGE clones, as well as high density PAC filters, were obtained from the Resource Centre of the German Human Genome Project, Berlin; or from a local copy of the RPCI–1 library.
Gene Specific Probes
Probes are described in Gawin et al (1995) or taken from Genome Data Base.
GenBank accession numbers are as follows: TR2, AJ245600;3K, AJ245599.
PAC Contig Construction
The methods applied for PAC contig construction have been described in detail in Niederführ et al. (1998). PAC DNAs were prepared by standard alkaline lysis followed by phenol/chloroform extraction. Clone ends were isolated according to a linker-mediated PCR protocol (Kere et al. 1992) with minor modifications. PCR products were isolated from low-melting agarose gels and directly employed for random priming without further purification. Hybridizations to colony filters of the sublibrary were carried out according to Church and Gilbert (1984) with excess human placental DNA to block repetitive sequences. Hybridization results were stored in a MS Access database. Ordering and graphical representation of the data obtained were carried out using SAM (System for Assembling Markers,Soderlund and Dunham 1995).
DNA Fingerprinting and Computational Analysis
Fingerprinting was performed according to Coulson et al. (1986)with modifications as described in Niederführ et al. (1998). Briefly, PAC DNA was cut with HindIII, radio labeled, and digested with Sau3AI. Sizes of the gel separated DNA fragments were imported into FPC allowing the identification of overlapping fingerprints. These data were transferred to 11DB, a chromosome 11 database that is based upon AceDB (http://chr11.bc.ic.ac.uk).
Northern Blot Analyses
Multiple human tissue Northern blots with poly(A)+ mRNA were obtained from Clontech or Invitrogen. Murine Northern blots contained 20 μg of total RNA prepared according to Chomczynski and Sacchi (1987) from various murine tissues or total embryos. Blots were probed with radio labeled inserts isolated from IMAGE clones or other cDNA clones as described.
In Situ Hybridization to Paraffin Sections of Mouse Embryos
In situ hybridization to paraffin sections of mouse embryos was performed as described previously (Leimeister et al. 1998). Digoxigenin-labeled riboprobes of IMAGE clones were prepared by linearization of the respective cDNA clones and subsequent transcription with T3, T7, or SP6 RNA polymerases in the presence of digoxigenin–UTP. Five-micron sections of mouse embryos embedded in paraffin were mounted on polylysine coated slides. The sections were subsequently dewaxed, rehydrated, refixed in PFA, digested with proteinase K, and fixed again. After hybridization of the riboprobes to the slides over night at 72°C, the slides were washed, RNase treated, and incubated with anti-digoxygenin antibodies at 4°C for 16–20 hr. Detection was carried out using BM-purple substrate (Boehringer).
Acknowledgments
We gratefully acknowledge P. de Jong, B. Weber, and H. Stoehr for providing and transferring the RPCI-1 library, the German Resource Centre RZPD for providing PAC high density grid filters as well as PAC and IMAGE clones, C. Leimeister for helpful discussion about RNA in situ hybridization, the UK Medical Research Council and the Wellcome Trust for financial support (P.F.R.L), the Deutsche Forschungsgemeinschaft for financial support (M.G., Ge539/6), and the European Union for supporting the collaboration (Biomed 2).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL gessler{at}biozentrum.uni-wuerzburg.de; FAX 49-931-888-4150.
-
- Received July 8, 1999.
- Accepted August 25, 1999.
- Cold Spring Harbor Laboratory Press








