
The Family of Caenorhabditis elegans Tyrosine Kinase Receptors: Similarities and Differences with Mammalian Receptors
Abstract
Transmembrane receptors with tyrosine kinase activity (RTK) constitute a superfamily of proteins present in all metazoans that is associated with the control and regulation of cellular processes. They have been the focus of numerous studies and are a good subject for comparative analyses of multigene families in different species aimed at understanding metazoan evolution. The sequence of the genome of the nematode worm Caenorhabditis elegans is available. This offers a good opportunity to study the superfamily of nematode RTKs in its entirety and to compare it with its mammalian counterpart. We show that the C. elegans RTKs constitute various groups with different phylogenetic relationships with mammalian RTKs. A group of four RTKs show structural similarity with the three mammalian receptors for the vascular endothelial growth factors. Another group comprises RTKs with a short extracellular region, a feature not known in mammals; the genes encoding these RTKs are clustered on chromosome II with other gene families, including genes encoding chitinase-like proteins. Most of theC. elegans RTKs have no direct orthologous relationship with any mammalian RTK, providing an illustration of the importance of the separate evolution of the different phyla.
[The sequences in this paper have been submitted to GenBank under the following accession numbers: AF188748, AF188749, AF188750, and AF188751.]
Understanding the processes that have governed metazoan genome evolution is an important issue of modern biology that is largely nourished by the wealth of data generated by genome projects devoted to the analysis of selected key species. Comparison of the data obtained in several species has so far led to the general belief that the number of genes per genome was doubled after the separation of Protostomia and Deuterostomia and multiplied by three or four times in the chordate phylum after the separation from echinoderms. Such an increase in gene number could be due to tetraploidization events, as first proposed by Ohno (1970). Accordingly, it is believed that the vertebrate ancestors had single copies of genes now found in multiple copies in vertebrates due to these polyploidization events (Schughart et al. 1989; Lundin 1993;Garcia-Fernàndez and Holland 1994; Holland et al. 1994; Holland and Garcia-Fernàndez 1996; Sidow 1996; Bailey et al. 1997;Coulier et al. 1997; Spring 1997; Pébusque et al. 1998). The number of genes found in vertebrates is roughly fourfold that found in nonvertebrates, and many single copy genes in nonvertebrates have a direct correspondence (orthology) with a gene family in vertebrates. Recent findings on fish genomes have indicated that many gene families comprise even more members than in mammals, again supporting large-scale duplications as the essential driving force toward diversity (Aparicio et al. 1997; Holland 1997; Amores et al. 1998;Finnerty and Martindale 1998; Postlethwait et al. 1998; Prince et al. 1998; Wittbrodt et al. 1998). To this, individual local duplications add some complexity for some gene families in which newly duplicated paralogs are positively selected and gradually increase in number (Kirschner and Gerhart 1998). These families with maximal evolvability are encoding molecules involved in environment-oriented defense and recognition processes such as immunoglobulin, major histocompatibility antigen, and olfactory receptor gene families.
Investigating various species of triploblastic metazoans will provide information about their common or specific evolution. In particular, the availability of the nematode worm Caenorhabditis elegansgenome sequence allows for the first time the closest possible look at a present-day metazoan genome and putative transcriptome and proteome (The C. elegans Sequencing Consortium 1998).
We are particularly interested in examining multigene families inC. elegans and comparing them with the mammalian homologs. In this paper we have studied the tyrosine kinase receptors (RTKs). RTKs are type I transmembrane proteins that are involved in the control and regulation of several key cellular processes during development and in adult life. They are constituted of an extracellular ligand-binding region of variable size made of various domains, a transmembrane hydrophobic domain and an intracellular region bearing a kinase domain (TK), which is sometimes split in two parts (TK1 and TK2). Based on their structure and ligand-binding specificities, several classes of RTKs have been distinguished in mammals. In three classes, the extracellular region is made solely of immunoglobulin-like domains. To date, two groups of RTKs have been described in C. elegans: orthologs of RTKs known in mammals (e.g., DAF-2, EGL-15, KIN-8, LET-23, and VAB-1) (Table 1) and RTKs identified only inC. elegans (e.g., KIN-15 and KIN-16) (Morgan and Greenwald 1993). Some RTKs are absent in C. elegans (e.g., RET) but present in mammals and in Drosophila melanogaster, and others are present in mammals (e.g., PDGFR and VEGFR) but have not yet been described in the fruit fly nor in the worm. Few of the C. elegans RTKs have characterized or potential ligands (Hill and Sternberg 1992; Burdine et al. 1997). Conversely, some ligands have not yet been associated with receptors (Roubin et al. 1999).
RTK Classes in Mammals and C. elegans
RTKs are an excellent model for studying evolution of cellular processes in metazoans: First, they represent large families of modular proteins; Second, they are major representatives of the intercellular regulatory pathways that are specific of metazoans and Third, they belong to the most widespread contingency-generating agents of eukaryotic cells that are the protein kinases (Kirschner and Gerhart 1998). Furthermore, because tyrosine kinases have been under close scrutiny for many years in various species, they are associated with a wealth of information, and most of the genes encoding these proteins are probably identified in mammals; these molecules thus provide a good starting point for comparative studies of multigene families in different species.
We describe here the organization of 21 new C. elegans genes that encode putative RTKs phylogenetically related to various mammalian RTK classes.
RESULTS
Identification of RTK Genes by Exploration and Annotation of theC. elegans Sequence
The availability of the complete genomic sequence of the nematodeC. elegans allowed the identification of new putative RTK genes following a keyword search on the Wormpep database and a TBLASTN search on the genomic sequence with either the entire sequence or the tyrosine kinase domain sequence of receptors belonging to 13 classes of RTK described in mammals (see Table 1). The protein sequences indexed in the databases as containing a tyrosine kinase domain were checked for the presence of this domain by SMART search. Only the sequences with a canonical tyrosine kinase domain (and not with a serine/threonine type or of undetermined specificity) were used. Prior to further analyses, we made some annotations with respect to the available sequences (see Methods).
Figures 1 and 2 show the distribution in the C. elegans genome of the genes cited in the text. A total of 28 protein sequences, including the corrected T17A3.1, T17A3.8, W04G5.6A, W04G5.6B, and R09D1.12 sequences (see Methods), were thus available for subsequent analysis (Tables 1 and2).
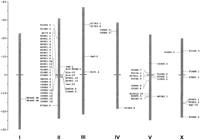
Distribution of RTK genes (at right of the chromosomes) and chitinase genes (at left) in the C. elegans genome. The genetic distances are indicated on a scale bar at left.
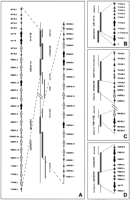
Details of chromosome localization of predicted C. elegans RTK genes described in this paper. Symbols are as follows (black arrows) RTK genes; (black arrowheads) RTK pseudogenes; (gray arrows) chitinase genes; (gray arrowheads) other genes. (A) clustering of genes encoding known and putative RTKs with a short extracellular region and chitinase-like genes on chromosome II. (B) Predicted RTK genes on chromosome III. (C) Predicted RTK and chitinase-like genes on chromosome V. (D) Predicted and known RTK genes on chromosome X.
Known and Putative C. elegans RTK Genes and Deduced Proteins
Phylogenetic Analysis of RTKs in C. elegans
A phylogenetic tree based on the alignment of the tyrosine kinase domain of RTKs was constructed; the sequences used were those identified by the C. elegans Sequencing Consortium (1998) and already included in ACeDB and the modifications that we made (see Methods). Thus, in addition to seven characterized nematode RTKs (i.e., DAF-2, EGL-15, LET-23, VAB-1, KIN-8, KIN-15, and KIN-16), we included 21 new putative RTKs.
As seen in Figure 3, DAF-2, EGL-15, LET-23, and VAB-1 had the expected phylogenetic relationship with their known mammalian orthologs: the insulin and IGF1 receptors, the fibroblast growth factor receptors, the receptors of the EGF receptor/ERBB family, and the ephrin receptors, respectively (Table 1). KIN-8 and the F11D5.3 putative protein shared a common ancestor with the mammalian ROR, TRK, MUSK, and discoidin receptor families. F11E6.8 shared a common ancestor with the MET/RON receptor family. The mammalian RTKs from classes III, IV, and V (Ig RTKs) grouped together and with class IX and X RTKs.
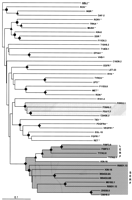
Phylogenetic analysis of known and predicted tyrosine kinase receptors of C. elegans bases on tyrosine kinase domain alignment. Proteins constituting monophyletic families (LERF, SERF, and four other RTKs) are indicated by shaded boxes. Symbols indicate the bootstrap value [(●) >500; (▴) >700; (♦) >900; 1000 replicates were done; statistical significance is reached with a value of 700]. The sequence of the human nonreceptor type tyrosine kinase ABL1 (underlined) is used as outgroup. Human sequences are with an asterisk (*), and accession nos. are as follows: ABL1 (X16416); DDR (Q08345); EGFR (P00533); EPHA1 (M18391); FGFR1 (M63888); INSR (M32972); MET(P08581); MUSK (AF006464); PDGFRA (P16234); RET (P07949); RON (X70040); ROR1 (M97675); ROS (M34353); TIE1 (X60957); TRKA (M23102); TYRO3 (P55146); UFO (P30530); VEGFR1 (X51602). A phylogenetic tree including more sequences can be found on our web site (http://olan.marseille.inserm.fr/u119/home.html).
Twenty-one known or predicted C. elegans RTKs did not share a recent common ancestor with mammalian RTKs, and some of them formed distinct phylogenetic families (Fig. 3, shaded boxes). A grouping of 13 RTKs (designated as LERF and SERF in Fig. 3; described below) was supported by a bootstrap value of >900/1000. Among these 13 RTKs were the KIN-15 and KIN-16 kinases (Morgan and Greenwald 1993). Four RTKs (F59F3.5, F59F3.1, T17A3.8, and T17A3.1) constituted a distinct monophyletic family (bootstrap value of >700). Four other RTKs (F09A5.2, F09G2.1, F08F1.1, and C24G6.2) grouped together (bootstrap value of >900).
Structural Features and Organization of C. elegans RTKs
Based on structural features, the 28 RTKs encoded by these known or predicted sequences could be divided into two groups.
In the first group, we classified the seven known or potential orthologs of mammalian RTKs (DAF-2, EGL-15, LET-23, VAB-1, KIN-8, F11D5.3, and F11E6.8). Based on a similar domain architecture and the phylogenetic analysis, F11D5.3 (Table 1) may be considered as the potential ortholog of collagen receptors DDR and TKT, RTKs with factor V–factor VIII domains in their extracellular region, also called discoidin-type RTKs. The sequence of the tyrosine kinase domain of F11E6.8 (Table 1) is very similar to that of the tyrosine kinase domain of class VI RTKs; however, the predicted protein is very short, with an extracellular region of only 40 amino acids lacking a signal peptide. Search in the genomic sequence did not reveal any similarity with the extracellular region of MET family of RTKs. We considered this putative protein as a wrong GENEFINDER prediction and a potential ortholog of class VI RTKs. No significant match and no modular architecture corresponding to RTKs from the other classes were found (Table 1).
Twenty-one predicted or characterized proteins with no extensive similarity to a particular class of mammalian RTKs constitute the second group. When their sequence and domain composition were compared, three different types could be identified (Table 2; Fig. 3).
- 1.
- One type contains two already characterized proteins, KIN-15 and KIN-16 (Morgan and Greenwald 1993), and seven other proteins. The extracellular region of KIN-15 and KIN-16 is very short (between 40 and 50 amino acids). We will hereafter refer to this family as the “short extracellular region” family (“SERF”). Such an unusual structure for a RTK has not been described in mammals. The other seven SERF proteins (C08H9.5, M01B2.1, R09D1.12, R09D1.13, W04G5.6A, W04G5.6B, and ZK938.5) have not yet been characterized in C. elegans. Thus, a total of nine putative RTKs belong to this family. By using the PSORT II and SMART tools, which give information on various sequence features related to protein sorting signal, we identified a putative peptide signal and a transmembrane region in the majority of the putative proteins belonging to SERF.
- 2.
- A second type is composed of four putative molecules (F59F3.1, F59F3.5, T17A3.1, and T17A3.8) containing an extracellular region of ∼850 amino acid residues. They will be referred to as the “long extracellular region” family (“LERF”). In the protein sequence of LERF RTKs, except in T17A3.8 that contains an erroneous GENEFINDER prediction for the extracellular region (see Methods), six immunoglobulin-like domains were identified by Pfam analysis using the fragment search option (we will see later that they actually have seven immunoglobulin-like domains, but the fourth one is distinct). Their tyrosine kinase domain was found to be bipartite, with 40–50 amino acid residues separating two subdomains.
- 3.
- A third type was represented by eight predictions that are very heterogeneous in their extracellular region (Table 2). Some predictions, such as F08F1.1, have a very short extracellular region lacking a signal peptide and may represent a wrong GENEFINDER prediction. Some others (F09A5.2, F09G2.1, R151.4, and T14E8.1) have a large extracellular region but do not contain any domain described to date. Two other protein predictions have in their extracellular region one (C24G6.2) or three (C16D9.2) fibronectin type III domains. The T10H9.2 protein prediction contains a low-density lipoprotein-a (LDL-a) domain, never described in mammalian RTKs.
C. elegans Putative RTKs with LERF Resemble Mammalian VEGFRs
The sequences of the extracellular region of the proteins with the LERF were aligned with the different classes of RTK containing five or seven immunoglobulin-like domains. The highest percentage of identity was obtained in the alignment with the extracellular regions of class V RTKs. These Ig RTKs are the three VEGFRs (Mustonen and Alitalo 1995;Fournier et al. 1997); their ligands are the vascular endothelial growth factors (Fig. 4). A remarkable conservation of the cysteines involved in the immunoglobulin-like disulfide bonds of the VEGFRs is seen in this alignment (not shown). Moreover, a region highly similar to the related fourth immunoglobulin-like domains of VEGFRs, which lacks a disulfide bond, is also seen in an equivalent location in the putative proteins of the LERF family. Some positions of this immunoglobulin-like domain containing aromatic amino acids are conserved among C. elegans and mammals. The immunoglobulin-like domains with disulfide bonds in the VEGFRs are structurally constituted by seven β-sheets and belong to the C2-type domain (Bork et al. 1994). Like in the mammalian VEGFRs, all immunoglobulin-like domains except the fourth one are made of seven β-sheets in the predicted secondary structure of the C. elegans LERF RTKs.
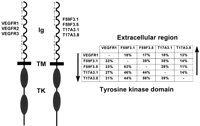
(Left) A schematic representation of the vascular endothelial growth factor receptors (class V RTKs) and the four C. elegansRTKs of the long extracellular region type; (Ig) Immunoglobulin-like domain (cysteine residues are shown), (TM) transmembrane domain, (TK) tyrosine kinase domain. (Right) The percentages of similar amino acids shared by these RTKs.
RTKs are characterized by the presence of a highly hydrophobic transmembrane domain made of an α-helix. In the LERF RTKs, similar regions with highly hydrophobic α-helices were identified upstream of the tyrosine kinase domain in all members and at equivalent positions (Table 2). Like the kinase domain of mammalian class III, IV, and V RTKs, the catalytic domain of the predicted LERF proteins is split by a kinase insert. Its size is 40–50 amino acids, different from the kinase insert of class IV RTKs (15 amino acids) and class III and V RTKs (70 amino acids).
Genomic Distribution of the Genes Encoding the New Predicted RTKs
The four genes encoding the LERF RTKs are located in tandem on either chromosome III or X (Fig. 2B, D). T17A3.1 and T17A3.8, the two genes from chromosome III (Fig. 2B), are oriented in an opposite direction, 6.5 kb apart. On chromosome X, F59F3.1 and F59F3.5 are in the same transcriptional orientation; no putative gene has been evidenced in the 4.5-kb region separating these genes by GENEFINDER (Fig. 2D).
The genes encoding the SERF RTKs are mostly found on chromosome II in two clusters separated by 0.5 Mb (Fig. 2A). One cluster, mapping at the approximate genetic position 0.99 and covering the M176 and R09D1 cosmids, includes the kin-15, kin-16, R09D1.13, and R09D1.12 genes, whereas the second cluster, at genetic map position 1.47 and covering the ZK938 and C08H9 cosmids, includes the ZK938.5, C08H9.5, and C08H9.8 genes. Three genes of this type are not located on chromosome II: W04G5.6A and W04G5.6B, located on chromosome I (Fig. 1), and M01B2.1, located on chromosome V (Fig. 2C).
A major feature of the RTK genes located in two clusters on chromosome II is their tandem organization. In the first cluster, thekin-15 and kin-16 coding sequences are separated by 529 bp and are transcribed unidirectionaly (Fig. 2A). Another pair of genes, R09D1.13 and R09D1.12, are also transcribed in the same direction but are 7.8 kb apart. In the second cluster, the ZK938.5 and C08H9.8 RTK genes are also organized in tandem. Similarly, but on chromosome I, the W04G5.6A and W04G5.6B genes are in the same transcriptional orientation and separated by only 0.7 kb.
Most of the C. elegans RTK Sequences Are Transcribed
To determine roughly how many of the predicted RTK genes correspond to expressed genes, we looked for the presence of ESTs in the DNA Data Bank of Japan. Seventeen genes have corresponding ESTs or complete cDNAs in this database (Table 2). We looked by means of reverse transcription–polymerase chain reaction (RT–PCR) for expression of the RTK genes of the short and long extracellular region families that do not have a corresponding EST or cDNA in the database. In addition to F59F3.1, for which ESTs exist in the database, we found that two other LERF genes (F59F3.5 and T17A3.1) and one SERF gene (R09D1.12) are expressed. Thus, a minimum of 20 RTK-encoding genes from a total of 28 putative genes have transcribed RNA.
Other Multigenic Families Are Located on Chromosome II
The two clusters on chromosome II containing genes predicted to encode RTKs also contain a large number of genes encoding chitinases (on cosmids M176, R09D1, and T19H5—genetic map position 0.99—and cosmids ZK938 and C08H9—genetic position 1.47) (Fig. 2A); guanylate cyclases (predictions AH6.1, R134.1, and R134.2 at approximate genetic position 1.01), olfactory receptors (from sra-1 tosra-9 on the AH6 cosmid, also at approximate genetic position 1.01), and cytochrome P450 molecules (one member on cosmid ZK1320 at approximate genetic position 1.13 and eight members on cosmid T10B9 at position 1.41) are also present in this area. A tyrosine kinase–chitinase-like genes association was also found on chromosome V. One gene coding for a SERF RTK (M01B2.1) and two genes coding for chitinase-like proteins (K08F9.3 and M01B2.6) are located very closely, in a region of chromosome V covered by 0.5 genetic map units.
The most prominent example for repetitive duplications of transcribed sequences in this region is the family of chitinases. Among the 40 chitinase genes or predicted chitinase-like genes (Table3), 25 are located on chromosome II along with SERF genes. Three other chitinase-like genes, T10D4.3, F15A4.8, and T13H5.3, map on chromosome II but far away from the tyrosine kinase–chitinase-like clusters, at position −20.28, 0.24, and 16.97, respectively (Fig. 1). The 12 other chitinase-like genes are located elsewhere in the genome: two on chromosome IV (C45E5.2 and C45E5.3 at the approximate genetic map position 23.98), nine on chromosome V (T05H4.7 located at the approximate genetic map position −1.11; T01C4.1 at 0.62; F07G11.9 and F10G2.5, representing two different predictions for the same genomic region, between 0.63 and 0.64; C08B6.4 and C51E3.8 at 3.18–3.19; R10D12.15 at 10.47; K08F9.3 at 20.47; and M01B2.6 at 21.11), and one on chromosome X (C04F6.3) (Fig. 1).
Chitinase Family in C. elegans
Based on both amino acid sequence differences and tridimensional structure (Henrissat 1990; Davies and Henrissat 1995; Henrissat and Romeu 1995), chitinases constitute two families of glycosyl hydrolases: family 18 (which contains chitinases from viruses, bacteria, fungi, animals, and classes III and V from plants), and family 19 (chitinase classes I, II, and IV from plants). Sequence analysis of the 40 chitinase or chitinase-like coding genes showed that they encode two types of proteins: a very large number of predicted proteins similar to chitinases of class II (family 18) and four predictions (K08F9.3, T05H4.7, R10D12.15, C08B6.4) similar to chitinase class IA. We conducted a phylogenetic analysis of these different chitinases. As shown in Figure 5, the chitinase-like proteins encoded by genes located in clusters on chromosome II shared a recent common ancestor (chromosome II, shaded boxes). The chitinase genes from class IA located on chromosome V were also grouped in one branch.
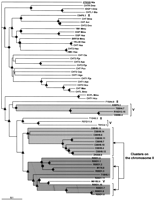
Phylogenetic analysis of chitinase-like proteins of C. elegans. Sequences analyzed in this tree are identical to those listed in Table 3 except for eight sequences that show an incomplete chitinase domain and have not been used in the phylogenetic analysis. Chromosomal localizations of C. elegans chitinase genes are indicated at right; the labeled shaded box indicate the two clusters localized on chromosome II (see Fig. 1). Symbols for bootstrap values are as in Fig. 3. The sequence X78325 (underlined) ofNicotiana tabacum is used as outgoup. Species abbreviations used for the phylogenetic tree are as follows: (Aea) Aedes aegypti (yellow fever mosquito); (Aga) Anopheles gambiae(african malaria mosquito); (Avi) Acanthocheilonema viteae(animal filarial nematode); (Bmo) Bombyx mori (silkmoth); (Bta) Bos taurus; (Bma) Brugia malayi (agent of Brugian lymphatic filariasis); (Bpa) Brugia pahangi (filarial nematode of domestic cats); (Csp) Chelonus sp. (braconid wasp); (Cte) Chironomus tentan (nonbiting midges); (Dme)Drosophila melanogaster (fruit fly); (Hcu) Hyphantria cunea (fall webworm); (Hsa) Homo sapiens (human); (Mse)Manduca sexta (tobacco hornworm); (Mmu) Mus musculus(mouse); (Nta) Nicotiana tabacum (common tobacco); (Ovo)Onchocerca volvulus (agent of river blindness); (Pco)Phaedon cochleariae (mustard beetle); (Pja) Penaeus japonicus (Kuruma prawn); (Wba) Wuchereria bancrofti(agent of lymphatic filariae).
DISCUSSION
Identification of Novel RTKs in C. elegans
A combined search of the C. elegans genome conducted along key words and through alignment (BLAST) allowed the identification of 28 putative protein-coding sequences with similarity to RTKs. Seven of these sequences were known RTKs (DAF-2, EGL-15, KIN-8, KIN-15, KIN-16, LET-23, and VAB-1) and 21 were new putative RTKs. Among the 28 RTKs, a first group of seven proteins is represented by the C. elegansorthologs of mammalian RTKs (Table 1); two protein predictions (F11D5.3 and F11E6.8) identified in this study represent candidate orthologs of mammalian discoidin receptors and receptors of the MET family, respectively.
Among a second group of 21 putative proteins, eight are very heterogeneous in their extracellular region; they lack similarity with any extracellular domain described to date or contain domains never described in mammalian RTKs, such as an LDL-a domain (Table 2). A subgroup of 13 putative RTKs was subdivided into two types according to the structure of their non-tyrosine kinase portion: Nine molecules containing a SERF were of the first type, and four putative RTKs containing a LERF were of the second type. Together they constitute a separate subgroup that does not share a recent common ancestor with any particular mammalian RTK class. It cannot be excluded that this is due to a rapid evolution of the genes, as is thought to be the case for two-thirds of protein-coding genes in C. elegans (Mushegian et al. 1998; Roberston 1998), even if the tree topology does not support it.
SERF RTKs have only 20–80 amino acids in their extracellular region. The previously described (Morgan and Greenwald 1993) KIN-15 and KIN-16 molecules, as well as seven other members, belong to this group. The function of KIN-15, KIN-16, and R09D1.12, for which the presence of messenger RNA was found, is unknown.
None of the putative LERF proteins has been described in C. elegans. They resemble the three mammalian VEGFRs. Like the VEGFRs, they contain an extracellular region composed of seven type C Ig domains (the fourth being a pseudo domain because it lacks the typical cysteine residues), a transmembrane domain, and an intracellular region composed of two tyrosine kinase subdomains separated by a kinase insert. From the phylogenetic tree built on the alignment of the tyrosine kinase domains, it seems that the two RTK types (LERF and SERF) described here have evolved very closely (Fig. 3). The loss of most of the extracellular domain from a seven immunoglobulin-like ancestor may have been the mechanism of apparition of the SERF RTKs. Further local duplications in cis or trans would have allowed the growth of this family up to nine members. The limited length of the branches and a high bootstrap value found in the phylogenetic analysis suggest that some of these duplications could be recent (C08H9.5 and ZK938.5 genes; see Fig. 3).
RTKs Resembling Mammalian VEGFRs Exist in C. elegans
Up to now, VEGFRs have only been described in vertebrates. Such molecules have not yet been recognized in Mollusca and Arthropoda; however, a genome from these phyla has not yet been completely sequenced.
In mammals, the three VEGFRs described to date participate in the development and architectural organization of both the circulatory and blood systems during embryogenesis and are involved in the regulation of cell permeability (Mustonen and Alitalo 1995). The existence of four molecules in the worm, which does not possess per se a circulatory system, is intriguing but may be a simple example of how the same molecules can be adapted to different roles in different species while performing the same basic processes. Chemotaxis, cell migrations, and pervasive growth are important processes of angiogenesis. The LERF receptors may be similarly involved in these processes in the worm, even if the cells in which they are expressed have a different differentiation status. It is also possible that the identification of the function of potential VEGFRs in C. elegans may point to new functions for this class of RTKs in vertebrates. We did not find any C. elegans sequence with significant similarity with any of the six known mammalian VEGFs. The identification of potential ligands of the RTKs with a LERF may provide some clues about their function and possible related function in mammals.
Possible Scheme of Evolution of RTKs in the C. elegans Lineage
Because of the remarkable conservation of domain architecture between C. elegans LERF RTKs and mammalian VEGFRs, convergent evolution may not be the cause of C. elegans VEGFR-like existence, although it cannot be ruled out. Based on the phylogenetic analysis and chromosome localization, we propose that the four LERF RTKs derive from a common ancestor through a series of cis, then trans and finally, cis duplications. They do not group with the mammalian VEGFRs. This is in favor of an independent expansion of these families after the separation from the last common ancestor of Protostomia and Deuterostomia. The egl-15 gene, encoding an FGFR, is, like F59F3.1 and F59F3.5, located on an overlapping cosmid on chromosome X, at position 15.79 (Fig. 2D). This suggests that the original linkage group that contained duplicated Ig RTK genes may have been maintained in the nematode lineage, whereas it has been disrupted in mammals where genes encoding class IV, on the one hand, and class III and VI RTKs, on the other hand, are separated (Rosnet and Birnbaum 1993; Pébusque et al. 1998).
The nine genes coding for the SERF RTKs are clustered on chromosomes I, II (in close proximity of chitinase-like genes), and V. From the relationships of these genes and their genome distribution, we propose a scenario of evolution implying large-scale duplications and local duplications. We hypothesize that a couple of tyrosine kinase–chitinase genes were represented in an “ancestral” linkage group that represented the precursor of a tyrosine kinase–chitinase-like cluster located on chromosome II. A second precursor cluster was generated in the close proximity of the first. Successive local duplications in cis allowed the expansion of tyrosine kinase and chitinase-like genes. This separate evolution of each cluster on chromosome II by the means of an earlier duplication of an ancestral linkage group followed by local amplification of genes is suggested by the topology of the trees of chitinase-like and tyrosine kinase genes. The phylogenetic analysis of the chitinases-like protein sequences revealed that the molecules located on each of the two chromosome II clusters form a separate branch and that both branches are grouped separately from the other chitinase genes of different species. Among the chitinase-like genes located on the other chromosomes, only M01B2.6 (on the same cosmid as MO1B2.1 SERF RTK gene) branches with the genes of one chromosome II cluster supporting the hypothesis of a partial trans duplication.
For chitinase-like proteins as for kinases, some duplication events could be recent, suggesting that the concerted expansion process may be ongoing. The presence of genes encoding olfactory receptors in the same clusters is interesting. Robertson (1998) has previously suggested an ongoing process of rapid evolution of olfactory receptor genes with extensive gene duplications, movement, and diversification. We propose below two nonexclusive types of possible explanation for this expansion.
Chitinases are glycosyl hydrolases that catalyze the degradation of chitin, a polymer of N-acetylglucosamine. In plants and insects, they are an important inductive host defense mechanism against fungi. It could be that chitinase-like proteins from C. elegans are also implicated in defense against parasites. In this case, the expansion of the chitinase family would be dependent on a positive selection mechanism, and passive “hitchhiking” by this mechanism may explain the combined expansion of RTK genes. However, some predictions for chitinase-like sequences yield proteins that lack the chitin-binding domain or the catalytic domain, and, at present, their role remains unknown.
A large region containing multiple copies of RcC9, RcD1, Rc35, and Rc123 DNA repetitive elements is located between the two clusters of tyrosine kinase–chitinase genes from chromosome II. These elements, showing an internal sequence organization resembling that of minisatellite sequences described in mammals, have a tendency to cluster at some positions on the chromosomes of C. elegans(Naclerio et al. 1992). The supraperiodic patterns, which are different in the closely related species of C. elegans,Caenorhabditis briggsae, and Rhabditis maupasi, are constant between arrays with different chromosomal locations suggesting an active mechanism of homogenization (crossing-over, gene conversion) (Naclerio et al. 1992; La Volpe 1994). Because in human cell lines, minisatellite sequences promote homologous recombination (Wahls et al. 1990), an increased rate of crossing-over promoted by the presence of repeated sequences, associated with possible slippage in the homologous pairing due to lower complexity of the region containing the repeated sequences, might have resulted in the progressive increase of copies number of genes surrounding the region. An example of gene expansion associated with a particular type of complex satellite structures has been shown in the human genome (Eichler et al. 1998).
Conclusions
The identification and comparison of orthologs of human proteins in the nematode may yield interesting clues as to the respective functions of proteins in the two species. Furthermore, the particular organization of genes in C. elegans, such as kinase and chitinase genes, may provide clues as to the mechanisms that have driven their typical, rapid evolution by duplication and, thus, mechanisms that influence evolution. These may be associated with positive selection as the resulting proteins could be involved in defense mechanisms or with instability of a genomic region due to repetitive elements. Both mechanisms may have worked together because evolutionary pressure may have favored expansion over contraction of the region.
In metazoan evolution, each lineage, having evolved independently over long periods of time, shows some extent of specificity with regard to genome organization (Ruddle 1997). Independent and specific evolution may be frequent. A good example is that of HOX genes in teleosts (Aparicio et al. 1997; Amores et al. 1998). We show here an example of independent evolution in a multigene family of RTKs in theC. elegans lineage. Examples in C. elegans will certainly be numerous, and several have been reported already (Robertson 1998; Jansen et al. 1999; Sluder et al. 1999). This shows that this type of evolution concerns various types of genes and various types of species. In C. elegans, the apparently rapid processes of genetic duplications and movements within the genome may greatly favor this independent evolution.
METHODS
Database Searches
To identify putative RTKs in C. elegans, two approaches were used. First, a search in the last release (no. 16) of Wormpep database using different combinations of the key words =elegans=, =tyrosine=, =kinase=, and =receptor= was done by means of the Entrez search tool (http://www.ncbi.nlm.nih.gov/Entrez/). We thus identified the GENEFINDER predictions indexed as putative RTK or only as tyrosine kinases. Second, a C. elegans-specific TBLASTN search (http://www.sanger.ac.uk/Projects/C_elegans/blast_server.shtml) using tyrosine kinase domain sequences was used for the identification of some genomic regions coding for such a domain and missed by GENEFINDER.
The sequences recovered by the two approaches larger than 400 amino acids were further analyzed for motif or domain composition using Pfam (http://www.sanger.ac.uk/Software/Pfam/) and SMART (http://coot.embl-heidelberg.de/SMART/) tools. This eliminated some serine/threonine kinases or undetermined proteins that were recovered from the initial searches. Then, to eliminate the potential nonreceptor type tyrosine kinases, a search in the PSORT II program (http://psort.nibb.ac.jp) was done. Two sequences (CO8H9.8 and T01G5.1) lacking a consensus ATP-binding site were discarded. For the C24G6.2 gene, there are two alternative predictions of putative transcripts, C24G6.2A (coding for a protein with a large extracellular region with fibronectin type III domains) and G24G6.2B (coding for a protein with a short extracellular region lacking any described domain).
The information for cosmids, genes, and gene products (Wormpep database) of C. elegans is available through the ACeDB database (http://www.sanger.ac.uk/Projects/C_elegans/webace_front_end.shtml).
Searches for expressed sequence were done in the DNA Data Bank of Japan maintained by Yuji Kohara (http://www.ddbj.nig.ac.jp/).
Sequence Analysis and Alignment
Sequence similarity searches were done using the BLASTP and TBLASTN algorithms (Altschul et al. 1990, 1997). The accession numbers for sequences in mammals and C. elegans are listed in the legend to Figure 3 and in Tables 2 and 3, respectively. The protein sequences were aligned using the CLUSTAL W (Thompson et al. 1994) program followed by manual edition. The protein sequence analysis and the alignments were confirmed by searches with the PSORT II (http://psort.nibb.ac.jp), Pfam, and SMART tools. Alignments were done with the entire protein sequences or after partitioning into distinct domains.
Annotations
Annotation 1
The T17A3.1 predicted protein contains a truncated tyrosine kinase domain (with the TK1 domain only). A region presenting a significant similarity to both kinase insert (KI) and TK2 domain, referred to as F40G9.13 gene by GENEFINDER, was identified downstream of the putative stop codon of the predicted T17A3.1, in the neighboring F40G9 cosmid (see Fig. 2B). We considered T17A3.1 as an erroneous prediction and corrected the protein sequence by the addition of the missing KI and TK2 region identified on cosmid F40G9. A short region presenting a weak similarity with the extracellular region of F59F3.1 was also identified on cosmid F40G9 but in an opposite orientation to the T17A3.1 transcript. We considered this region lacking a GENEFINDER prediction as a pseudogene.
Annotation 2
In the region of the T17A3.8 transcript, we identified two short sequences with high similarities to the one encoding the putative extracellular region of F59F3.1, which were not predicted by GENEFINDER. We corrected the deduced protein sequence of T17A3.8 by including these two sequences. We identified by BLAST search on the genomic sequences an exon not predicted by GENEFINDER coding for a region downstream of the ATP-binding site. On the same cosmid, the gene prediction T17A3.10, which is oriented in the same direction as T17A3.8 (see Fig. 2B), has a similarity with the region contained between amino acid positions 450 and 850 of putative RTKs F59F3.1 and F59F3.5. It was considered as a probable pseudogene because we were unable to identify upstream and downstream sequences with similarity to the remaining of an RTK molecule.
Annotation 3
When we used the Pfam module recognition tool, we found that one of the genes, namely W04G5.6, contained sequences encoding two tyrosine kinase domains that, when analyzed by TBLASTN, happened to be very similar. We considered these two regions as belonging to two different transcripts transcribed in the same direction that were missed by the GENEFINDER prediction. They will hereafter be referred to as W04G5.6A and W04G5.6B (see Fig. 1).
Annotation 4
Sequence analysis of the cDNA fragment amplified by RT–PCR using R09D1.12 primer pair showed that exon four of the R09D1.12 gene is 75 bp longer in its 3′ end than predicted. The corrected protein sequence, by addition of 25 amino acids in the tyrosine kinase domain, was used for phylogenetic analysis.
Phylogenetic Analyses
Phylogenetic trees were inferred using neighbor-joining algorithms (Saitou and Nei 1987) of the CLUSTAL W phylogenetic package (Thompson et al. 1994) to determine the evolutionary relationships among sequences. Default parameters of CLUSTAL W were used. The results were analyzed using the bootstrap method (1000 replicates) to provide confidence levels for the tree topology (Felsenstein 1985). The construction of trees was done with TREEVIEW (Page 1996).
For the phylogenetic analysis of RTKs, we used only the tyrosine kinase domain of proteins; due to the length variability of the kinase insert region between different proteins, these were eliminated. The large gaps were also eliminated. The tyrosine kinase domain was defined as follows: For vertebrate tyrosine kinases, the sequences used were those defined by the Pfam program; and for C. elegans kinases, the domain was identified by SMART and aligned with the alignment existing for the vertebrate sequences in Pfam. This alignment can be found on our Web site (http://olan.marseille.inserm.fr/u119/home.html). The tree was rooted using the kinase domain of human ABL1, a cytoplasmic tyrosine kinase.
The chitinase domains of well-characterized chitinases and of chitinase-like proteins from different species of mammals, insects, and worms were used for the phylogenetic analysis of chitinases-like proteins. A plant chitinase was used for rooting the tree.
Expression Analyses
RT reactions were performed from 2 μg of total RNA extracted from mixed-staged worm preparations by lithium chloride precipitation (MacLeod et al. 1981), using random hexanucleotides and SuperScript II Reverse Transcriptase (GIBCO BRL) according to manufacturer's instructions. The equivalent of 200 ng of reverse transcribed RNA and 50 ng of total DNA as control were used for PCR detection of messengers using the following primers for genes encoding RTK: R09D1.12F, 5′-CCCAAGATTAACTCGATCGATCG-3′; R09D1.12R, 5′-TCCTTCAATGAACTCCGAAACG-3′; F59F3.5F, 5′-TACAGCAAAGTGGTCCAGG-3′; F59F3.5R, 5′-CTGTCATTTGGCTCATCG-3′; T17A3.1F, 5′-CAACGGATTATCGGATTCAG-3′; and T17A3.1R, 5′-TTGAGCTGCATATCCTTTCCTA-3′.
The following conditions were used for PCR amplification: initial denaturation at 95°C for 5 min, 35 cycles of 30-sec denaturation at 95°C, 30-sec annealing at 55°C (for R09D1.12 and T17A3.1) or 52°C (for F59F3.5), 1-min extension at 72°C, and a final extension at 72°C for 10 min. The fragments were gel-purified, cloned using pGEM-T Easy kit (Promega), and sequenced at Génome Express (Grenoble, France) using an automated sequencer (Applied Biosystems 373).
Acknowledgments
This work has been supported by INSERM and Institut PaoliCalmettes. We thank J. Ewbank, D. Maraninchi, C. Mawas, N. Pujol, and M.J. Santoni for helpful discussions and encouragement.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
NOTE ADDED IN PROOF
The KIN-8 receptor tyrosine kinase has been renamed CAM-1 (Forrester et al. 1999. Nature 400: 881–885).
Footnotes
-
↵4 Corresponding author.
-
E-MAIL birnbaum{at}marseille.inserm.fr; FAX 33 4 91 26 03 64.
-
- Received May 4, 1999.
- Accepted August 17, 1999.
- Cold Spring Harbor Laboratory Press








