
A Multilocus Genotyping Assay for Candidate Markers of Cardiovascular Disease Risk
- Suzanne Cheng1,9,
- Michael A. Grow1,
- Céline Pallaud2,
- William Klitz3,
- Henry A. Erlich1,
- Sophia Visvikis2,
- John J. Chen3,4,
- Clive R. Pullinger5,
- Mary J. Malloy6,7,
- Gérard Siest2, and
- John P. Kane6,8
- 1Department of Human Genetics, Roche Molecular Systems, Inc., Alameda, California 94501 USA; 2Centre de Médecine Préventive, 54501 Vandoeuvre-lès-Nancy, France; 3School of Public Health and 4Department of Integrative Biology, University of California at Berkeley, Berkeley, California 94720 USA; 5Cardiovascular Research Institute, 6Department of Medicine, 7Department of Pediatrics, and 8Department of Biochemistry and Biophysics, University of California at San Francisco, San Francisco, California 94143 USA
Abstract
A number of chronic diseases, including cardiovascular disease, appear to have a multifactorial genetic risk component. Consequently, techniques are needed to facilitate evaluation of complex genetic risk factors in large cohorts. We have designed a prototype assay for genotyping a panel of 35 biallelic sites that represent variation within 15 genes from biochemical pathways implicated in the development and progression of cardiovascular disease. Each DNA sample is amplified using two multiplex polymerase chain reactions, and the alleles are genotyped simultaneously using an array of immobilized, sequence-specific oligonucleotide probes. This multilocus assay was applied to two types of cohorts. Population frequencies for the markers were estimated using 496 unrelated individuals from a family-based cohort, and the observed values were consistent with previous reports. Linkage disequilibrium between consecutive pairs of markers within theapoCIII, LPL, and ELAM genes was also estimated. A preliminary analysis of single and pairwise locus associations with severity of atherosclerosis was performed using a composite cohort of 142 individuals for whom quantitative angiography data were available; evaluation of the potentially interesting associations observed will require analysis of an independent and larger cohort. This assay format provides a research tool for studies of multilocus genetic risk factors in large cardiovascular disease cohorts, and for the subsequent development of diagnostic tests.
Multiple genetic and environmental risk factors appear to contribute to common diseases such as cardiovascular disease and cancer. Although specific genetic causes have been identified among certain families with a history of disease, the association of these genes with disease in the general population is not fully understood. One reason is that genetic predisposition to such diseases can result from the cumulative effect of common allelic variants, variants that individually confer only a modest increased risk. Thus, one challenge is to identify the common multilocus profiles that confer a high risk for disease; a second challenge is to understand how environmental factors modulate expression of a genetic predisposition to disease.
A growing number of genetic variants have been implicated in the development of complex diseases. As these candidate genes are identified, there is an increasing need for assays capable of simultaneously genotyping multiple loci. Studies focused on single markers can be used to assign relative risk values, but this approach provides only a limited context for evaluating genetic risk factors. Studies encompassing multiple markers provide a broader context that is critical to assess information on candidate markers for multifactorial diseases, and multilocus assays can greatly facilitate the necessary genotyping process. Multilocus results can provide insight into mechanisms of disease susceptibility and identify key subsets of predictive markers that are clinically informative. These informative genetic markers can then be used to supplement routine biochemical assays for patient care, for example, in lieu of protein activity or concentration measurements that are difficult to make or show significant intra- and interindividual variability independent of disease state.
In developing a prototype multilocus genotyping assay, we focused on cardiovascular disease (CVD), a leading cause of death worldwide. Monogenic disorders, such as familial hypercholesterolemia and hypertrophic cardiomyopathy (for reviews, see Hobbs et al. 1992; Day et al. 1997; Bonne et al. 1998), have been identified among some families. Established risk factors for disease in the general population include age, gender, diabetes mellitus, obesity, high serum cholesterol levels, and hypertension, as well as cigarette smoking and physical inactivity (Pasternak et al. 1996). These factors, however, do not explain all premature CVD cases (Hoeg 1997). A number of these established factors have genetic components, and as yet unknown risk factors may be primarily genetic. In addition, recent evidence indicates that genetic factors influence patient responsiveness to therapeutic intervention, both dietary (Humphries et al. 1996) and pharmaceutical (Kuivenhoven et al. 1998).
The “CVD35” assay described here is comprised of 35 biallelic sites within 15 genes representing pathways implicated in the development and progression of atherosclerotic plaques: lipid metabolism, homocysteine metabolism, blood pressure regulation, thrombosis, and leukocyte adhesion (Table 1). The panel includes well-known polymorphisms in the apolipoprotein E (apoE; for review, see Mahley 1988) and angiotensinogen (AGT; Jeunemaitre et al. 1992) genes, mutations such asapoB Gln-3500 (Soria et al. 1989) and factor V Leiden (Bertina et al. 1994), and more recently identified sequence variations in the methylene tetrahydrofolate reductase (MTHFR; Frosst et al. 1995; Goyette et al. 1995) and E-selectin (ELAM; Wenzel et al. 1996) genes. The CVD35 assay uses pooled polymerase chain reaction (PCR; Mullis and Faloona 1987; Saiki et al. 1988) primer pairs to coamplify 27 targets from genomic DNA in two reactions. Amplified fragments within each PCR product pool are then detected colorimetrically with sequence-specific oligonucleotide probes immobilized in a linear array on nylon membranes (Saiki et al. 1989). Probe sequences have been optimized carefully to permit genotyping of all sites under a single assay condition. Therefore, large cohorts can be typed rapidly at all 35 sites, providing an extended database for evaluating the disease association of these markers, and a multilocus context for evaluating new candidate markers. We have applied this multilocus assay to a population-based cohort to estimate allele frequencies and intragenic haplotypes, and to a lipid clinic-based cohort to model a case-control study.
Targets Genotyped by the CVD35 Assay
RESULTS
CVD35 Assay
A three-primer, two-probe system for apoE was used as the basis for the CVD35 assay; the specificity of this system has been described previously (Cheng et al. 1998). No Arg-112/Cys-158 alleles were detected among any of the 1400 control or cohort samples genotyped, consistent with previous studies of the apoE gene (Houlston et al. 1989). In future versions of this assay, the allele-specific primers for codon 112 will be replaced by probes to simplify genotyping of the apoE marker (data not shown).
The PCR products range from 95 to 535 bp in size. As shown in Figure1, nearly all of the PCR products in each of the final multiplexes (14 in multiplex A, 13 in multiplex B) could be clearly distinguished by gel electrophoresis. Although the largest product bands appeared relatively weak in fluorescence intensity, these yields were sufficient for detection by the immobilized probes.
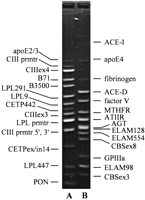
Agarose gel image illustrating the PCR product pool resulting from coamplification of multiple loci in the CVD35 research assay. The molecular weight standard is HaeIII-digested ΦX174. The PCR products in lane A (multiplex A) range from 95 to 395 bp; those in lane B (multiplex B) range from 105 to 535 bp. Not all of the PCR products are clearly visible, and one low molecular weight nontarget band is visible in lane A.
Detection of the amplified alleles is illustrated in Figure 2, as well as the specificity of the probe panel; for example, the results distinguish each of the three possible genotypes atLPL(−93) (Fig. 2A: strips three to five), PON192(Fig. 2A: strips one to three), AGT235 (Fig. 2B: strips one to three), and factor V 506 (Fig. 2B: strips one, two, and four). Figure 2C shows the third strip (strip B2) for two different individuals; of the ∼720 unrelated individuals genotyped from all sources, only one variant allele was detected among these four candidate markers (data not shown).

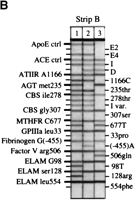
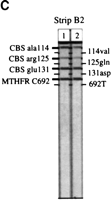
Representative probe strips demonstrating the identification of different alleles. Genomic DNA samples were amplified with either the multiplex A (A) or the multiplex B (B,C) primer pool, then hybridized to the corresponding immobilized probe strips, as described under Methods. Five examples each of probe strips A and B are shown; only two examples of probe strip B2 are shown. For the purpose of illustrating a range of different genotypes, the individuals typed here in A do not correspond to those inB or C. On each vertical strip, alleles amplified from the original sample are indicated by horizontal lines. The template guides at the left and right identify the allele detected at each probe position. With the exception of apoE3, ACE-I, andACE-D, the less prevalent genetic variant for each marker is listed on the right. Differences in the relative efficiencies of amplification and probe hybridization contribute to the variation in the actual intensity among the loci. (In actual size, each strip is ∼8 cm long.)
Samples representing a subset of 303 families from the Stanislas cohort (Siest et al. 1998) were used to assess the performance of the CVD35 assay in a large-scale genotyping effort. Eight families were excluded from the haplotype analysis because of inconsistencies of genotypes between parents and offspring, although the unrelated parents were included in the analysis of allele frequencies. In addition, four samples were omitted as a result of weak second allele signals for several markers. Because the assay was designed to yield comparable signals for both alleles in heterozygotes, these weak signals may have resulted from sample contamination. When available, all questionable samples will be retyped using new DNA preparations. A total of 1190 samples were used for subsequent analyses of the allele frequencies for all markers and haplotypes within the apoCIII, ELAM, andLPL genes.
Genotyping data for apoE, ACE, and LPL447, which had been obtained previously through independent methods, were used to evaluate the accuracy of the CVD35 assay for these three markers. No discordant results were noted for LPL447; those few samples yielding discordant genotype results for apoE and ACEwere investigated further. For apoE, detection of thee4 allele by the CVD35 assay was problematic if insufficient template DNA was used for amplification; this difficulty should be corrected in future versions of the assay that no longer rely on codon 112-specific primers (data not shown). Three initially discordant results were traced to the specific aliquots tested.
For the ACE I/D marker, one D allele and twoI alleles that had been identified by capillary electrophoresis were not detected by the CVD35 assay. This particularACE-D allele was detectable if an alternative primer pair was used, suggesting the presence of a novel sequence variation within the default priming sites. One of the undetected ACE-I alleles was also amplifiable with an alternative upstream primer, although with poor efficiency, and gel electrophoresis revealed a truncated insertion of ∼50 bp in size (data not shown); this ACE-I allele had been detected previously based on the 3′ insertion junction sequence. The second undetected ACE-I allele was identified correctly after single-target amplification with the original primer pair, or using a multiplex amplification with an alternativeACE-I-specific primer spanning the 3′ insertion junction, as in Evans et al. (1994; data not shown). Failure of the CVD35 assay to amplify the full-length Alu element from a wild-type sequence in a multiplex reaction appeared to be unique to this sample. With the 3′ junction-specific primer, the ACE-I target was reduced from 533 to 155 bp; this primer was used to confirm allD/D genotypes within the UCSF cohort (data not shown). Use of the ACE-I-specific primer spanning the 3′ insertion junction in future assays should address these few difficulties.
Allele Frequencies
No variant alleles were observed within either cohort for 6 of the 35 sites: LPL nucleotide (−39); CETP codon 442;CBS codons 125, 131, and 307; and MTHFR nucleotide 692. Within the Stanislas cohort, a single CBS Val-114 allele and no apoB codon Gln-3500 alleles were detected. The genotypes of 496 unrelated parents from this group were used to estimate population frequencies for each of the remaining 28 markers; these data (Table 2) extend the previous report based on 455 individuals (Cheng et al. 1998). The observed frequencies are consistent with previous reports for caucasian populations (as cited in Table 2). Four markers (CBSIle/Thr-278, CBS del/ins, ELAM 98, and ELAM128) appeared to differ (χ2 > 3.84) from expectations based on Hardy–Weinberg equilibrium, and complete concordance in genotypes between the two CBS markers and between the two ELAM markers was also noted. This may be type I error, given the number of loci tested; in general, the observed allele frequencies did predict the observed genotype frequencies.
Allele Frequencies (pA, pa) Observed among Unrelated Individuals within the Population-Based Stanislas Cohort
Within the UCSF cohort of 142 individuals, no variant CBSVal-114 alleles were observed. Four carriers of the apoBGln-3500 mutation were noted within this clinic-based cohort, in contrast to the population-based Stanislas cohort. One carrier of theCBS Thr-278 allele without the 68-bp insertion was also detected; this genotype was confirmed by sequencing (data not shown). One sample initially yielded a null result for the ACE target, but subsequently was assigned the I/I genotype with use of the insertion-junction-specific primer. All ACE D/Dgenotypes were confirmed by reanalysis with the junction-specific primer (data not shown). Allele frequencies are given in Table 2. One marker (ELAM554) appeared to differ (χ2 = 4.14) from expectations based on Hardy–Weinberg equilibrium, but given the small sample size and number of loci, this may reflect type I error; in general, the observed allele frequencies predicted the observed genotype frequencies.
Linkage Disequilibrium
Using 1100 chromosomes from 275 families, three loci were examined for haplotypes defined by multiple sites typed within each gene:ELAM (three sites), LPL (four sites), andapoCIII (six sites). Linkage disequilibrium estimates between proximal marker sites are presented in Figure 3; the haplotype data will be explored in greater detail elsewhere (W. Klitz et al., in prep.). Allele frequencies among this subset of the Stanislas cohort were comparable to those listed in Table 2.
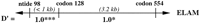
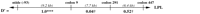
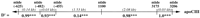
Linkage disequilibrium values (D′) estimated from the intragenic haplotypes for the ELAM, LPL, and apoCIII genes that were observed among 275 families from the Stanislas cohort. Above each gene represented by a horizontal line, the genotyped sites are labeled by nucleotide (ntide) or codon position. Distances between adjacent sites are given in kilobases; the spacing is not to scale. (†)P > 0.1; (*) P < 0.05; (***)P < 0.001.
Maximum disequilibrium (D′ = 1.0) was observed among the three sites genotyped within the ELAM gene. The lower statistical significance associated with codon 554 was due to the low frequency of the Phe-554 allele. Codon Phe-554 was observed only on chromosomes carrying the G98 and Ser-128 alleles. Although complete concordance in genotypes between the ELAM 98 and 128 sites was noted in this study, chromosomes bearing only one of the two variants have been reported (Wenzel et al. 1996).
Maximum linkage disequilibrium was also observed between theLPL promoter site (−93) and codon 9. Three chromosomes were observed with the (−93)G (less frequent allele) and Asp-9 (more frequent allele) haplotype; all others were concordant in genotype between the two sites. Codons 291 and 447 appeared to be in modest linkage disequilibrium, although statistical significance was not achieved due to the low frequency of the Ser-291 allele (0.016). The strong disequilibrium between promoter site (−93) and codon 9 among caucasians has been reported previously (Hall et al. 1997). The suggestion of greater recombination rates upstream of codon 291 and between codons 291 and 447 (exons 6 and 9), as inferred from the linkage disequilibrium results, is consistent with recently reported data (Clark et al. 1998; Nickerson et al. 1998).
For the apoCIII gene, linkage disequilibrium was greatest within the promoter and exon 4 (markers 3175, 3206), regions with marker sites separated by fewer than 150 bp. Within the promoter, interestingly, linkage disequilibrium was not maximal between the (−482) and (−455) sites, separated by only 27 bp. Disequilibrium was low between the promoter region and site 1100, which are separated by 1.5 kb, yet strong disequilibrium was observed between sites 1100 and 3175 (exons 3 and 4), a 2-kb separation. The relatively infrequent 3175-G variant did appear to be in strong disequilibrium with the promoter variants (data not shown), as had been reported previously (Dammerman et al. 1993). Strong linkage disequilibrium between sites 1100 and 3206 has also been previously noted (Xu et al. 1994).
Although the likelihood of detecting association with disease is expected to be greatest with allelic variants of demonstrated functional significance, this information may not be readily available. In the absence of clear evidence as to the most functionally significant variations within a single gene, linkage disequilibrium data assist in determining the most informative sites to genotype for disease association studies. Redundant sites may then be replaced by new candidate markers. Linkage disequilibrium data may also lead to hypotheses tracing the evolution of haplotypes that may be associated with disease.
Disease Association
The variant allele frequencies observed within the lipid clinic-based UCSF cohort are listed alongside those of the population-based Stanislas cohort in Table 2. Although the Stanislas and UCSF cohorts were not specifically matched for population substructure, higher frequencies of apoE e2 and e4, apoB Gln-3500, and apoCIII 1100T and 3206G alleles (P < 0.05) were observed within the clinic-based UCSF cohort, consistent with previous reports associating these variants with elevated lipid levels; none of the nonlipid-related markers showed a nominally significant difference in frequencies between the two cohorts. The overall trend of increased frequencies for reportedly disease-associated alleles that was observed in the UCSF cohort might be expected among these individuals who are at higher risk for coronary events than the general population.
The UCSF cohort was comprised of 142 unrelated caucasians for whom angiograms had been quantitated and scored by the Gensini method (Gensini 1975). These scores were used to subdivide the cohort into quintiles that represented differing severities of coronary arterial occlusion. No significant deviations from Hardy–Weinberg equilibrium were noted within these quintiles (data not shown). The Gensini-based quintiles did not show significant correlation with total, low-density lipoprotein (LDL), or high-density lipoprotein (HDL) cholesterol levels, although there was an unexpected, suggestive trend toward lower average very low-density lipoprotein (VLDL)-triglyceride (TG) and VLDL-cholesterol levels with increasing Gensini score (data not shown).
Disease association with the allelic variants of 15 markers was explored among female-only (FQ1 vs. FQ5) and the combined gender (Q1 vs. Q5) quintiles, as described under Methods. Although the small size of this UCSF cohort limited the statistical power to detect one- and two-locus effects on risk for CVD, the intent of this analysis was to demonstrate how the CVD35 assay could be used to evaluate disease association and genotype interactions with a case-control study design. Given the exploratory nature of these preliminary analyses, no formal statistical correction for multiple testing was applied.
The markers were first considered individually for association with disease. The test for apoB codon 71 among women yielded a nominally significant difference in frequency between the extreme Gensini quintiles (12 carriers of the Ile-71 allele among 20 individuals in FQ1, 4 carriers among 18 in FQ5; uncorrected, two-tailedP < 0.03). These results are potentially interesting, but no conclusions can be drawn in light of the small sample size available. Previous evidence for association of the apoBIle-71 site with plasma lipoprotein levels has been mixed (Young et al. 1987; Tikkanen et al. 1988).
Multilocus data are of particular value in enabling evaluation of combinations of markers for their association to complex disease. Although only large effects would be expected to yield statistically significant results with this limited sample size, we sought to explore this opportunity by considering two-locus effects. As shown in Table3, analysis for two-locus effects within the UCSF cohort yielded 14 pairs of variant alleles that showed nominally significant associations (uncorrected P < 0.05) with angiographic scores in the combined gender or female-only quintile comparisons. One marker pair, GPIIIa Pro-33 withATIIR 1166C, yielded a possibly predisposing association in both the combined gender and female-only quintile comparisons; the small cohort size did not permit direct evaluation of the role of gender. This preliminary study did suggest a number of potentially interesting two-locus effects, such as increased risk for disease associated with having two hypertension gene variants, ATIIR11166C and AGT Thr-235. A particularly high relative risk was estimated if CBS Thr-278 (associated with hyperhomocysteinemia; Hu et al. 1993) was paired with either apoE e4 or apoCIII promoter variants (associated with hypertriglyceridemia; Dammerman et al. 1993). In contrast, when thisCBS variant was paired with ATIIR1 1166C (hypertension pathway; Bonnardeaux et al. 1994), the effect appeared to be protective. Overall, the number of nominally significant (uncorrected P < 0.05) marker pairs just exceeded expectations given a type I error rate of 5%. Analysis of larger cohorts would provide the necessary power to detect true effects of clinical relevance, leading to hypotheses that could be tested subsequently in independent cohorts.
Pairs of Variant Alleles Observed to Have Suggestively Significant Association with the Severity of Atherosclerosis as Represented by Gensini Scores
DISCUSSION
With this immobilized probe assay format, large cohorts can be assayed rapidly for multiple biallelic sites, providing the necessary epidemiological data for evaluation of these markers in association with disease or therapeutic response. An additional advantage of this technology is the relative ease with which the panel of targets can be modified to include new markers of interest. The assay described here is currently being expanded to type >60 sites in 36 genes, and could be expanded even further. One limitation of this approach is that sequence-specific probes will not identify new mutations or polymorphisms; only those new sequence variations resulting in unusually weak signal intensities would be detected. Furthermore, this format does not detect variable number tandem repeat polymorphisms, and higher density probe arrays are more appropriate for detection of specific mutations in genes such as the LDL receptor gene, for which >600 mutations, including large deletions, have been reported (University College of London 1999). Our multilocus assay can be adopted more readily by individual laboratories, however, particularly for candidate gene evaluations similar to those described here, as compared with genome-wide scanning efforts using high-density arrays. The use of minisequencing on primer arrays with 33P incorporation to simultaneously genotype 12 variable sites was reported recently (Pastinen et al. 1998); this approach is also promising for rapid analysis of large cohorts.
With larger cohorts, multiple regression or logistic regression methods have greater power to identify those combinations of genotypes that are most clinically informative with regard to disease phenotypes and endpoints. Alternative analytical approaches for multilocus genotype data sets may also reveal interesting associations. Extensive (n > 10,000) epidemiological and intervention studies such as the Framingham Heart Study (Dawber et al. 1951), Women's Health Initiative (The Women's Health Initiative Study Group 1998), and Multiple Risk Factor Intervention Trial (The MRFIT Research Group 1982) may offer the greatest power to detect multilocus risk factors, but smaller cohorts of carefully characterized individuals should also be informative for factors having significant impact on disease. Even with relatively large cohorts, direct evaluation of disease risk associated with combinations of four or more genotypes may be difficult, and inferences may need to be drawn from analyses of smaller subsets of markers. As increasing numbers of markers are analyzed, the issue of multiple testing must also be addressed to provide appropriate statistical interpretion of the results. This issue arises whether samples are genotyped using a multilocus assay, as described here, or through a series of single-locus studies.
Understanding the molecular basis of genetic predisposition to common multifactorial diseases such as cardiovascular disease will depend on the joint efforts of those performing genome-wide scans to identify candidate loci in regions detected through linkage studies and those studying specific mutations and polymorphisms through association studies. Functional studies will also be critical to identify the genetic variations contributing to disease development. The assay described here was designed to provide multilocus genotype information for CVD, but this format can be applied to other diseases such as asthma, bipolar disorder, and osteoporosis. Given the complexity of these diseases, well-defined cases and phenotypes will be essential components of studies seeking to provide insight into disease development from the complex genetic data. Genotype data can then guide the development of algorithms incorporating genetic contributions to calculate aggregate scores of risk, expanding on the approach developed by the Framingham Heart Study investigators for coronary heart disease (Wilson et al.), for example. Clinically informative subsets of these research markers may then form the basis of panels for diagnostic or prognostic use in patient care.
METHODS
Primers
The sites targeted for PCR amplification are listed in Table 1. Primers were synthesized with 5′ biotinylation using the cyanoethoxyphosphoramidite method (1-μmole scale) on an Applied Biosystems 394 DNA Synthesizer (Perkin-Elmer, Foster City, CA). The use of allele-specific primers at codon 112 combined with probes for codon 158 to genotype apoE alleles has been described (Cheng et al. 1998). Primers for the CBS exon 8, factor V Leiden, and MTHFR targets were published previously (Hu et al. 1993;Goyette et al. 1995; Ridker et al. 1995); the forward primer forCBS exon 8 was later relocated further upstream, to eliminate duplication of the 68-bp insertion sequence (Tsai et al. 1996). The remaining primer sequences were selected with the assistance of two software packages, Oligo (v. 5.0, National Biosciences, Plymouth, MN) and Amplify (v. 1.2, W. Engels, University of Wisconsin, Madison).
Two PCR pools were developed: Multiplex A consisted of 14 biotinylated primer pairs designed to amplify the e2 and e3alleles of apoE, and targets within the apoB, apoCIII, CETP, LPL, and PON genes. Multiplex B consisted of 13 biotinylated primer pairs designed to amplify the e4 allele ofapoE, and targets within the ACE, ATIIR1, AGT, CBS, MTHFR, GPIIIa, fibrinogen, factor V, and ELAM genes. To the extent possible, PCR targets were chosen to be within the 100- to 400-bp size range and to permit resolution of all products by agarose gel electrophoresis. Gel analysis was then used to guide the optimization of PCR conditions. Primer concentrations were adjusted for generally comparable yields of all targets, and ranged from 0.04 to 0.75 μm.
As others have reported (Houlston et al. 1989; Hixson and Vernier 1990), amplification of the apoE region, which is relatively high in GC-bp content, was most efficient in the presence of DMSO. Even in the presence of DMSO, however, the apoCIII promoter target was amplified most effectively if divided into two separate amplicons of 163 and 165 bp.
Unexpectedly weak probe signal intensities necessitated primer redesign for the AGT and GPIIIa targets. Reducing the size of each amplicon resulted in much stronger probe intensities, suggesting the possibility that the longer amplicons were able to form stable secondary structures that inhibited probe binding (data not shown). TheAGT target was reduced from 360 to 171 bp; the GPIIIatarget was reduced from 312 to 131 bp.
Oligonucleotide Probes
Two probes were designed for each biallelic site, to detect and distinguish between the variant sequences. Most of the markers required discrimination of single base differences. To confirm successful amplification for the two largest PCR targets, probes were also designed for invariant regions of apoE and ACE. Candidate probe sequences were selected initially using published guidelines (Thein and Wallace 1986), with the assistance of the MELT program by J. Wetmur (Mt. Sinai School of Medicine, New York, NY; see also Wetmur 1991) for calculation of dissociation temperatures. Sequences were then modified to meet sensitivity and specificity requirements under the assay temperature and buffer conditions. Concentrations of the final 70 probes were chosen to achieve signal balance between alleles at each variable site, and for generally comparable intensities among all of the loci. Probes were conjugated at their 5′ ends to bovine serum albumin (BSA) by methods similar toTung et al. (1991), then applied in a linear array to sheets of backed nylon membrane using a Linear Striper and Multispense2000 controller (IVEK, N. Springfield, VT). Each sheet was cut into strips between 0.35 and 0.5 cm in width. The probes on “Probe Strip A” corresponded to the targets amplified by the multiplex A primer pool; “Probe Strips B and B2” corresponded to the targets amplified by the multiplex B primer pool.
Control DNA Templates
Total genomic DNA from three cell lines was used for preliminary experiments: Molt-4 (GM02219C from the Human Genetic Mutant Cell Repository, Coriell Institute, Camden, NJ), KASO11 (no. 9009 from the 10th International Histocompatibility Workshop; Dupont 1987), and CRK (kindly provided by the Clinical Immunogenetics Laboratory, Fred Hutchinson Cancer Center, Seattle, WA). Genomic DNA samples previously characterized at individual sites by other methods were generously provided by G. Assmann and H. Funke (Westfälisches Wilhelms-Universität, Münster, Germany) for ACE, apoB3500, apoE, CETP405, LPL9, LPL291, LPL447, andMTHFR677; P.F. Bray (Johns Hopkins University, Baltimore, MD) for GPIIIa; F. Chehab (University of California, San Francisco, CA) for factor V Leiden; R.M. Krauss and P. Blanche (Lawrence Berkeley Laboratory, Berkeley, CA) for apoB3500, apoCIII3206, and apoE; and B. Shane (University of California, Berkeley, CA) for MTHFR677. Single-stranded templates containing the point mutations in CBS exons 3 and 8 were prepared on an Applied Biosystems 394 DNA Synthesizer, then converted to double-stranded templates by PCR, using the appropriate primer pairs from the multiplex primer pools. For the remaining markers, variant alleles that were identified during development of the assay itself were confirmed by sequencing using Dye Terminator and dRhodamine Terminator Cycle Sequencing Kits with an ABI Prism DNA Sequencer (Perkin-Elmer). All of these samples were used as controls to guide the optimization of probe sequences and concentrations for specificity and sensitivity.
Additional Reagents
MicroAmp tubes for PCR, dNTPs (N = A, G, C, U), and AmpliTaq Gold DNA polymerase were obtained from Perkin-Elmer. Deaza-dGTP was obtained from Boehringer Mannheim Biochemicals (Indianapolis, IN; now Roche Molecular Biochemicals). For higher volume assays, PCRs were performed in 96-well Thermowell Polypropylene Plates with Sealing Mats (Corning Costar, Cambridge, MA). Typing Trays (20-well capacity, amber lid), denaturation solution (1.6% NaOH), SSPE concentrate (20× sodium phosphate solution with NaCl, EDTA), SDS concentrate (20%), streptavidin–horseradish peroxidase conjugate (SA–HRP), substrates A (0.01% H2O2 in citrate solution) and B (0.1% 3,3′,5,5′-tetramethylbenzidine in 40% dimethylformamide) for color development, and citrate concentrate (40×) were obtained from Roche Diagnostic Systems (Branchburg, NJ). For manual assays, color development reagent was prepared by mixing five volumes of substrate A per volume of substrate B; for automated assays, substrate A was reduced to four volumes per volume of substrate B.
PCR Amplifications
Approximately 50 ng of total genomic DNA was used for each assay, 25 ng for each multiplex A and multiplex B reaction. In addition to the primer pools, each 50-μl reaction contained 20 mmTris-HCl [0.2 m stock (pH 8.3) at 25°C], 50 mmKCl, 8.5% DMSO (vol/vol), 0.1 mm dATP, 0.1 mmdCTP, 0.07 mm dGTP, 0.03 mm 7–deaza-dGTP, 0.2 mm dUTP, 1.7 mm MgCl2 or MgOAc2, and 7 units of AmpliTaq Gold. The final concentration of 8.5% DMSO was chosen to enable reliable amplification of theapoE alleles with minimal adverse impact on the yields of other products such as the β-fibrinogen promoter region, which is relatively high in AT-bp content. Deaza–dGTP was also incorporated to facilitate amplification of regions high in GC content. Deoxy–UTP was included for compatibility with the use of uracil N-glycosylase to eliminate PCR product contamination (Longo et al. 1990). Samples were amplified in a Perkin-Elmer GeneAmp PCR System 9600 using a 2.4-hr thermal cycling profile: an initial hold of 94°C for 12.5 min; then 33 cycles of 96°C for 15 sec, 60°C for 1 min, and 72°C for 1.25 min; and a final extension step of 68°C for 5 min.
During assay development, 3- to 5-μl aliquots were run on horizontal agarose gels using 3% NuSieve, 1% SeaKem GTG agarose (FMC BioProducts, Rockland, ME) in TBE (89 mm Tris-borate, 1 mm EDTA) with ethidium bromide. ΦX174 RF DNA/HaeIII fragments and 123-bp DNA ladder (GIBCO BRL, Gaithersburg, MD) were used as molecular weight standards.
Allele-Specific Detection
The assay was initially developed at 50°C, and the final 52°C assay temperature reflects a compromise made to improve specificity at the apoCIII (−625) site. This marker is the presence (more frequent allele) or absence (less frequent allele) of an A:T bp between a G:C bp doublet and quartet within a generally GC-rich region. Sufficient discrimination between these alleles was achieved only by introducing G:T mismatches into the deletion-specific probe sequence; these relatively stable mismatches (Thein and Wallace 1986) destabilized the region sufficiently to reduce cross-hybridization with wild-type PCR product while maintaining sensitivity for the variant allele. Improved discrimination between apoCIII (−625) alleles was also observed at assay temperatures >52°C, but the signal intensities from probes for other markers were adversely affected by these higher temperatures (data not shown).
Therefore, detection of amplified alleles was performed at 52°C using a water bath rotating at 50–60 rpm (Hot Shaker Plus; Bellco, Vineland, NJ). Probe strips were first washed to remove unbound probe in 2× SSPE (0.36 m NaCl, 0.02 mNa2HPO4, 2 mm EDTA, adjusted to pH 7.4 with NaOH), 0.5% SDS. Twenty-microliter aliquots of the biotinylated PCR product pools from multiplex A and B reactions were denatured with equal volumes of denaturation solution, then added to Typing Tray wells containing 3 ml of hybridization buffer (4× SSPE, 0.5% SDS) and a correspondingly labeled probe strip A or probe strip B. Probe strip B2 was included with strip B. After 20 min at 52°C, the hybridization solution was replaced with fresh buffer containing 10 μl of SA-HRP and the strips were returned to the water bath for 5 min. This enzyme conjugate solution was then replaced with the stringent wash buffer (2× SSPE, 0.5% SDS), and the strips were returned to the water bath for 12 min. The washed strips were equilibrated in 50 mmNa-citrate at room temperature on a rotating (50–60 rpm) platform (Gyrotory Shaker Model G2; New Brunswick Scientific, Edison, NJ), then agitated in color development reagent for 8–10 min at room temperature. Developed strips were rinsed with distilled water, aligned on a flat surface next to a guide identifying the allele detected by each probe line, and photographed using type 559 or 55 film from Polaroid (Cambridge, MA). Genotype interpretations were made manually and independently by two individuals. Given this protocol, at least 40 DNA samples per day can be genotyped by one individual. An SLT ProfiBlot IIT (Tecan US, Research Triangle Park, NC) can also be used to automate the hybridization, stringent wash, and color development steps for 12 samples (24 wells) at a time. This level of automation can be used to increase the throughput of one individual to at least 75 samples per day.
Test Cohorts
To estimate population frequencies, the assay was used to genotype a subset of 1190 samples from 286 families of the Stanislas cohort recruited from families within eastern France (Siest et al. 1998). DNA was prepared from whole blood by the method of salting-out (Miller et al. 1988). These samples had been genotyped for apoE, LPL447, and ACE by methods described previously (Hixson and Vernier 1990; Evans et al. 1994; Salah et al. 1997).
The UCSF cohort was a composite cohort of 142 unrelated caucasian individuals recruited from clinics within the San Francisco Bay area (California, USA). These individuals had been recruited on the basis of a family history of disease, hyperlipidemia, or a treadmill test indication for angiography. Total cholesterol levels ranged from 162 to 548 mg/dl, with an average of 323 ± 69 mg/dl. DNA was prepared from whole blood either by the method of Bell (Bell et al. 1981) or using the Puregene DNA Isolation Kit (Gentra Systems, Inc., Minneapolis, MN). Each DNA sample was associated with a Gensini score, which assigns greater weight to proximal lesions identified through quantitative angiography (Gensini 1975). Fifty of the samples were from men with an average age of 45.5 ± 8.5 years at the time of angiography, and Gensini scores ranging from 5 to 135. Ninety-two samples were from women with an average age of 54.0 ± 11.4 years, and Gensini scores ranging from 0 to 120. Some of these individuals had already been genotyped for apoE and apoB3500 by methods described previously (Hixson and Vernier 1990; Pullinger et al. 1995).
Allele Frequencies and Linkage Disequilibrium Analysis
Population frequencies were estimated from allele counts among 496 unrelated parents from the Stanislas cohort for whom all 35 sites had been genotyped. Allele frequencies were also calculated for the UCSF cohort. Deviation from Hardy–Weinberg equilibrium was assessed using the χ2 statistic.
Intragenic haplotypes for multiple markers within the apoCIII, LPL, and ELAM loci were estimated using the Family Analysis Program (v. PL1; M. Neugebauer and M.P. Baur; Neugebauer et al. 1984) from 275 families (1100 chromosomes) within the Stanislas cohort. This data set included families for whom markers on probe strip B2 had not been genotyped because of the rarity of variation at these four markers; therefore, this data set included samples that were not counted in the estimation of population frequencies. The intragenic haplotypes were used to estimate pairwise linkage disequilibrium values (D′; Lewontin 1964; Klitz et al. 1995) between consecutive sites within each locus.
Evaluation of Disease Association
The UCSF cohort was divided into quintiles based on the Gensini scores. The first combined gender quintile (Q1) contained the 28 lowest scores (0–8), and the fifth quintile (Q5) contained the 28 highest scores (35–135). The male subset was deemed insufficient in size for separate analysis, but the female subset was considered separately: female-only quintile 1 (FQ1) contained the 20 lowest scores (0–7); FQ5 contained the 18 highest scores (36–120).
A preliminary analysis of disease association was undertaken for a subset of 15 markers: apoE, apoB71, all apoCIII sites except 3175, CETP405, PON192, ACE, ATIIR, AGT, CBS278, MTHFR677, GPIIIa, andfibrinogen. For the remaining markers, the observed variant allele frequencies were deemed too rare for such an analysis. In addition, nearly complete linkage disequilibrium was observed betweenapoCIII sites (−625) and (−455); therefore, these two were subsequently treated as one marker. Disease association was examined by comparing the extreme quintiles, interpreting Q1 and FQ1 as individuals having little or no disease, and Q5 and FQ5 as individuals having the most severe disease. Heterozygous and homozygous carriers of the variant alleles were counted together, with the exception ofACE. The ACE alleles were grouped in two ways, consistent with combining either carriers of the reported risk allele (D/D, I/D) or carriers of the less frequent allele (I/I, I/D). For apoE, e3/e4 ande4/e4 were counted together; no e2/e4 genotypes were observed in any of the quintiles used for this analysis. For single sites (more frequent allele A, less frequent allelea), the odds ratios corresponded to the risk associated with carriers of the less frequent allele (Aa or aagenotypes) relative to the AA genotype. For pairwise combinations of sites, odds ratios were calculated for the risk associated with having variant alleles at two sites compared with just one site. Odds ratios were calculated with Haldane's correction when necessary (Haldane 1955). P values were calculated using Fisher's two-tailed exact test (Sokal and Rohlf 1995). These analyses were intended to be exploratory; therefore, no formal correction for multiple testing was applied.
Acknowledgments
We are indebted to our collaborators for their expert advice and generous gifts of characterized DNA samples: G. Assmann and H. Funke (Westfälische Wilhelms-Universität), S. Humphries and I. Day (University College of London Medical School), R. Krauss and P. Blanche (Lawrence Berkeley National Laboratory), P. Bray (Johns Hopkins Medical School), F. Chehab (University of California, San Francisco), and B. Shane (University of California, Berkeley). This work would not have been possible without the support of the Oligo Synthesis and Sequencing Groups at Roche Molecular Systems, and we thank A. Turck and J. Novotny for their technical advice. We also thank R. Higuchi, J. Sninsky, and T. White for their enthusiastic support.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.








