
Ribonuclease k6: Chromosomal Mapping and Divergent Rates of Evolution within the RNase A Gene Superfamily
Abstract
We have localized the gene encoding human RNase k6 to within ∼120 kb on the long (q) arm of chromosome 14 by HAPPY mapping. With this information, the relative positions of the six human RNase A ribonucleases that have been mapped to this locus can be inferred. To further our understanding of the individual lineages comprising the RNase A superfamily, we have isolated and characterized 10 novel genes orthologous to that encoding human RNase k6 from Great Ape, Old World, and New World monkey genomes. Each gene encodes a complete ORF with no less than 86% amino acid sequence identity to human RNase k6 with the eight cysteines and catalytic histidines (H15 and H123) and lysine (K38) typically observed among members of the RNase A superfamily. Interesting trends include an unusually low number of synonymous substitutions (K s) observed among the New World monkey RNase k6 genes. When considering nonsilent mutations, RNase k6 is a relatively stable lineage, with a nonsynonymous substitution rate of 0.40 × 10−9 nonsynonymous substitutions/nonsynonymous site/year (ns/ns/yr). These results stand in contrast to those determined for the primate orthologs of the two closely related ribonucleases, the eosinophil-derived neurotoxin (EDN) and eosinophil cationic protein (ECP), which have incorporated nonsilent mutations at very rapid rates (1.9 × 10−9and 2.0 × 10−9 ns/ns/yr, respectively). The uneventful trends observed for RNase k6 serve to spotlight the unique nature of EDN and ECP and the unusual evolutionary constraints to which these two ribonuclease genes must be responding.
[The sequence data described in this paper have been submitted to the GenBank data library under accession nos. AF037081–AF037090.]
Ribonuclease k6 (RNase k6) is a recently identified member of the RNase A superfamily, a group of otherwise divergent proteins defined by specific elements of sequence homology to the prototype, Ribonuclease A. In previous work we have shown that human RNase k6 is encoded by a single-copy gene, with mRNA transcripts expressed in numerous somatic tissues (Rosenberg and Dyer 1996). Within this superfamily, RNase k6 is most closely related to the two eosinophil ribonucleases, the eosinophil-derived neurotoxin (EDN/RNase 2) and eosinophil cationic protein (ECP/RNase 3), although mRNA encoding RNase k6 was detected in peripheral blood neutrophils and monocytes, but not eosinophils. From an enzymatic perspective, recombinant RNase k6 was found to be a relatively efficient ribonuclease, with catalytic activity only ∼40-fold lower than that of recombinant EDN (Rosenberg and Dyer 1995a, 1996); in contrast, the catalytic actvity of recombinant ECP was found to be ∼2000-fold lower than that of recombinant EDN (Rosenberg and Dyer 1997).
The RNase A superfamily has been the subject of numerous studies that have elucidated patterns of evolution at the molecular level (Beintema et al. 1986, 1988; Benner and Allemann 1989; Fitch and Beintema 1990;Confalone et al. 1995; Jermann et al. 1995; Rosenberg et al. 1995;Trabesinger-Ruef et al. 1996). At present, this superfamily includes at least four distinct sequence lineages (Riordan and D’Alessio 1997) and two species-limited clusters (Palmieri et al. 1985; Watanabe et al. 1988; Sasso et al. 1991; Batten et al. 1997; Larson et al. 1997), as well as the two most rapidly evolving functional coding sequences known among primates (Rosenberg et al. 1995). The basis for this remarkable expenditure of evolutionary energy remains uncertain.
As part of our ongoing interest in the evolution of the RNase A gene superfamily, we have focused our attention on RNase k6; we describe here the isolation and characterization of ten nonhuman primate orthologs of the human RNase k6 gene. In contrast to the rapid rates observed for both EDN and ECP (Rosenberg et al. 1995), we have found that nonsilent mutations have been incorporated into genes of the RNase k6 lineage at a relatively conservative pace. In addition, we have determined the chromosomal localization of human RNase k6 by HAPPY mapping (Dear and Cook 1989, 1993; Walter et al. 1993; Dear 1997; Dear et al. 1997), and have used this information to position RNase k6 with respect to other members of this family along the long (q) arm of human chromosome 14.
RESULTS
Chromosomal Localization of RNase k6 by HAPPY Mapping
A HAPPY map of human chromosome 14, including 1001 novel sequence tagged sites (STSs), has been reported recently by Dear and colleagues (Dear and Cook 1989, 1993; Walter et al. 1993; Dear 1997; Dear et al. 1997). The novel STSs were generated by PCR amplification of genomic sequences located between Alu sites. These inter-Alusites were mapped to ∼100-kb resolution by determining their cosegregation frequency in radiation-sheared, size-selected fragments of genomic DNA. The plasmid artificial clone (PAC) 11h15, isolated from the RPCI1 human PAC library (Ioannou and de Jong 1996) was identified as containing the RNase k6 coding sequence. This clone was also found to include five specific HAPPY markers (Fig. 1A) that span ∼100–120 kb within the q11 region of human chromosome 14. One of these markers, h14a1599, is found within the gene encoding angiogenin, another member of the RNase A gene superfamily. Taken together, these results indicate that angiogenin and RNase k6 lie within a PAC distance (∼100 kb) of one another.
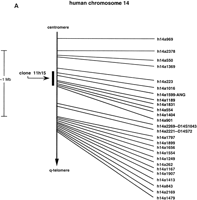
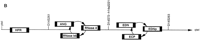
(A) Chromosomal localization of human RNase k6 by HAPPY mapping. Shown is a region of the HAPPY map of human chromosome 14 (Dear and Cook 1989, 1993; Walter et al. 1993; Dear 1997; Dear et al. 1997). The markers, STSs listed at the right are encoded by inter-Alu segments of genomic DNA; those located within one PAC distance of a previously defined marker (D14S1043, D14S72) are also noted. Clone 11h15 from the de Jong RPCI1 library (Ioannou and de Jong 1996) includes the gene encoding human RNase k6 and the five HAPPY map markers h14a1016, h14a1599, h14a1189, h14a1831, and h14a554, within a span that corresponds to the the q11 region of chromosome 14. Marker h14a1599 is found within the gene encoding angiogenin (marked ANG). The GenBank accession nos. for the STSs are as follows: h14a1016, G35789; h14a1599, G36057; h14a1189, G35876; h14a1831, G36118; h14a554, G35567. (B) Relative positions of the six human RNase A family members on chromosome 14. Assembling the information presented in Awith that available from the combined maps of the NCBI. (http://www.ncbi.nlm.nih.gov), the most likely linear arrangement of these ribonucleases can be inferred; those listed in circular fashion cannot be placed in a definitive order with respect to one another with the information available. (HPR) Human pancreatic ribonuclease/RNase 1; (ANG) angiogenin/RNase 5; (RNase 4) ribonuclease 4; (RNase k6) ribonuclease k6; (EDN) eosinophil-derived neurotoxin/RNase 2; (EDNp) eosinophil-derived neurotoxin pseudogene; (ECP) eosinophil cationic protein/RNase 3. The diagrams are not drawn to scale.
With this information, together with information provided from a composite of the OMIM, RH Consortium, and Whitehead maps (http://www.ncbi.nlm.nih.gov), the relative positions of these ribonucleases with respect to one another can be inferred (Fig. 1B).
Isolation and Characterization of Primate Orthologs of RNase k6
By use of genomic DNA templates from various primate species, ∼400-bp amplification products were generated with primers complementary to the human RNase k6 sequence (Rosenberg and Dyer 1996). All genomic fragments isolated encode ORFs with no less than 86% amino acid sequence identity to human RNase k6 (Table 1). As anticipated from the accepted phylogeny of these primate species, sequences within one family (e.g., Great Apes, Old World monkeys, or New World monkeys) were more similar to one another than to those of another superfamily, and increasing divergence was observed with increasing time elapsed since species divergence (Sibley and Ahlquist 1984). Also, as anticipated, the amino acid sequence of the nonprimate ortholog, RNase k2, originally isolated by Irie and colleagues (Niwata et al. 1985; Irie et al. 1988) from the bovine species B. taurus, differed significantly from all the primate isolates shown. Interestingly, several of the conservative amino acid changes maintained constancy within family groupings. Specifically, residue 1 was identified as tryptophan (W) in humans, Great Apes and Old World monkeys, and found to be isoleucine (I) in all three New World monkey sequences evaluated. Similar patterns were seen for V61 and F101 (Fig. 2A).
Percent Nucleotide and Amino Acid Sequence Identities: Ribonuclease k6 Genes

(A) Alignment of the primate orthologs of RNase k6. The DNA sequences encoding the polypeptides shown were generated by PCR from genomic DNAs as described. Each DNA sequence encodes an ORF that includes the eight canonical cysteines (shaded boxes), H15, H123, and K38 corresponding to residues found in the active site of RNase A (numbered), and the CKXXNTF motif (bracketa) common to all members of this superfamily. The amino acid sequence shown in the lowermost line denotes residues at which the eleven orthologs are identical (lettered) or divergent (*); residues included in boxes are common to all RNase A superfamily members (Riordan and D’Alessro 1997). Glycosylation sites initially identified in the human sequence are indicated by the brackets marked b.Each DNA sequence also encodes a 23 amino acid signal sequence (not shown); the residues demarcated by bracket c were included within the 3′ primer sequence. Common names for each genus/species include H. sapiens, human (GenBank accession no. U64998);P. troglodytes, chimpanzee (AF037081); G. gorilla,gorilla (AF037088); P. pygmaeus, orang-utan (AF037082);C. aethiops, African green monkey (AF037090); M. talapoin, talapoin (AF037087); P. hamadryas, baboon (AF037083); M. mulatta, rhesus monkey (AF037089); A. trivirgatus, owl monkey (AF037084); S. sciureus, squirrel monkey (AF037085); S. oedipus, cottontop tamarin (AF037086). The DNA sequence of human RNase k6 has been corrected from that originally reported (Rosenberg and Dyer 1996), resulting in the conversion of R53 to A. (B) pIs of the RNase k6 orthologs were calculated by the PEPTIDESORT algorithm of the Wisconsin Genetics Computer Group Program on-line at the National Institutes of Health.
An alignment of the encoded amino acid sequences of both human and nonhuman primate RNase k6 orthologs are shown in Figure 2A. Each sequence contains the eight appropriately spaced cysteines that characterize most members of this superfamily, as well as the histidine and lysine residues analogous to those found in the catalytic site of RNase A. Similarly, all primate orthologs include the CKXXNTF sequence bracketing the catalytic lysine (K38), a motif found to be invariant among the divergent members of this superfamily (Rosenberg and Dyer 1996; Riordan and D’Alessio 1997). Of the two glycosylation sites initially identified in the human RNase k6 sequence, the first shows a limited degree of divergence, whereas the second is conserved throughout. Each sequence also includes a 23-residue signal sequence (not shown); subsequent analyses were limited to the sequence of the mature protein. The amino termini of the primate sequences were identified by comparison with the amino terminus of the bovine RNase k2 protein as determined by peptide sequencing (Niwata et al. 1985; Irie et al. 1988).
Isoelectric points (pI) calculated for each primate sequence are listed in Figure 2B. The pIs range from a low of 8.43 (for the New World monkey, S. scuireus) to a high of 9.49 (for the Great Ape,P. pygmaeus), which are all within the range typically seen for ribonucleases of this superfamily. Interestingly, the trend of increased pI in higher primates is analogous to that observed among primate orthologs of both EDN/RNase 2 and ECP/RNase 3 (Rosenberg and Dyer 1995a).
Molecular Evolution of RNase k6; Quantitative Analysis
Values for nonsynonymous substitution per nonsynonymous site (K a) and synonymous substitution per synonymous site (K s), comparing each ortholog with the human sequence were calculated as described by Li (1993) and are listed in Table 2. As anticipated, the values forK a increase with increasing time elapsed since species divergence. Interestingly, the values calculated forK s among the three New World monkeys do not diverge to the extent one might expect, resulting in elevatedK a /K s ratios for all three New World monkey sequences.
Evolutionary Analysis of Primate RNase k6
Values obtained for both Ka and Ks for the human-New World monkey coding sequence pairs were compared with 13 similar coding sequence pairs reported previously (Rosenberg et al. 1995). Similar to results reported earlier (Graur 1985; Li et al. 1985; Ticher and Graur 1989;Wolfe and Sharp 1993; Mouchiroud et al. 1995), we observed a distinct correlation between K a and K s for the pairs evaluated (Fig. 3;r 2 = 0.4). Whereas all three points for RNase k6 pairs fell below the line, indicating relatively lower K s values even at the given lowerK a, the degree of divergence was not statistically significant.
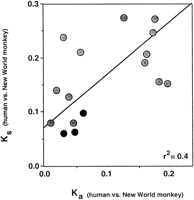
Synonymous substitution per synonymous site (K s) vs. nonsynonymous substitution per nonsynonymous site (K a) for 16 human–New World monkey coding sequence pairs. The values calculated for RNase k6 pairs are indicated with black circles. The shaded circles represent values calculated for 13 coding sequence pairs originally reported by Rosenberg et al. (1995)including ECP, EDN, interleukin-3, CD59, protamine P2, protamine P1, SRY, insulin a-chain, interferon-γ; complement C4a, insulin b-chain, c-myc proto-oncongene, and EGF-like growth factor. (See Rosenberg et al. 1995 for Genbank accession nos. for these sequence pairs).
Human and New World monkey RNase k6 sequences were subjected to relative-rate testing with a bovine RNase k6 ortholog as the outgroup (Table 3). In all three instances, positive values are obtained when the K s calculated for the New World monkey-bovine pair (K ac) is subtracted from that calculated for the human-bovine pair (K ab), suggesting diverging patterns of synonymous substitution (see Discussion).
Relative Rate Analysis
In Figure 4, the values for K a are plotted against years of divergent evolution, by use of dates of species divergence determined by Sibley and Ahlquist (1984). The slope of this line, representing the rate of incorporation of nonsilent mutations, was calculated to be 0.40 × 10−9nonsynonymous substitutions/nonsynonymous site/year (ns/ns/yr). This value can be compared with those calculated in the series reported byRosenberg et al. (1995), which includes 63 human/non-human primate pairs from 26 independent functional coding sequences. The rate determined for RNase k6 is most similar to those reported for interferon-gamma (0.48 × 10−9 ns/ns/yr) and apolipoprotein A1 (0.42 × 10−9 ns/ns/yr), and is only slightly higher than the series average (0.37 × 10−9ns/ns/yr).
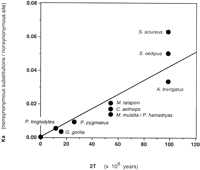
Nonsynonymous substitution per nonsynonymous site (K a) vs. years of divergent evolution. The values for K a are listed in Table 2, and the years of divergent evolution (2T) are as per Sibley and Ahlquist (1984). Common name for each genus/species are as listed in the legend to Fig. 2. BothK a and 2T for the Old World monkeys M. mulatta and P. hamadryas are identical and are thus represented by a single point. The slope of the line representing a best fit through these data points is 0.40 × 10−9ns/ns/yr, with a correlation coefficient (r 2) of 0.93.
In Table 4, the rate determined for RNase k6 is compared with those determined for RNase 2 (EDN) and RNase 3 (ECP), two members of the RNase A superfamily previously shown to be incorporating nonsilent mutations more rapidly than any other functional sequences studied among primates (Rosenberg et al. 1995). Despite the fact that RNase k6 is a close relative of both EDN and ECP, the rates of sequence divergence differ dramatically (see Discussion).
Rates of Nonsynonymous and Synonymous Substitution of Three Distinct Ribonucleases of the RNase A Gene Superfamily
DISCUSSION
As part of this work, we have isolated and characterized 10 primate orthologs of human RNase k6, a member of the RNase A superfamily (Rosenberg and Dyer 1996). The sequences encode complete ORFs, all with the appropriately spaced cysteines and catalytic histidines and lysine identifying them as members of the RNase A superfamily. The amino acid sequence similarities among these isolates also place them within the RNase k6 sequence lineage. The degree of amino acid sequence divergence among the various human/nonhuman primate RNase k6 pairs is consistent with the accepted phylogeny of these primate species. Interestingly, although the genes encoding primate orthologs of the related RNases EDN and ECP have incorporated nonsilent mutations at very rapid rates (1.9 × 10−9 and 2.0 × 10−9 ns/ns/yr, respectively), the genes encoding primate orthologs RNase k6 have remained relatively stable (0.40 × 10−9 ns/ns/yr). The fact that RNase k6 does not have a dynamic evolutionary history underscores the unique nature of the EDN/ECP lineage, and the distinct evolutionary constraints to which these two genes must be responding. These constraints have been shown to promote the development of increased toxicity and enhanced ribonuclease activity of ECP and EDN, respectively (Rosenberg and Dyer 1995a, 1997), features of particular significance to the roles played by these proteins in eosinophil-mediated host defense (Spry 1988;Makino and Fukuda 1993; Domachowske and Rosenberg 1997; Domachowske et al. 1998).
Whereas the rate of nonsynonymous substitution in the RNase k6 lineage is relatively conservative, several interesting features of specific primate orthologs merit comment. It is interesting to note that the New World monkey sequences (A. trivirgatus, S. scuireus, andS. oedipus) are more highly divergent from one another (Table1; Fig. 2). An assessment of the relative ribonuclease activities of these proteins in recombinant form may provide some insight into the functional significance of this observation, specifically whether divergence is actively promoted or simply tolerated under the given set of evolutionary constraints. Although theK a/K s ratios for the New World monkey/human pairs (Table 2) are relatively high, suggesting that divergence is promoted, the unusually low values forK s render this conclusion somewhat suspect. Additionally, it is not clear whether this degree of divergence is an idiosyncratic feature of the New World monkeys, or whether this pattern persists among more distant species; evaluation of prosimian and nonprimate mammal sequences may serve to clarify this issue.
The values calculated for synonymous substitutions per synonymous site (K s) among the New World monkey/human RNase k6 pairs appear to be unusually low, ranging from ∼30%–50% below the average (0.18 synonymous substitutions per synonymous site (ss/ss) calculated for a series of 13 independent human-New World monkey coding sequence pairs (Rosenberg et al. 1995). Although seemingly independent variables, several groups have reported correlations between Ks and Ka for interspecies coding pairs (Graur 1985; Li et al. 1985; Ticher and Graur 1989; Wolfe and Sharp 1993; Mouchiroud et al. 1995). To examine our results in this context, we compared K a andK s for each of the aforementioned human–New World monkey coding sequence pairs, as shown in Figure 3. Whereas all threeK s values calculated for RNase k6 pairs fall below the best-fit line, and thus are low even in the setting of lowK a, the difference does not reach statistical significance. To investigate this phenomenon further, we performed a relative rate test, using the sequence of bovine RNase k6 (Rosenberg and Dyer 1996) as an outgroup (Table 3). Interestingly, the results of relative rate analysis suggest that the K s values for New World monkeys are unusually low, as the synonymous substitution probability calculated for the human-bovine RNase k6 coding pair (K ab) exceeds that calculated for all three New World monkey-bovine pairs (K bc), even before generation time is taken into account. The explanation for this finding is not readily apparent. Although synonymous substitutions are generally perceived as silent, mutations in specific sequences may introduce subtle alterations in gene structure and/or stability that are not considered in this type of analysis.
The calculated pIs for each of the RNase k6 coding sequence display a range typical for members of the RNase A superfamily, including the trend upward in higher versus lower primates analogous to that observed for the related RNases EDN and ECP (Rosenberg and Dyer 1995a). Increased cationicity has been associated with enhanced cytoxicity in the EDN/ECP lineage; whether this finding is replicated in the RNase k6 lineage remains to be evaluated.
Finally, we have used the HAPPY mapping technique (Dear and Cook 1989,1993; Walter et al. 1993; Dear 1997; Dear et al. 1997) to localize the gene encoding RNase k6 to within ∼120 kb within the q11 region of human chromosome 14. All six human members of the RNase A superfamily have been localized to chromosome 14 (Kurachi et al. 1985; Hamann et al. 1990; Rosenberg and Dyer 1995b), a configuration most likely to be the result of multiple gene duplication events. Although the evolutionary history of these RNase A family loci cannot be traced with assurance, the duplication that gave rise to distinct genes encoding EDN and ECP has been traced to the divergence of Old World from New World monkeys (Rosenberg et al. 1995). As there may be as yet undiscovered members of the RNase A superfamily, a more thorough evaluation of this region of human chromosome 14 is warranted.
METHODS
Localization of RNase k6 by HAPPY Mapping
The HAPPY map of human chromosome 14, recently described by Dear and colleagues (Dear and Cook 1989, 1993; Walter et al. 1993; Dear 1997; Dear et al. 1997) includes 1001 STSs, most mapped to within 100-kb resolution on the basis of cosegregation in vitro after controlled irradiation and size selection of human genomic DNA. By use of RNase k6-specific oligonucleotide primers, 5′-CAGCCAAGTCCTCTCCAATGCAACAGGG-3′ (nucleotides 118–145, GenBank U64998) and 5′-CTGGGCAGCAGCACTATAGCGGCACTGG-3′ (nucleotides 363–336), the PAC clone 11h15 was isolated from the de Jong RPCI1 human PAC library (Ioannou and de Jong 1996), which included both the gene encoding RNase k6 and five specific HAPPY map markers (Fig. 1).
Isolation of Primate RNase k6 Orthologs by PCR
Oligonucleotide primers complementary to the human sequence included 5′-ATGGTGCTATGCTTTCCTCTTCTTTTACTG-3′ (nucleotides 1–30, GenBank U64998) and 5′-TTAGAGAATACTATCTAAGTGTACAGGAAC-3′ (nucleotides 453–424). Genomic DNAs from chimpanzee (Pan troglodytes), orangutan (Pongo pygmaeus), cottontop tamarin (Saguinus oedipus), owl monkey (Aotus trivirgatus), green monkey (Cercopithecus aethiops) and rhesus monkey (Macaca mulatta) were purchased from BIOS Labs, New Haven, CT. Genomic DNAs were also isolated from cell lines purchased from American Type Tissue Collection, Rockville, MD, including ROK, gorilla (Gorilla gorilla); DPSO 14474, squirrel monkey (Saimiri sciureus); FRhK-4, rhesus monkey (M. mulatta); MT1.k, talapoin (Miopithecus talapoin); and 26CD-1, baboon (Papio hamadryas). Concentrations and cycling temperatures of the individual PCR reactions were as described previously (Rosenberg et al. 1995; Batten et al. 1997). Amplification products of ∼400 bp were isolated and subcloned into the PCR II TA cloning vector (Invitrogen, San Diego, CA) and subjected to automated sequencing analysis (ABI Prism, Applied Biosystems, Foster City, CA). Sequence analysis and comparisons were performed with the assistance of Sequencher (Gene Codes Corporation, Ann Arbor, MI) and ABI Prism software (Perkin Elmer, Foster City, CA) and algorithms within the Wisconsin Genetics Computer Group program available on-line at the National Institutes of Health.
Determination of Rates of Synonymous and Nonsynonymous Substitution
Sequences of the primate RNase k6 orthologs were compared with the human RNase k6 sequence, and values of synonymous (K s) and nonsynonymous (K a) substitution probability were calculated as described (Li 1993). Values for K a and K s were plotted against years of divergent evolution (Sibley and Ahlquist 1984) and slopes of the best-fitting lines determined by least squares method with correlation coefficients (r2) included.
Acknowledgments
We thank Harry L. Malech and John I. Gallin for their ongoing support of the work in our laboratory.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL hr2k{at}nih.gov; FAX (301) 402-4369.
-
- Received January 7, 1998.
- Accepted April 22, 1998.
- Cold Spring Harbor Laboratory Press








