
Analysis of EST-Driven Gene Annotation in Human Genomic Sequence
Abstract
We have performed a systematic analysis of gene identification in genomic sequence by similarity search against expressed sequence tags (ESTs) to assess the suitability of this method for automated annotation of the human genome. A BLAST-based strategy was constructed to examine the potential of this approach, and was applied to test sets containing all human genomic sequences longer than 5 kb in public databases, plus 300 kb of exhaustively characterized benchmark sequence. At high stringency, 70%–90% of all annotated genes are detected by near-identity to EST sequence; >95% of ESTs aligning with well-annotated sequences overlap a gene. These ESTs provide immediate access to the corresponding cDNA clones for follow-up laboratory verification and subsequent biologic analysis. At lower stringency, up to 97% of annotated genes were identified by similarity to ESTs. The apparent false-positive rate rose to 55% of ESTs among all sequences and 20% among benchmark sequences at the lowest stringency, indicating that many genes in public database entries are unannotated. Approximately half of the alignments span multiple exons, and thus aid in the construction of gene predictions and elucidation of alternative splicing. In addition, ESTs from multiple cDNA libraries frequently cluster over genes, providing a starting point for crude expression profiles. Clone IDs may be used to form EST pairs, and particularly to extend models by associating alignments of lower stringency with high-quality alignments. These results demonstrate that EST similarity search is a practical general-purpose annotation technique that complements pattern recognition methods as a tool for gene characterization.
Similarity search has been less successful as a tool for gene identification in genomic sequence than pattern-based methods such as GRAIL (Uberbacher et al. 1996) and GeneParser (Snyder and Stormo 1995), largely because of the limited pool of mRNA sequences available for use as a probe. Recently, however, this technique has become more feasible with the rapid accumulation of collections of expressed sequence tags (ESTs), single-pass end sequences from cDNA clones randomly selected from multiple libraries. Large-scale EST sequencing projects have been undertaken with the goal of representing a substantial fraction of all human genes as ESTs (Adams et al. 1995;Williamson et al. 1995; Hillier et al. 1996), and have produced over one million ESTs in publicly available databases (Boguski et al. 1993;Adams et al. 1995), as well as a number of proprietary collections. A similar project for mouse ESTs has generated >200,000 sequences to date, and initiatives for other organisms, while operating on a smaller scale, constitute a growing fraction of the public EST dataset as well.
ESTs represent 200–500 nucleotide gene signatures that provide information not derivable from pattern recognition techniques alone. In principle, similarity to an EST is a highly reliable indicator that a sequence is associated with a gene or pseudogene, because cloning of non-mRNAs into the cDNA libraries from which ESTs are derived is presumed to be rare. In practice, this assumption remains to be tested. The cDNA clones associated with most human ESTs are also publicly available, so that identification of an EST often provides rapid access to a laboratory reagent useful for further characterizing a potential gene of interest. Other information associated with ESTs, such as the library of origin, can supply useful information about expression patterns within an organism, or conservation of structure across species. ESTs are also particularly valuable reagents for detecting alternative splicing and polymorphisms and for locating the 3′ ends of genes, which can be used to distinguish related genes from each other (Boguski and Schuler 1995). None of these tasks are well handled by pattern recognition techniques, which rely heavily on coding potential as a predictive metric, generally producing a single gene prediction from a set of exons.
Computational analysis will be critical in providing a framework for further characterizing sequence generated by large-scale sequencing efforts. Because the need for high throughput requires that much of this initial annotation be generated automatically, the reliability of the techniques used must be examined with care. This annotation must be sufficiently reliable to guide investment of laboratory resources in further investigation of predicted genes. The principal purpose of gene identification techniques, then, will be to predict the presence of a gene in a region of genomic sequence, and, where possible, provide the means to obtain or design reagents, such as cDNA clones or PCR primers, for use in the laboratory. This laboratory follow-up will remain the gold standard for elucidation of gene structure, regulation, and function.
A growing number of anecdotal reports underscore the utility of ESTs in the characterization of human genes at both the mRNA (Clark et al. 1994; Cerretti et al. 1995; Greene et al. 1996; O’Dowd et al. 1996;Wilson and Majerus 1996) and genomic (Chen et al. 1996; Heiss et al. 1996; Lamerdin et al. 1996) levels. In the former case, ESTs of interest are often recognized by a moderate degree of sequence similarity to a gene or motif of known function. Cross-species relationships are often critical components of this process, and systematic efforts have been made to identify ESTs containing conserved sequences (Bassett et al. 1995; Banfi et al. 1996). In the large-scale analysis of genomic sequence, however, high-stringency sequence comparison plays a crucial role, because ESTs that are very similar to a genomic query sequence provide higher confidence in the identification of a gene ab initio. In addition, the data may supply information about expression in various contexts, and can illuminate structural variations that may not have been described previously, because they are rare forms of the mRNA, or are expressed in a context not available for study (Wolfsberg and Landsman 1997). Directed extension of gene predictions with low-stringency results, especially involving sequences from different species, will be useful in filling out these initial predictions.
We have, therefore, undertaken to evaluate the potential of nucleotide similarity to ESTs as a computational probe for expressed regions in human genomic sequence. In particular, we have focused on its potential as a gene-finding technique in large sets of sequences, and on properties that are especially relevant to systematic first-pass annotation. In this context, the most important characteristic is high positive predictive value, so that the resulting annotation forms a reliable resource, and does not lead to the frequent misapplication of laboratory efforts. Once an area of interest has been so identified, additional similarity data, obtained from both high and low stringency analysis of nucleotide and predicted amino acid sequences, can add further information about gene structure, variation, and potential function (Bedian et al. 1997; Wolfsberg and Landsman 1997). Such information may be used not only to further biologic evaluation of specific genes, but as the basis for preliminary investigations of genome-wide structure and function.
RESULTS
Construction of Genomic Test Sets
To assess the effectiveness of ESTs as a probe for transcribed regions, we compiled three test sets of genomic sequences. The first, designed to include as many sequences as possible, was based on all human genomic sequence entries of length 5 kb or more identified in the Genome Sequence Database (GSDB). This produced a test set (set AS) comprising 774 sequences, in which all or part of 634 genes were annotated. The average mRNA size for these genes was 8350 nucleotides (median 5340), with an average of 7 exons per gene, and an average exon size of 245 nucleotides (median 136). Although this set is still quite small compared with the estimated human gene complement of 60,000–100,000 (Antequera and Bird 1993; Fields et al. 1994) and does not represent a random sample of all human genes, it is, to our knowledge, the largest collection of human gene structures that can be obtained at present.
The second set was a subset of the first, in which we selected only entries containing whole genes (set WG). This set included 226 entries containing 287 genes, with an average mRNA size of 9770 nucleotides (median 5670), an average of eight exons per gene, and an average exon size of 236 nucleotides (median 135).
Finally, we constructed a third set of benchmark sequences (set BE) that had been extensively characterized experimentally, and whose annotation reflected, insofar as we could determine, an exhaustive transcript map of the genomic sequence. Comprehensive annotation of this sort is essential to identify false-positive and false-negative results of any gene-finding technique. This is particularly difficult to assess in many public sequences, because annotation is often restricted to transcripts that form the focus of the accompanying references. Variant or additional transcripts in the sequence may remain unidentified, or, if identified, uncharacterized or unpublished. For example, even in regions such as the HLA gene cluster, where an extensive body of work has accumulated, available annotation does not yet completely describe the gene content of the sequence (Bedian et al. 1997). Therefore, it was necessary to restrict this set to a small number of sequences for which there was clear evidence of exhaustive transcript mapping: ∼170 kb from the DiGeorge syndrome minimal critical region (Gong et al. 1996; DGCR, IC accession nos. L77569,L77570, AC002522; GSDB accession nos. GSDB:S:75553, GSDB:S:75554, and GSDB:S:1725789), which was examined by use of cDNA library screening, exon amplification, and RT–PCR between GRAIL-predicted exons, and the extensively studied human β-globin gene cluster (IC accession no. U01317; GSDB accession no. GSDB:S:1257806). This set comprised 17 genes, five of which are intronless and do not contain a clear ORF; it is not known whether these are partial clones of noncoding genes or genes with small coding regions, or whether they represent sterile transcripts arising from the gene-dense DGCR. The average mRNA size of 9500 nucleotides (median 1650) was somewhat smaller than that for set WG, as was the average number of exons per gene, four. However, the average exon size of 330 nucleotides (median 170) was larger than that for set WG. Both of the latter differences arise because of the presence of the DGCR single-exon transcripts, as well as the three-exon structure of the β-globin gene family members.
Definition of Stringency Classes
To better understand the behavior of ESTs as a gene-finding probe, we used several stringency classes for interpretation of similarity results (Table 1). The identity thresholds for different classes ranged from 95%, just below the estimated level of sequencing error in ESTs (Nishikawa and Nagai, pers. comm.), to 70%, just above an estimate of the identity in untranslated portions of transcripts conserved between human and mouse (Makalowski et al. 1996). Although it is certainly possible that cross-species conservation might produce positive results at any of these identity thresholds (Cerretti et al. 1995), we expected that the majority of alignments, particularly at higher levels of identity, would be with human ESTs. We also wished to determine how often an alignment involved the entire EST, and how often it was limited to a short region, as one might expect from reuse of functional motifs or crossover between coding exons of related genes. Therefore, at each level of identity we constructed two stringency classes, the first of which also imposed the requirement that a large part of the EST be involved in an alignment with the genomic sequence that met the identity threshold, whereas the second did not impose this requirement. These coverage requirements are indicated by the suffix L (long) and S (short), respectively, in the name of each stringency class. For stringency class 95L, we imposed the additional requirement that the alignment be contiguous along the EST, because we were interested in identifying ESTs that were identical to the genomic query sequence within the limits of sequencing error. Gaps in the alignment along the genomic sequence were always accepted, as this is a natural consequence of mRNA splicing. A similar requirement for contiguous coverage along the EST was imposed for stringency class 70L, to filter out alignments that consisted of multiple short patches of low-grade similarity; these were accepted as part of class 70S.
Definitions of Stringency Classes
Identification of Annotated Genes
Each sequence in the genomic test sets was used to search all of dbEST as described below, resulting in the alignment at different stringency thresholds of between 1748 and 257,409 ESTs with genomic sequences (Table 1). EST matches to annotated genes were determined, and the results are summarized in Figure 1. At the 95% identity threshold, 70%–80% of the annotated genes were detected by an EST aligning with at least one exon; this is consistent with previous results with larger test sets of mRNAs (Aaronson et al. 1996; White and Kerlavage 1996). Not surprisingly, the fraction of genes detected is somewhat higher for set WG than for set AS. Multiple functional classes of gene product (e.g., hormones or cytokines, structural proteins, and enzymes involved in intermediary metabolism) were represented among both genes identified and genes missed in different stringency classes (data not shown).
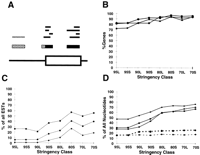
Correlation of EST results with annotated genes. (A) Determination of overlap between EST alignments and annotated genes. The bottom line shows the exon/intron structure of a portion of a hypothetical gene. The top tier of bars denote individual EST alignments; those in black are scored as overlapping the gene, whereas those in gray are scored as complete misses. Themiddle tier shows the projected CRs derived from these alignments; nucleotides in the black portions of CRs are scored as falling within an annotated exon, whereas those in the hatched portions are scored as falling outside of known excons. (B) Fraction of all annotated genes that were identified by at least one EST alignment. (▪) Data for set AS; (•) data for set WG; (♦) data for set BE. (C) Fraction of all EST alignments that do not overlap annotated exons. (D) Fraction of nucleotides in projected EST alignments that do not fall within annotated exons. EST alignments with each genomic query sequence were projected onto the query sequence as described in the text. Solid lines denote fraction of nucleotides from all projected CRs; broken lines denote fraction of nucleotides from projected CRs at least partially overlapping annotated exons.
Relaxation of the identity threshold to 90% yields an increase of ∼10% in the number of genes identified. This was primarily caused by the presence of additional ESTs that were excluded at the 95% identity level, rather than crossover of ESTs among closely related members of gene families. We expect that the gain in sensitivity is the result of a combination of two phenomena. First, a sequencing error rate slightly above average, or isolated sequencing errors in small exons, may pull the identity level of the alignment for an exon below 95%. A 90% threshold will be less sensitive to these effects, as well as to edge effects produced by extension of BLAST alignments across exon boundaries. Second, relaxation of the requirement for contiguous coverage of the EST in class 90L permits a number of ESTs with small internal gaps to be scored as positive. These gaps may represent divergent regions skipped in BLAST alignments, sequencing errors or artifacts in cDNA clones, deletions occurring during cDNA library construction, insertional polymorphisms in the human population, or small differences in structure between closely related genes.
Further decreasing the identity threshold to 80% detects only 5% more of the annotated genes, suggesting that the 90% identity threshold provides sufficient freedom to account for most sequencing errors and allelic variation, but remains above the level of identity existing between members of gene families. It is of interest that at the 90% and 80% identity thresholds, relaxing the length threshold leads to the identification of a substantial number of additional genes, particularly in set AS. This may occur because the 90S and 80S classes are less sensitive to the fragmentation of an alignment by indels into smaller HSPs, each of which may be pulled below the identity threshold by a small number of mismatches. Although this may prevent the accumulation of extensive coverage in high-identity HSPs, some of the resulting HSPs are likely to retain high identity levels, and therefore satisfy the requirements for the classes with lower coverage thresholds. It is also possible that the lower coverage thresholds permit the detection of genes based in conservation of small functional domains among related genes, whereas the 90L and 80L classes require more substantial conservation of gene structure. At each downward step in stringency, the increase in sensitivity is primarily the result of inclusion of new ESTs. However, crossover events—detection of a new gene at a given stringency by one or more ESTs that detected a different gene at a higher stringency—do occur more frequently in the steps from 90% and 80% identity to lower levels than they occur at steps down from the 95% threshold. As expected, crossover events are more common at steps down from the 90S classes than the 90L class; there were too few crossover events from the 80S class to 70L and 70S to assess the effect of the length threshold in this case (data not shown).
Estimation of False-Positive Rates
Because we were interested in using this technique as a tool to guide further investment of laboratory resources, we attempted to assess the false-positive rate in two ways. For experiments such as library screening or sequencing using cDNA clones associated with ESTs, the critical question is whether the EST correctly identifies one or more exons, or is an artifact arising from intronic or intergenic sequence. As a first measure of reliability, we examined the frequency of complete misses, that is, cases in which an EST aligned with the genomic sequence but did not overlap an annotated gene at all (Fig.1C). This provides an upper bound for the chance that an EST would be erroneously selected for laboratory follow up. At the highest stringencies, overlap is nearly complete in set BE, whereas ∼6% of the ESTs identified by set WG do not overlap known genes. The overall proportion of 5′ and 3′ ESTs in these stringency classes approximately matches that of dbEST as a whole (data not shown). One might expect that use of alternative coding exons within a gene would be reflected primarily in 5′ ESTs and alternative polyadenylation in 3′ ESTs, whereas ESTs arising from unannotated genes and pseudogenes would more closely reflect the polarity distribution of the entire EST database. Therefore, although all of these phenomena undoubtedly contribute to the complete misses we observed, we suggest that unannotated genes may be the principal source of these ESTs. Not surprisingly, the fraction of discordant ESTs produced by set AS is greater even for the high stringency classes. 5′ ESTs are not recruited more frequently by this set than by set WG (data not shown), again suggesting that unannotated genes, rather than incomplete annotation of genes on the basis of partial cDNAs, underlie most of these ESTs.
The proportion of complete misses rises with the relaxation of the length threshold at 90% identity, and does so even more sharply at 80% identity. The existence of these additional alignments may provide further evidence for the presence of short conserved motifs in genomic DNA. Because these data are based on alignments to ESTs, such regions presumably occur within genes and pseudogenes, but their functional significance is unclear. It is unlikely, however, that they arise from a class of unknown or inadequately masked interspersed repeat elements, because in the 80S stringency class, which contains the most complete misses, <30% of these ESTs align with loci in more than one genomic sequence, and <6% align with loci in more than five genomic sequences.
The drop in the fraction of complete misses from the 80S class to the 70L and 70S class likely arises because of the greater length threshold imposed in the 70% identity classes. Further, there is little difference in the results for stringency classes 70L and 70S, probably because the difference between these classes involves only contiguity of the alignment, not its overall length.
Experiments such as RT–PCR or oligonucleotide hybridization, which use short primers synthesized directly from the sequence, depend on the accuracy of a specific region in an alignment, rather than on the EST as a whole. Therefore, we also estimated the false-positive rate as the fraction of nucleotides from the query sequence that participate in EST alignments but fall outside annotated exons (Fig. 1D). This provides an upper bound for the rate at which ESTs falsely identified a region of genomic sequence as part of an exon, because it also includes sequence scored as nonexonic as a result of missing annotation for a true gene. Set BE provides an estimate of the ideal false-positive rate at each level of stringency. At the 95% identity threshold, ca. one in four nucleotides falls outside an annotated exon; this fraction rises as stringency is relaxed so that in all test sets a majority of the nucleotides in EST alignments fall outside annotated exons. At high stringencies, however, the fraction of contiguous ranges (CRs; the projections of overlapping segments of EST alignments from a given search onto the genomic sequence) that do not overlap exons at all is much lower (data not shown). Because there are several ways to identify regions more likely to have detected a true gene, it is also useful to examine the per-nucleotide false-positive rate for those CRs that do overlap known exons. For set BE, this is a stable 17% ± 1% across all stringency classes. As expected, the values are somewhat higher for the other test sets, but do not exceed 27% for any stringency class.
There are many possible reasons nonoverlapping regions might arise, not all of which represent errors in gene identification. These include the presence of undiscovered genes within or beyond a known gene, alternative exon, splice site, or polyadenylation site usage in known genes, complete or partial pseudogenes, spurious incorporation of genomic DNA into cDNA clones from which ESTs were derived, and imprecision in alignment of an EST with genomic sequence at an exon/intron boundary. Errors in reverse transcription, such as priming on hnRNA or on poly(A) tracts in repeat elements and pseudogenes, can also lead to identifications that are accurate reflections of the cDNA structure, but do not correspond to normal expression of an underlying gene.
Clustering of ESTs Over Annotated Genes
To better understand the relationship between ESTs and expressed regions of genomic DNA, we have further analyzed those alignments involving ESTs that overlap known genes. Our intent was twofold: to identify criteria that might be used to recognize alignments most likely to represent actual genes, and to obtain as much additional information as possible about gene structure and expression from EST alignments.
The clustering of multiple EST alignments may supply additional information about exon structure, potential peptide products, and possibly variations in expression of a predicted gene. First, it reduces the likelihood that the model is based on a rare artifact such as cloning of genomic DNA into a cDNA library. Second, multiple ESTs may also identify patterns consistent with alternative exon usage (Bedian et al. 1997). Third, the positions of ESTs from each end of different cDNA clones may reveal different portions of the gene, and, by comparison of cDNA size to separation of ESTs on the genomic sequence, provide clues about exon/intron structure. Fourth, the presence of ESTs from multiple sources may provide a coarse initial impression of a gene’s expression pattern. Finally, clustering of ESTs on long genomic sequences facilitates the develop ment of gene indices (Merck 1996), because it permits genome-directed assembly of nonoverlapping ESTs.
Therefore, we have examined separately the clustering of EST alignments over known genes and new CRs (Fig. 2). Over 81% of the genes identified at the 95% identity threshold, and over 90% of the genes identified at the lower identity thresholds, were detected by ESTs from more than one cDNA clone. (The single exception, class 80L for set BE, represents identification of one additional gene by a single EST.) The number of ESTs in a particular stringency class similar to a single gene varied from one to >5000; a typical distribution (class 90L for set WG) is shown in Figure 2B. In contrast, >42% of the new CRs resulted from alignment with a single EST.
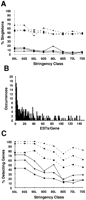
Clustering of ESTs over genes. (A) Solid lines indicate the fraction of annotated genes identified by a single EST. Broken lines indicate the fraction of all CRs not overlapping an annotated gene that are defined by a single EST. (▪) Data for set AS; (•) data for set WG; (♦) data for set BE. (B) Number of ESTs overlapping each annotated gene in set WG that was identified by ESTs from the 90L stringency class. The 41 largest clusters, each a different size in the range 156 to 2195 ESTs, are not shown. (C) Solid lines indicate fraction of singleton ESTs by overlap that detect a gene; broken lines indicate fraction of multi-EST clusters by overlap that detect a gene.
For the purpose of first-pass annotation, the inverse of this analysis is particularly important: Given a significant alignment between the genomic sequence and one or multiple ESTs, what is the likelihood that a gene has been identified (Fig. 2C)? As one might expect, this depends on the criteria used to cluster ESTs. If one requires that the alignments involving all members of a cluster overlap with each other along the genomic sequence, the frequency with which a singleton does not detect an exon ranges from 29% to 89%. Therefore, the presence of a singleton by overlap is not in itself a strong indication that the alignment is a false-positive identification. However, if gene annotation is used to cluster all ESTs overlapping a given gene, then a singleton alignment is much less likely to identify a known gene: This occurs for <40% of the genes in sets WG and BE at any stringency, and <5% for classes 90L, 95S, and 95L. These data indicate that if criteria other than overlap are used to construct gene predictions, the presence of only a single EST alignment may provide much stronger evidence that the prediction is not correct. This principle must not be applied too rigidly, however, as it is expected that some genes will be underrepresented in EST collections. This is particularly true of genes with a very low level or narrow window of expression; information about the tissue source and size of the parent library, and possible functional assignment by amino acid similarity, may help to assess this possibility in specific cases.
Although the presence of a lone EST by overlap is not necessarily a strong a priori predictor of accuracy, the clustering of multiple ESTs is, particularly at high stringency. The presence of two or more overlapping EST alignments corresponds to identification of an annotated gene 44%–80% of the time in set AS (>75% for classes 95L, 95S, and 90L), 60%–96% in set WG (>95% for classes 95L, 95S, and 90L), and 74%–100% in set BE (>99% for classes 95L, 95S, and 90L). These values do not change significantly if per-gene clustering is used instead of overlap clustering. Under either strategy, then, clustering of ESTs can be used as a triage criterion for selecting gene predictions for more intensive follow up: Although a singleton does not necessarily rule out the presence of a gene, the clustering of multiple ESTs is a strong indicator of a correct gene identification. Moreover, multiple ESTs may permit the construction of a more complete gene prediction for further study, as discussed below.
Because cDNA library amplification and normalization may increase the representation of a given clone in a library from which ESTs are generated, two ESTs with different clone IDs may actually represent the same original mRNA. The origin of clones in different cDNA libraries, however, provides clear evidence of their independence. It has the further advantage that, like clone IDs, it is usually recorded automatically as ESTs are generated, but because the library of origin is generally constant for all ESTs in a particular sequencing run, assignment is less subject to tracking error than clone ID. For example, in our analysis of 120,996 cDNA clones in this study, we identified only five instances in which the ESTs with the same clone ID had differing library IDs. Information about library of origin for the ESTs in a cluster also provides an initial estimate of conservation and expression for a gene prediction, in a manner analogous to the laboratory zoo blot or multi-tissue Northern or PCR analysis. Factors such as the tissue source and quality of the library, as well as whether it has been normalized, will also be important for interpreting an EST’s contribution to this type of estimate.
To determine whether this criterion was useful in practice, we examined the distribution among cDNA libraries of those ESTs identifying annotated genes in this study. Seven hundred eighty-one libraries were represented, contributing from 1 to 24,026 ESTs (average, 207, median, 19) to the results for a particular stringency class. A summary of the data by stringency class is presented in Figure 3. Once again, we found that in the great majority of cases, if a known gene is identified by any ESTs, it is identified by ESTs from multiple cDNA libraries, confirming that coarse expression profiles can often be derived from EST clusters. It should be noted, however, that a number of the cDNA libraries contributing ESTs to dbEST overlap in tissue/cell type and developmental stage, so these data overestimate to some extent the breadth of the expression profiles that can be generated from ESTs.
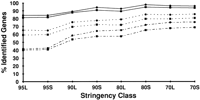
Diversity of cDNAs identifying annotated genes. For each stringency class, the fraction of all identified genes that were detected by ESTs arising from >1 cDNA library (—), 5 or more cDNA libraries (- - -), or 10 or more cDNA libraries (– - –) is shown. (▪) Data for set AS; (•) data for set WG.
Expression profiles are particularly valuable when they demonstrate cross-species conservation, which may provide a starting point for functional analysis of a predicted gene. In addition, cross-species alignments are less likely to arise from cloning artifacts such as tissue contamination (excepting libraries made from somatic cell hybrids). Although human ESTs were the principal contributors to alignments in this study, detecting >97% of the genes identified in each stringency class, ESTs from 73 species were represented. Not surprisingly, mouse was the second most common species, with ESTs present in 3%–86% of the alignments with known genes. Overall, ESTs from 9 species detected >10%, and 16 species >5%, of the genes identified in a given stringency class. Most genes identified in stringency classes 90L and higher were detected by ESTs from a single species, whereas 2–3 species/gene was more common in classes 90S and below; in all cases, <10% of genes were detected by ESTs from 4 or more species.
Use of cDNA Clone Information to Establish Inter-EST Relationships
When using ESTs for gene finding, it is often difficult to determine which sets of nonoverlapping alignments between ESTs and the genomic sequence should contribute to a single gene prediction. At the most basic level, one might simply use proximity as a criterion, by collecting all ESTs whose alignments fall within a certain distance of each other along the genomic sequence into a gene prediction. Although this is effective in regions of low gene density, it fails to address the possibility of interleaved or adjacent genes. However, a significant increase in resolution may be obtained by considering the polarity of ESTs as well. Because most ESTs (including all ESTs from the I.M.A.G.E./W.U. initiative) are derived from oligo(dT)-primed cDNA clones, 3′ ESTs effectively mark the 3′ ends of genes, often forming a cluster of heavily overlapping alignments near a polyadenylation site. Alternative polyadenylation sites for a single gene may appear as separate clusters of 3′ ESTs that lie very close to each other, or that are each linked to 5′ ESTs upstream of all of the clusters. The position of alignments involving 5′ ESTs depends on the length of the parent cDNA clone, so they are less likely to overlap with each other, but may form a pattern upstream of a 3′ cluster that helps to define the extent of a gene. In the absence of overlapping genes, this may be sufficient to distinguish among genes in a region.
Among all ESTs in dbEST whose polarity is known, there is an overall preponderance of 5′ ESTs (55% 5′, 27% 3′, 18% unknown). We have examined the polarity of ESTs aligning with genomic sequence, to see whether the methods used here introduce a bias in the polarity of ESTs scored as significant. In the two large test sets, the distribution of ESTs in search results approximates that of dbEST as a whole (data not shown). Moreover, >66% of the genes identified in set AS, and >85% in set WG, align with at least one 5′ and 3′ EST, confirming the utility of this technique in setting initial boundaries for gene predictions. Interestingly, of the remaining genes, from 4-fold to 15-fold more were detected solely by 5′ ESTs than by 3′ ESTs. This is significantly greater than the ratio of unpaired 5′ ESTs to unpaired 3′ ESTs in the database: For those ESTs produced by the W.U./Merck/I.M.A.G.E. project, this ratio is 1.6. The excess of 5′-only identifications may be due, in part, to the greater likelihood that 5′ ESTs will fall within a coding region, and therefore cross over between conserved regions of related genes. Because such crossovers do not occur frequently at high stringencies, however, other factors must contribute to this phenomenon as well.
It should be possible to link pairs of 5′ and 3′ ESTs derived from a single cDNA clone by straightforward database queries. Although this identifies only a subset of the ESTs arising from a gene, it is very unlikely to create false associations between ESTs from interleaved or nested genes. Moreover, additional ESTs that overlap with either of the original pair may be included in the gene prediction with high confidence, because few instances of distinct genes with overlapping exons have been demonstrated. When we examined our results, however, we found several limitations to the practical application of this approach. Approximately 17% of all ESTs meeting threshold criteria for alignment have no associated clone ID and are therefore excluded immediately. In the two large test sets, 60%–67% of the clones identified by the remaining ESTs, comprising 45%–61% of the ESTs, did not have both 5′ and 3′ ESTs in the database (ranges indicate values for different stringency classes). Therefore, only 39%–55% of the ESTs can be linked in this way.
When we examined the data for these ESTs, we found that in a substantial number of cases, only one member of an EST pair produced an alignment with genomic DNA meeting the threshold criteria for a particular class, although both ESTs were present in the database (Table 2). As expected, the proportion of pairs for which one member was missed was highest at the 95% identity threshold, and there was a particular tendency to miss the 5′ member at this level. Similar observations have been made in a prior study (Wolfsberg and Landsman 1997). However, at less stringent identity thresholds, the frequency with which only one end of a clone was scored as positive remained relatively high. The best results overall were obtained at the 90% identity level; the strongest effect of the length threshold was seen at the 80% identity cutoff.
Linking of ESTs by cDNA Clone ID
A certain number of discordant EST pairs are expected in cases in which one member consists of repeat sequence, is derived from a genomic region beyond the end of the query sequence, or has been assigned an incorrect clone ID. However, these reasons were not sufficient to explain the results we had observed. In particular, we were concerned that the inability to introduce gaps into alignments would force BLAST 1.4.8 to break into multiple high-scoring segment pairs (HSPs) those alignments in which the overall identity between an EST and the genomic sequence was high, but many small indels were present. This might occur, for instance, because of alternative readings of compressions or low quality traces from a sequencing gel. The smaller HSPs would more easily be pulled below the identity threshold for a stringency class by a small number of mismatches, leading to exclusion of the EST from that class. Therefore, we selected for further analysis 3815 of the cases from all test sets and classes where an EST had not been scored positive for the stringency class into which the EST from the other end of the same clone fell. For each genomic test sequence in this subset, we performed a search and analysis against the 3815 ESTs using either blastn version 1.4.8, as done previously, or WU-BLAST 2.0 (Gish 1997), which permits gaps in alignments, as the search engine.
The repeated BLAST 1.4.8 searches identified ≤5% of the missed ESTs for all classes except 80S and 70S in set WG, and classes 95L, 95S, 90L, and 80L in set AS, indicating that the depth of the original searches was adequate to identify most positive ESTs from these classes in dbEST. The recovery rate for the remaining classes ranged from 11%–28%, suggesting that especially in lower stringency searches, it may be necessary to screen very large numbers of alignments to recover all ESTs of interest. In contrast, use of the WU-BLAST 2.0 algorithm led to the recovery of an average of 27% (range 8%–54%) across all stringency classes of entries missed in the BLAST 1.4.8 searches by use of sequences in sets AS and WG. Interestingly, however, when WU-BLAST 2.0 was used as the engine for dbEST searches with all of the query sequences from the test sets, the overall success rates for linking members of EST pairs were similar to those seen for BLAST 1.4.8 (data not shown). We expect that this occurs because in addition to identifying members of EST pairs missed by BLAST 1.4.8, WU-BLAST 2.0 recruits additional ESTs, and hence additional pairs, into a stringency class, in which one EST is missed for reasons other than small indels.
This observation suggests that missed ESTs may be recovered by allowing for an overall reduction in stringency. When we recomputed the results for this set of missed ESTs, taking into account ESTs from the stringency class whose identity threshold is one step lower, an average of 66% (range 32%–97%) of the missed ESTs are recovered across all stringency classes in the results from sets AS and WG by use of either BLAST 1.4.8 or WU-BLAST 2.0. In each class except 90L and 80L, the incremental recovery was greater with BLAST 1.4.8 than with BLAST 2.0; in the latter two classes, WU-BLAST 2.0 recovered an additional 23%–37% of the missed ESTs. These results indicate that once a gene has been found using high-stringency alignments, significant additional information may be gained by recruiting lower-stringency alignments by clone ID linking. This is effective when using either the widely available BLAST 1.4.8 or the currently experimental WU-BLAST 2.0 as a search engine.
At least part of the remaining missed ESTs may be accounted for by masking of repeats in the original searches. For instance, of the ESTs missed in class 90L for set WG, 1.4% consist nearly completely of repeat sequence, and 5.3% included at least one segment of repeat sequence ⩾35 nucleotides long. It may, therefore, be useful when constructing gene predictions to specifically determine whether a missed EST contains significant repeat sequence. If so, an attempt may be made to align it to the genomic sequence without masking, by use of very high stringency to minimize the chance of spurious alignment at similar repeats.
Determination of Gene Structure
Alignments between ESTs and genomic sequence also aid in elucidating the exon/intron structure of genes. A single EST that aligns with multiple widely separated regions in the genomic sequence is almost certain to have arisen from a spliced transcript. Approximately half of all ESTs aligning with annotated genes identify multiple exons in this fashion. For both set AS and set WG, little variation was seen across stringency classes; the range of values was 53% ± 7%. A typical distribution of the number of exons detected by single ESTs is shown in Figure 4.
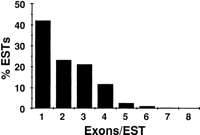
Detection of multiple exons by ESTs. The fraction of all EST alignments detecting genes from set WG in stringency class 90L that span the indicated number of exons is shown.
It is considerably more difficult to predict gene structure by use of multiple independent EST alignments. Because there are strong constraints on the size of internal exons in mammalian genes (Berget 1995), distance between ESTs may be used to detect probable introns. However, 3′ terminal exons may be much longer, and it is impossible to determine simply on the basis of distance whether a pair of EST alignments both fall within the final exon of a gene or span the last intron. This problem arises frequently in EST-based gene predictions, because the cDNA libraries from which most ESTs have been generated preferentially include the 3′ portions of transcripts. Nonetheless, a significant amount of information about internal exons is returned by EST alignments (Fig. 5). As the stringency thresholds for the alignments are relaxed, the total fraction of exons detected by ESTs increases from 43% to 83% in the larger test sets (data not shown). The corresponding values are higher for set BE (70%–94%), but this may simply reflect the smaller average gene size in this set.
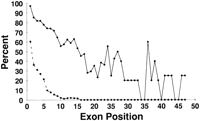
Position of exons identified by ESTs. Results shown are for EST alignments in stringency class 90L detecting exons from genes in set WG. The abscissa indicates the relative position of an exon within a gene, expressed as an offset from the 3′ end of the gene, with the value 1 corresponding to the final exons. (Solid line) The ordinate indicates the fraction of annotated genes having an exon at that position, for which the specified exon was identified by at least one EST. (Broken line) The ordinate indicates the fraction of all ESTs that detect the specified exon in an annotated gene.
Even when an EST split across multiple exons is not present, other methods may prove useful in estimating exon structure. Where two ESTs are linked by a common cDNA clone of origin, the distance between them along the genomic DNA may be compared with the estimated size of the cDNA clone, and the presence of an intron detected, although not its boundaries. The locations of EST alignments may also be compared with results from gene finders that analyze coding potential along the sequence, and consistent predictions constructed.
DISCUSSION
The data presented here demonstrate that current EST databases constitute an effective general-purpose probe for gene detection in human genomic sequence by use of straightforward similarity search techniques. With appropriate use of stringency thresholds to filter search results, it is possible to detect >80% of all known genes in genomic sequences at least 5 kb long, whereas 99% of the EST alignments, 83% of the CRs, and 68% of the nucleotides overlap known exons in sequences whose transcription map is well annotated. This is reasonably close to the sensitivity of many existing gene recognition tools on the basis of various pattern-matching techniques (Burset and Guigo 1996). In addition, the high positive predictive value provided by ESTs does not appear to degrade significantly on long sequences; this has been a concern in preliminary use of some existing gene finders. These characteristics are particularly important for large-scale annotation, where the primary goal is to detect genes and direct laboratory follow-up (Bailey et al. 1998).
Because of the requirements in large-scale annotation for high throughput and consistent data, it is also important to develop methods for automated sequence analysis. Rule-based systems are particularly useful in this regard; the data presented here suggest several principles as the foundation for such an approach to EST-driven annotation. Initial gene identification is best made at higher stringencies, such as the 90L class described here, to maintain high specificity while detecting as many genes as possible. Additional criteria, such as clustering of ESTs from multiple cDNA libraries and the presence of ESTs spanning multiple exons, may be used to further select regions of particular interest and to nucleate gene predictions for further study. Clone-of-origin relationships permit recruitment of additional ESTs, which may lead to better definition of gene structure; it is useful at this step to consider lower stringency data that are linked to high-stringency events. The resulting gene predictions present evidence about the location of genes, may yield initial information about polymorphism, alternative exon usage, and expression patterns, and provide a link to cDNA reagents to assist in laboratory investigation.
EST-driven methods carry with them a number of weaknesses as well. The greatest of these is the incomplete coverage of rare genes in EST collections. The good overall sensitivity found in this study is reassuring, as is the observation that genes of many different functional types are identified by ESTs, but it is still likely that a number of genes, particularly those with small tissue or developmental windows of expression, will be absent from EST databases for the foreseeable future. This may, to some extent, be compensated by the presence of ESTs from related members of gene families; it will be instructive to explore further the utility of low-stringency nucleotide- and amino acid-based similarity search, of both orthologous cDNA libraries and those from related species. It is also useful to repeat analyses frequently, because the size and coverage of available EST databases increases constantly. Once a gene is detected, focused application of alternative methods, such as Smith-Waterman alignments combined with splice signal prediction, may prove useful for refining its structure. Misassigment of identifiers in EST databases may also introduce noise into gene predictions. Even in the absence of false-positive components, EST-based gene predictions are likely to provide only partial gene structures, as the underlying cDNA clones do not generally reflect the full length of an mRNA. Finally, it remains difficult to interpret singleton EST alignments within a genomic region.
There are several areas for further study, then, that will assist in taking full advantage of EST-based annotation strategies. It will be essential to integrate similarity-based techniques and traditional pattern-matching methods, as each complements the other in many important respects. For example, ESTs are often effective in identifying the 3′ end of a gene, whereas gene finders that rely heavily on measures of coding potential have difficulty identifying 3′ UTRs. Conversely, pattern-matching may prove more effective in the 5′ portions of genes, which oligo(dT)-primed cDNA clones often fail to reach. Peptide-based alignment methods may also be effective in increasing sensitivity by better detecting conserved functional domains, and in providing a better starting point for prediction of gene function. Such searches, however, are much more compute-intensive than nucleotide comparisons; except in specialized centers, their use may be practically restricted to regions of special interest.
Careful evaluation and characterization of computational annotation remains important as part of a systematic approach to large-scale sequence annotation, because these data will, in many cases, be the primary source of guidance for investment of laboratory resources. To do this effectively, though, it will be necessary to increase the number of benchmark sequences available as test beds for annotation systems. The annotation in most currently available sequence entries from databases such as GSDB or DDBJ/EMBL/GenBank generally reflects data obtained as part of a focused biologic investigation, rather than exhaustive characterization of a particular region. However, the critical characteristic of benchmark sequences is that the annotation of their gene content is as complete as possible; ideally, the locations of all genes, as well as alternatives for exon usage, have been established by use of reliable laboratory techniques. This makes it possible to accurately assess both the positive and negative predictive power of gene finding methods. The limitations of our set BE underscore the need for a greater number of benchmark sequences to be generated as an early part of genome sequencing efforts. Several large genomic sequences have been published recently with annotation including a transcript map, but these have still been limited to confirmation of specific computational predictions (e.g., Ansari-Lari et al. 1997; Frazer et al. 1997). Transcript mapping has also been performed in a number of disease-associated regions (e.g., Heiss et al. 1996; Hu et al. 1997; Ruddy et al. 1997); when genomic sequence becomes available for these regions, they will provide starting points for benchmark sequences as well. There has been some initial effort in the genomics community to facilitate such analyses (Bidaud 1997), but the production, collection, and updating of benchmark sequences must be encouraged on a much larger scale. In many cases, it may be necessary to perform several cycles of computational prediction and laboratory verification to define the transcript map of a region.
Much work remains to be done in the design of systems for high-throughput, rigorous annotation of genomic sequence. It has, however, become generally accepted that high-quality, consistent annotation will constitute an important step in the process of making sequence data useful to biologists studying gene function and regulation. Similarity-based methods, particularly those that are EST-driven, will form an important component of such annotation systems for species in which significant EST data are available. This study has focused on human genomic sequences, because Homo sapiens is the mammalian species having the largest number of ESTs available at present, but we expect that the principles derived from these results will generalize well to other species with similar genome structure. With some modifications (e.g., to account for larger exon sizes) this approach is likely to be useful for more distantly related species as well. These principles provide a solid basis both for the interpretation by individual investigators of single search results, and for rule-based systems functioning as part of an integrated approach to genome-scale annotation.
METHODS
Computational Resources
Computation was performed on a collection of Sun SPARCstations and UltraSPARCs running the Solaris 2.4 or 2.5 operating system. Software tools constructed as part of this project were written in C, Perl 5, SICStus Prolog, and Sybase Transact SQL. Publicaly available software tools were compiled locally where possible, otherwise executable images were obtained from the authors. Public sequence databases were available via local flatfile copies, which were updated from servers at the U.S. National Center for Biotechnology Information (NCBI) daily, and periodically regenerated completely to insure synchronization with the master versions.
EST Sequences
The complete dbEST database (Boguski et al. 1993) was used as the target for EST similarity searches. During the time the searches were performed, this comprised ∼1.4 million EST sequences, of which ∼875,000 were derived from human cDNAs. The entire sequence of each EST was used in similarity searches.
Genomic Test Sequences
Test sequences were identified by querying the GSDB (Keen et al. 1996) periodically between April 1996 and February 1997 for human sequence entries 5000 nucleotides or longer for which the molecule sequenced was DNA or dsDNA. The IC accession number was then used to retrieve each entry as a flatfile from a local copy of GenBank. Entries were screened manually, and those that were actually mRNA sequences, organellar DNA, or proviral DNA were excluded, as were entries consisting solely of interspersed repeat elements. We also excluded entries containing the T-cell receptor genes, because these contain annotation for a large number of V-region segments representing a single type of gene that is likely to be under-represented in EST databases, and which, therefore, skewed the test results.
The feature table for each entry was analyzed by use of the SSP1 parser (Overton et al. 1994), as well as other software written specifically for this investigation, to identify gene-related features and define the extent of annotated genes. In cases where SSP1 failed or produced errors, results were reviewed manually. The location of a gene was considered to be the union of all exonic regions specified inexon, mRNA, CDS, 5′ UTR, and 3′ UTRfeatures. Where appropriate, information fromprim_transcript, polyA_signal,and polyA_site features was used to extend the first or last exon of a gene. For genes subject to alternative splicing, initiation of transcription, or polyadenylation, all alternatives were considered to be part of the gene. Where a single database entry contained multiple genes, the values of the gene, standard_ name, and product qualifiers were used to group features. Features in different entries with the same gene names were considered part of the same gene. Sequences from the DGCR were received directly from the investigators, and the locations of genes were annotated manually based on information provided by them. Where multiple entries contained genes with the same name and ⩾90% sequence identity, they were considered duplicates, and all but one were discarded.
Because in many cases the annotation did not unambiguously indicate whether a complete mRNA was present in an entry, we adopted as a working criterion for inclusion in set WG the presence of a complete coding region, with evidence, such as a polyA_signalfeature, that the 3′ end of the gene was present.
BLAST Similarity Searches
Prior to searching, interspersed and simple sequence repeats were masked off in the genomic query sequence by use of a copy of CENSOR (Jurka et al. 1996) version 1.1 kindly made available by the authors. Repeat templates consisted of a collection of human repeat elements (v. 5.0, Jurka et al. 1992), simple repeat elements (v. 3.0, Jurka and Pethiyagoda 1995), and the cloning vector pUC19. CENSOR was run once with a linear gap penalty and again with a logarithmic gap penalty to better identify partial repeats; the default alignment parameters (Conservative 2 set: DASHER window size of 150 with no overlap and a score threshold of 4.5; LOCAL score threshold of 25.0 and ratio threshold of 2.0) were used at each step. The masked sequences were then used as a query in a similarity search against all of dbEST by use of blastn version 1.4.8 (Altschul et al. 1990). BLAST was chosen as the search algorithm because significant HSPs involving all parts of the sequences are reported, rather than a single optimal alignment. This makes it possible to deal easily with the large gaps, corresponding to introns, which one expects will occur in an alignment of cDNA sequence to genomic sequence. The rapid execution of BLAST-based search engines relative to other common alignment algorithms also makes it the option of choice for large-scale annotation in laboratories without extensive computational resources. Furthermore, because we have focused on close nucleotide similarity as a criterion for gene identification, BLAST’s somewhat lower sensitivity to distant relationships than algorithms such as FASTA was not a significant liability. In all BLAST searches against dbEST, default values were used for parameters controlling alignments (E = 10 E2 = 0.024 M = 5 N = −4 W = 11). The output reporting parameter V was set to 1, and B was initially set to 250, and increased until the final 10 alignments in the output failed to reach the thresholds for any stringency class, or to a maximum value of 5250. This maximum was reached in searches with 96 of the genomic query sequences. In searches against the sample database of 3815 ESTs described below, B was set to 4000. After each search, sequence position numbering was corrected to account for BLAST’s deletion of masked regions, and a summary of each reported HSP was saved.
WU-BLAST2 Comparisons
Alignment of selected ESTs and genomic sequences was performed by use of the currently available implementation of wu-blastn version 2.0a10 (Gish 1997). Default parameters (identical to those for blastn 1.4.8 above, except B = 4000 Q = 10 R = 10) were used. Each genomic sequence was used as the query against a database of 3815 test ESTs, and the results for ESTs of interest were classified by use of the same rules as were used for the BLAST 1.4.8 search results.
Classification of Similarity Results
Alignments of an EST with a genomic query sequence were classified by use of a set of rules that considered the degree of identity between the two, and the fraction of the EST that was similar to the query sequence (Table 1). An alignment was included in a particular stringency class if it was a member of one of that class’s parents, or if it met the length and identity thresholds for that class directly. In determining whether a length threshold was met, each HSP or gapped segment pair (GSP) was considered to cover a continuous span along an EST from the starting position to the ending position of the segment. All HSPs or GSPs meeting a given identity threshold were used to determine whether that alignment met the length threshold for the stringency class under consideration.
Analysis of EST Origin
The clone ID, library of origin, and polarity (i.e., 5′ or 3′ end) of each EST studied were retrieved by direct query to the dbEST SQL server maintained by NCBI from theclone_uid, id_lib, andp_end fields, respectively, of theEST table. A suffix consisting of whitespace or the wordend was removed from the polarity value, if present, and the word prime was replaced with the symbol (′) to regularize the representation; if the resulting string was neither 5′ nor 3′, it was considered uninterpretable. Manual review of these uninterpretable values indicated that, with rare exceptions, they designated ESTs whose polarity was, in fact, not determined. ESTs were considered to have arisen from the same cDNA clone if they shared the same clone ID.
Correlation of EST Results with Gene Annotation
An EST was considered to identify an annotated gene at a particular identity threshold if any HSP or GSP in the BLAST-generated alignment between that EST and the genomic sequence overlapped any portion of that gene’s location. A similar criterion was used for identification of individual exons by an EST. Because polarity information was not available for all ESTs, we did not require that this overlap occur on the same strand. A cDNA clone was considered to identify an annotated gene if either of the ESTs arising from that clone identified the gene.
In places where EST alignments to the genomic sequence fell outside of annotated genes, all overlapping segments of EST-genomic alignments were projected into a single new CR on the genomic sequence, to approximate a portion of an exon in a potential unannotated gene. Each annotated exon was also considered to be a single CR in the genomic sequence.
Acknowledgments
We thank Juergen Haas for assistance with the SSP1 parser; Marcia Budarf, Weilong Gong, and Bruce Roe for providing access to data from the DGCR prior to publication; Jonathan Schug for a critical evaluation of the computational data; and Stephen Fischer, Jonathan Crabtree, and Mark Gibson for database and system support. We are also grateful to Sun Microsystems, Inc., for donating several processors for the E4000 on which the searches were run. This work was supported by grant DE-FG02-92ER61371 from the U.S. Department of Energy (G.C.O., D.B.S.), and grants R011-HG-01539-01 and R01-HG-01450-01 from the National Institutes of Health (L.C.B., G.C.O.). L.C.B. was supported by a fellowship from the Life and Health Insurance Medical Research Fund.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL bailey{at}www.cbil.upenn.edu; FAX (215) 573-3111.
-
- Received August 29, 1997.
- Accepted February 19, 1998.
- Cold Spring Harbor Laboratory Press








