
Homologs of the Yeast Longevity Gene LAG1 in Caenorhabditis elegans and Human
Abstract
LAG1 is a longevity gene, the first such gene to be identified and cloned from the yeast Saccharomyces cerevisiae. A close homolog of this gene, which we call LAC1, has been found in the yeast genome. We have cloned the human homolog ofLAG1 with the ultimate goal of examining its possible function in human aging. In the process, we have also cloned a homolog from the nematode worm Caenorhabditis elegans. Both of these homologs,LAG1Hs and LAG1Ce-1, functionally complemented the lethality of a lag1Δ lac1Δdouble deletion, despite low overall sequence similarity to the yeast proteins. The proteins shared a short sequence, the Lag1 motif, and a similar transmembrane domain profile. Another, more distant human homolog, TRAM, which lacks this motif, did not complement.LAG1Hs also restored the life span of the double deletion, demonstrating that it functions in establishing the longevity phenotype in yeast. LAG1Hs mapped to 19p12, and it was expressed in only three tissues: brain, skeletal muscle, and testis. This gene possesses a trinucleotide (CTG) repeat within exon 1. This and its expression profile raise the possibility that it may be involved in neurodegenerative disease. This possibility suggests at least one way in which LAG1Hs might be involved in human aging.
[The sequence data described in this paper have been submitted to GenBank under accession nos. AF105005–AF105009(LAG1Hs) and AF105010 (LAG1Ce-1).]
During the past 15 years, 15 genes that determine life span have been identified by mutation or overexpression in lower eukaryotes. Additionally, certain histocompatibility haplotypes have been associated with longevity in mice and humans. Association studies have also implicated several genes in human longevity that possess alleles that affect the risk of cardiac disease or Alzheimer’s disease (for review, see Jazwinski 1996a). The mammalian studies have yet to yield causal connections to longevity in contrast to the studies in lower organisms. The first longevity gene cloned as such from any species was the longevity-assurance geneLAG1 from the yeast Saccharomyces cerevisiae(D’mello et al. 1994). This gene is preferentially expressed in young yeasts. It encodes a protein 411 amino acids in length, which has several putative transmembrane domains. The molecular mechanism by which LAG1 determines yeast longevity is unclear at present.
The yeast life span is measured by the number of divisions an individual cell completes or, in other words, the number of daughters it produces, rather than by time (Mortimer and Johnston 1959; Muller et al. 1980). Despite the differences, yeasts display the fundamental features of aging found in higher organisms. They experience an exponential increase in mortality rate, and they manifest a variety of age changes, some of which are clearly decremental (Jazwinski 1996b). The yeast replicative life span is somewhat similar to the senescence of mammalian cells in tissue culture (Hayflick 1965). However, there are clear distinctions. All of the mammalian cells in culture cease dividing, unless they are immortalized (for review, see Smith and Pereira-Smith 1996). In yeast cultures, individual cells are mortal, but the daughters they produce have the potential for a full life span. Thus, the culture is immortal. This resembles closely the mortality of populations of aging metazoans (Finch 1990), rather than the senescence, in culture, of individual cells taken from multicellular organisms. Indeed, the yeast cell is the organism. Furthermore, yeast telomeres do not shorten with age (D’mello and Jazwinski 1991), unlike the telomere attrition that occurs in human cells as they senesce in culture (Harley et al. 1990).
The broad physiological principles underlying aging in species ranging from yeast to mice may be similar (for review, see Jazwinski 1996a). The control of metabolism and of stress appear to be important elements that determine life span. Fruit flies selected for delayed reproduction exhibit extended longevity (Luckinbill et al. 1984; Rose 1984). This is correlated with changes in glycogen and lipid metabolism (Service et al. 1985; Service 1987; Graves et al. 1992; Arking et al. 1993; Dudas and Arking 1995) and an expanded metabolic capacity, as measured by lifetime oxygen consumption and egg production (Arking et al. 1988). The long-lived flies display more frequent flight and greater flight duration (Graves et al. 1988). They are also more resistant to starvation, dessication, heat, and ethanol (Service et al. 1985;Service 1987; Graves et al. 1992). The postponed senescence in the fruit fly is associated with increased resistance to oxidative stress, which correlates with elevation of antioxidant enzyme activities (Arking et al. 1993; Dudas and Arking 1995).
The daf pathway in Caenorhabditis elegans is involved in a response to starvation, heat, and crowding, and it determines this worm’s adult life span (Kenyon et al. 1993; Larsen et al. 1995). Thedaf-2 gene, at the head of this pathway, encodes an insulin receptor homolog (Kimura et al. 1997), and its downstream effectordaf-16 codes for a transcription factor of the forkhead family that includes HNF-3 (Lin et al. 1997; Ogg et al. 1997). HNF-3 is known to participate in insulin-regulated metabolic gene transcription (Lai et al. 1990). Indeed, the daf-2 pathway affects the activity of metabolic enzymes (Vanfleteren and De Vreese 1995), and this results in accumulation of glycogen and lipid (Ogg et al. 1997). At the same time, activation of the daf-2 pathway results in the elevation of antioxidant enzyme activities and enhances resistance to oxidative stress (Larsen 1993; Vanfleteren 1993). This pathway also regulates resistance to thermal stress and ultraviolet light (Murakami and Johnson 1996).
Yeast mutants selected for resistance to starvation and cold stress are long-lived and resistant to other stresses (Kennedy et al. 1995). TheRAS genes, which are part of the nutritional sensor (Tatchell 1993), play a role in determining yeast longevity (Sun et al. 1994). Extension of life span in yeast results from an increase in metabolic capacity (Jazwinski 1996a), and the induction of certain metabolic enzymes including ones involved in glucose and lipid metabolism is associated with increased longevity (P.A. Kirchman, S. Kim, C.-Y. Lai, and S.M. Jazwinski, unpubl.). RAS2 modulates the response to a variety of stresses (Marchler et al. 1993). Its activity is required to resist the life-span-shortening effect of sublethal, chronic heat stress (Shama et al. 1998). This gene is required for resistance to ultraviolet radiation (Engelberg et al. 1994), and its expression parallels the biphasic profile of ultraviolet radiation resistance during the yeast life span (Kale and Jazwinski 1996).
There are similarities between the physiological processes associated with life extension in the invertebrate systems described above and the dietary restriction paradigm in mammals. Dietary restriction is the only known means by which mammalian life span has been extended reproducibly (Richardson and Pahlavani 1994; Masoro 1995). Dietary restrictions results in many metabolic changes, including lower blood glucose and insulin and higher glucocorticoid levels. Despite this, the animals use as much glucose and oxygen per weight as do animals fed ad libitum. Dietary restriction maintains antioxidant enzyme levels late in life (Xia et al. 1995). It also enhances resistance to heat stress because of maintenance of a robust heat-shock response (Heydari et al. 1993).
The similarities and differences between aging of mammals and yeasts can provide a lively topic for debate. Notwithstanding, the key question is whether there is any relationship between the molecular mechanisms of aging between these phylogenetically distant organisms. To address this question, it is essential to identify human homologs of yeast longevity genes and analyze the functions they support. It is then necessary to manipulate these genes in mammalian systems to study their effects on longevity. Here, we describe the first step on this path. We have cloned the human homolog of yeast LAG1. We show that the human gene can complement the yeast gene, a surprising result given the low conservation of amino acid sequence between them. We also present the genomic structure of the human homolog, LAG1Hs, its cytogenetic mapping, and phylogenetic relationship with other similar genes. In the process, a C. elegans homolog,LAG1Ce-1, has been characterized, and its features are shown here.
RESULTS
Search for LAG1Sc Homologs
A dozen genomic and cDNA libraries from human, mouse, rat, C. elegans, Podospora anserina, and Schizosaccharomyces pombe were screened using the yeast LAG1Sc gene as a probe at low to moderate stringencies or using degenerate PCR primers. These screenings did not reveal, beyond numerous false positives, any homologous clones from these libraries. However, a search of the GenBank database identified a homolog in S. cerevisiae(YKL008c), which we call LAC1, that had just been sequenced by the Yeast Genome Project. The predicted amino acid sequence ofLAC1 is 72% identical (81% similar) to the LAG1Scprotein sequence. Neither deletion of LAG1Sc nor ofLAC1Sc affects yeast viability. The availability of both theLAG1Sc and LAC1Sc amino acid sequences provided us with greater power in searching for homologous genes in the database. These searches revealed six proteins with significant homology to the yeast Lag1p and Lac1p. Two of the homologs were predicted products of hypothetical open reading frames (ORFs) from C. elegans. The two C. elegans genes were deposited in the database by theC. elegans Genome Project as putative ORFs, called CO9G4.1 (GenBank accession no. U42438) and KO2G10.6 (accession no. U40415). Both are on chromosome III. The predicted proteins share 29% identity (52% similarity) and 30% identity (55% similarity) with Lag1p, over 132 and 59 amino acids, respectively. Another two homologs were predicted products of the UOG-1 potential ORFs (accession nos.M62302 and M62301) from human and mouse (Lee 1991). These two potential proteins are 30% identical (47% similar) and 35% identical (52% similar) to Lag1p, over 123 and 84 amino acids, respectively.UOG-1 is transcribed on the same mRNA as GDF-1(growth and differentiationfactor-1), hence the name UOG-1(upstream ofGDF-1). The bicistronic message is very unusual and no function is known for UOG-1. The final two genes coding for proteins with significant homology to yeast Lag1p and Lac1p were the translocating chain-associating membrane (TRAM) proteins from human (accession no. X63679) and dog (accession no. X63678). These proteins are 24% identical (42% similar) and 25% identical (43% similar) to Lag1p, over 202 and 196 amino acids, respectively. TRAM proteins function in the processing of certain proteins in the endoplasmic reticulum (Gorlich et al. 1992). Subsequently, an additional homolog, LAG1Schp, was detected in S. pombe (accession no. U76608), whose predicted product is 39% identical (51% similar) to Lag1p, over 221 amino acids. The phylogenetic relationship of these proteins is depicted in the dendrogram in Figure 1. Apparently the TRAM genes from mouse and human are most distant evolutionarily from LAG1Sc, and the fission yeast and worm LAG1 genes are closest.
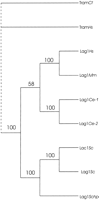
Dendrogram of the Lag1Sc protein homologs. The dendrogram was obtained by comparison of the deduced amino acid sequences of proteins with significant homology to Lag1p by BLAST. Numbers above the branches indicate the percentages of bootstrap analyses supporting the grouping at each node. (Cf) Canis familiaris, (Hs) Homo sapiens, (Mm) Mus musculus, (Ce) Caenorhabditis elegans, (Sc) Saccharomyces cerevisiae, (Schp)Schizosaccharomyces pombe.
A multiple sequence alignment of LAG1Sc, its homologLAC1Sc, the C. elegans homologs LAG1Ce-1(CO9G4.1), LAG1Ce-2 (KO2G10.6), and human homologLAG1Hs (UOG-1) reveals very limited sequence similarity at the protein level (Fig. 2A). However there is a 52 amino acid stretch (amino acids 246–297 from theLAG1Sc sequence) in all the proteins that showed an overall sequence identity of 23% and identity plus similarity of 52% in a five-way comparison (Fig. 2B). We call this the Lag1p motif. A search of the database with the Lag1p motif revealed no additional proteins. The TRAM proteins from human and dog do not display this motif (Fig.2B). Further evidence of homology comes from hydrophobicity plots that suggest that these LAG1 family members encode membrane proteins. A transmembrane prediction plot for each of these proteins is shown in Figure 3. Although they are not identical in all of their details, these proteins show substantial overall similarity in their transmembrane domain profiles. The Lag1p motif spans one of the several predicted transmembrane domains and adjacent hydrophilic regions in each of the predicted gene products. These similarities suggested that all of these proteins indeed resemble each other. TRAM protein, though lacking the Lag1p motif, displays some similarity in transmembrane domain profile (Fig. 3). The presence of several potential transmembrane domains in TRAM protein provides the possibility for some convergence of the amino acid sequence with Lag1p, outside the Lag1p motif.


Multiple sequence comparison of Lag1Sc protein and its homologs. (A) Five-way comparison of the full sequence of Lag1Sc with Lac1Sc, Lag1Ce-1, Lag1Ce-2, and Lag1Hs is shown. Residues identical in >50% of the compared sequences are on a black background; similar residues, based on evolutionary conservation, are on a gray background. (B) The region spanning amino acids 246–297 (Lag1p coordinates) was defined as the Lag1p motif. The corresponding sequences in the homologs, including in addition human and dog TRAM, have been compared, as in A. The percent identity and similarity with respect to Lag1p is tabulated.
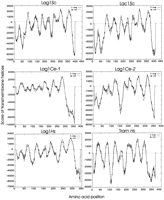
Transmembrane domain comparison of Lag1Sc protein and its homologs. Hydrophobicity increases above the horizontal, dotted line. Both orientations of the polypeptide chain i (inside → outside) ando → i are plotted.
Cloning of LAG1Ce-1, LAG1Hs, and Human TRAM
To further study the LAG1Sc homologs, one from C. elegans (LAG1Ce-1) and two from human (LAG1Hs and human TRAM) were cloned. The sequence of two independent cDNA clones of LAG1Ce-1 indicated clearly that the prediction of the last intron and exon by the C. elegans Genome Project was incorrect. A different splice site was actually utilized resulting in a slightly larger product (Fig. 4). The sequences of several full-length cDNA clones of LAG1Hs agreed with that found in the database. One of several cDNA clones of humanTRAM, whose sequence was confirmed, was chosen for further studies.

The sequence of Lag1Ce-1 protein. The splice site of the last intron in this gene is different (LAG1Ce-1) from that predicted by theC. elegans Genome Project (LAG1Ce-1D).
Functional Complementation Test
To provide further evidence that the cloned C. elegans and human genes were homologs of yeast LAG1, it was necessary to demonstrate that these genes could functionally replaceLAG1Sc. In a haploid, a deletion of both LAG1Sc andLAC1Sc resulted in an inviable strain. A diploid yeast strain, heterozygous for these deletions (marked by TRP1 andLEU2), was transformed with an expression vector carrying theLAG1 homolog. Following sporulation, the tetrads from individual diploid cells were dissected into their four haploid spores. Because deletion of both LAG1Sc and LAC1Sc is lethal in a haploid, any haploid colonies that form that are both Leu+ and Trp+ must be ones in which the deletion of LAG1Sc andLAC1Sc has been complemented by the homolog. Such complementation shows that the homolog can replace the yeast gene functionally. The nutritional phenotypes of haploid colonies formed from the spores showed two-to-two segregation of the LEU2 andTRP1 markers for both LAG1Hs transformants (Fig. 5A) and LAG1Ce-1 transformants (Fig. 5B). The tetrads had either two Leu+ Trp+ and two Leu− Trp− spores, or two Leu+ Trp− and two Leu− Trp+ spores. Diploids transformed with either of the homologs produced Leu+ Trp+spores. There were 15 Leu+ Trp+ spores out of a total of 48 spores dissected for the LAG1Ce-1 transformants and 14 out of 64 for LAG1Hs. In the controls, there were no spores that were both Leu+ and Trp+, indicating that a LAG1 homolog is required for growth of a strain carrying deletions of bothLAG1Sc and LAC1Sc. Diploids transformed with humanTRAM (Fig. 5C) showed no Leu+ Trp+ spores; only Leu+Trp−, Leu− Trp+, or Leu− Trp− spores survived. Tetrads having Leu− Trp− and Leu+ Trp+ segregants formed only two spores, both Leu− Trp−. Of 48 spores dissected, none were Leu+Trp+. The results indicate that the human TRAM gene cannot complement the LAG1Sc/LAC1Sc deletion.
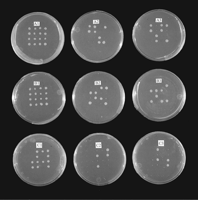
Tetrad dissection of LAG1 transformants. Tetrads are arranged vertically. (A) LAG1Hs transformants. (A1) Dissected spores from tetrads grown on a ura− plate. (A2) Colonies replicated from A1 grown on a ura− and leu−plate. (A3) colonies replicated from A1 grown on a ura− and trp− plate. Carbon sources were 2% galactose and 1% raffinose. (B) LAG1Ce-1 transformants. B1,B2, and B3 plates were the same as A1,A2, and A3, respectively. (C) HumanTRAM transformants. C1, C2, and C3plates were the same as A1, A2, and A3, respectively.
A further test was carried out to confirm the requirement for viability of either LAG1Ce-1 or LAG1Hs in yeasts deleted ofLAG1Sc and LAC1Sc. The haploid transformants, described above, were spread on leu−, trp−, ura+ plates in the presence of galactose and 5-fluoro-orotic acid (5-FOA). The presence of the URA3 gene results in the conversion of 5-FOA to a metabolic inhibitor (Boeke et al. 1987). Cells possessing theURA3-containing plasmid with the complementingLAG1Ce-1 (Fig. 6A) or LAG1Hs (Fig.6B) did not grow on 5-FOA. However, 80% of the haploid cells, plated at the same dilution, grew in the absence of 5-FOA and uracil forming ∼4000 colonies, indicating complementation of theLAG1Sc/LAC1Sc deletion by expression of the C. elegans or the human gene induced from the GAL1 promoter by galactose. As a control, the diploid transformants used to obtain the haploid were also spread on plates containing 5-FOA and uracil at the same cell number. Loss of the plasmid in the diploids allows some of the cells (1.6%) to grow. Diploids should grow after plasmid loss because only a single copy of LAG1Sc and LAC1Sc is deleted. In another control, the growth of the haploids was repressed on medium containing glucose rather than galactose, indicating the requirement for LAG1Ce-1 or LAG1Hs expression.
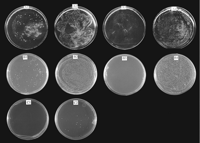
LAG1Ce-1 and LAG1Hs complement theLAG1Sc/LAC1Sc deletion. (A)LAG1Ce-1 transformants. (A1) Diploid cells on a leu−, trp−, ura+, and 5-FOA+ plate. (A2) Same cells as A1, but on a leu−, trp−, and ura− plate. (A3) Haploid cells on a leu−, trp−, ura+ and 5-FOA+ plate. (A4) Same cells as A3, but on a leu−, trp−, and ura− plate. (B) LAG1Hstransformants. B1–B4 were the same as A1–A4. (C) LAG1Hs transformants. (C1) Double (LAG1Sc and LAC1Sc) deletion haploid cells transformed with pJR1588 plasmid alone. (C2) Double deletion haploid cells transformed with pJR1588 containing LAG1Hs. Haploid double deletion cells were maintained by pRS416 containingLAG1Sc, which was replaced by the pJR1588 derivative on 5-FOA+ ura+ plates incubated for 6 days.
Another version of the complementation test was carried out forLAG1Hs. The plasmid pJR1588 with LAG1Hs under the control of the MET3 promoter was shuffled into the haploid deleted for LAG1Sc and LAC1Sc, which was maintained viable through the expression of LAG1Sc from its own promoter on a plasmid containing the URA3 gene. The empty pJR1588 vector was the control. Transformants were selected on medium lacking histidine, leucine, tryptophan, and methionine, which allows loss of the plasmid carrying LAG1Sc. They were then plated on medium lacking histidine, leucine, tryptophan, and methionine, but containing uracil and 5-FOA. No colonies were found on the control plate (Fig. 6C1). However, colonies arose at a frequency of 0.8% on the test plate (Fig. 6C2). This indicated that LAG1Hs complements the deletion of LAG1Sc and LAC1Sc in the population of cells that have lost LAG1Sc on the plasmid.
A more demanding functional complementation test was performed forLAG1Hs. The ability of the gene to perform longevity gene function was ascertained by determining the life span of yeast cells expressing the human homolog from the LAG1Sc promoter in a double deletion of LAG1Sc and LAC1Sc (Fig.7). LAG1Hs was 85% as effective asLAG1Sc in supporting the life span of the yeast strain with this double deletion. It is possible that altering the level of expression of LAG1Hs would be even more effective. There was no significant difference between the life spans of the strain complemented with LAG1Sc on a plasmid and the strain in whichLAG1Sc and LAC1Sc were intact (Fig. 7). Germinating haploid spores containing a double deletion of LAG1Sc andLAC1Sc undergo 5–6 divisions, in the presence or absence of human TRAM expression. This suggests that there is enough of the products of these yeast genes in the sporulating diploid cell to allow the resulting haploid cells to grow and divide to a limited extent without any further expression. It also indicates thatTRAM is not capable of complementing the double deletion.
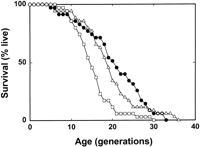
Life-span determination. Survival curves for the control strain [YPK9 containing the empty vector (circles)], the strain carrying the plasmid with LAG1Sc expressed from its own promoter (triangles), and the strain carrying the plasmid with LAG1Hsexpressed from the LAG1Sc promoter (squares) are shown. There is no significant difference between the life spans of the first two strains (P = 0.13). The life spans of the first and third strains (P = 0.001) and those of the second and third strains (P = 0.02) differ. The mean life spans were 21.4, 19.3, and 16.4 generations for these three strains, respectively.
Tissue and Cell-Type Specific Expression of LAG1Hs
The tissue expression profile of LAG1Hs was examined by hybridizing multitissue Northern blots using this gene as a probe (Fig.8). The expected transcript of ∼3 kb, which contains both LAG1Hs and GDF-1, was found in adult human brain, skeletal muscle, and testis, as well as in fetal human brain. The expression profile of LAG1Hs in several cell lines was also examined. The 3-kb transcript was found in two cultured cell lines from the nervous system, glioblastoma, and neuroblastoma cells. However, expression was not detected in normal human epidermal keratinocytes, normal human dermal fibroblasts, HeLa cells, or WI38 cells.

LAG1Hs expression in human tissues. (A) Adult. (Lane1) Heart, (lane 2) brain, (lane 3) placenta, (lane 4) lung, (lane 5) liver, (lane 6) skeletal muscle, (lane 7) kidney, and (lane 8) pancreas. (B) Adult. (Lane 1) Spleen, (lane2) thymus, (lane 3) prostate, (lane 4) testis, (lane 5) ovary, (lane 6) small intestine, (lane 7) colon, and (lane 8) peripheral blood leukocytes. (C) Fetal. (Lane 1) Brain, (lane2) lung, (lane 3) liver, and (lane 4) kidney.
Structure of Genomic LAG1Hs and Cytogenetic Mapping
Primers were designed to amplify a DNA fragment from the 3′ end of LAG1Hs based on the cDNA sequence. They were used to screen a human genomic DNA bacterial artificial chromosome (BAC) library by PCR. Two positive clones were identified. These were verified by sequencing PCR fragments derived from them.
The genomic structure of LAG1Hs was analyzed by sequencing the relevant region of one of the BAC clones. The clones obtained from subcloning of the restriction fragments of the BAC clone covered the entire region of LAG1Hs, including 4 kb of upstream promoter region. Primers for sequencing were designed based on the cDNA sequence. Primer walking was carried out. All the LAG1Hs cDNA regions and boundaries between exons and introns were sequenced, as well as the promoter region. The regions sequenced are presented in Figure 9. BLAST search using these sequences located overlapping sequences in the databases (LLNL Genome Center, unanalyzed raw data). Combining the sequences obtained from our primer walking and from the databases, a complete genomic map of LAG1Hs was constructed, as shown in Figure 9. The genomic region, including the entire transcribed portion, consists of eight exons and seven introns. It spans ∼30 kb, if the promoter region is included. Interestingly, the coding regions of LAG1Hs are highly dispersed with large introns such as 9.3 kb (intron 2) and 8.4 kb (intron 6), whereas the coding regions of downstream GDF-1 were separated only by a 592-bp intron.

Genomic structure of LAG1Hs. Numbers above are cDNA sequence coordinates (nucleotides). Numbers below are genomic DNA sequence coordinates (nucleotides). Translation start codon (ATG) is number 1. The transcription start site (−447) is indicated.
There is another feature that distinguishes LAG1Hs fromGDF-1. The 25 kb of LAG1Hs genomic DNA and the immediately adjacent 15 kb of upstream sequence have been the site of numerous Alu repeat insertions, totaling up to 56. The density of Alu elements in this genomic region is about eight-fold higher than by chance insertion alone. In contrast, GDF-1 is devoid of these repeats, even though the neighboring downstream region contains many Alu sequences. The Alu repeats inLAG1Hs are found exclusively in introns 2, 3, and 6. These are the largest introns in the gene, and they are 9.3, 3.8, and 8.4 kb, respectively. Forty-three Alu elements are more or less equally distributed among these three introns. The closest upstreamAlu resides 1.5 kb from the ATG translation start codon ofLAG1Hs. Scrutiny of the LAG1Hs ORF also reveals a significant sequence characteristic. Exon 1 possesses four repeats of the trinucleotide CTG located 177 bp from the ATG translation start codon.
We have mapped the transcription start site of the bicistronicLAG1Hs/GDF-1 mRNA. There was one start site identified, and it was located 447 bp upstream of the ATG translation start codon of LAG1Hs in a GC-rich region (Fig.10). We have searched the 300 bp upstream of the transcription start site for characteristic promoter elements. No typical TATA box was found. However, there were many predicted Cap signals. A search of the same region for potential transcription factor binding sites revealed several interesting matches. There were three Sp1, three glucocorticoid receptor (GR), two Zeste, and one each NF-κB and myogenin-binding sites among the potential sites found.
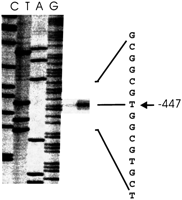
Transcription start site. The transcription start site was determined by primer extension as described in Methods. Lanes marked C, T, A, and G contain the bands from the sequencing gel in which the corresponding dideoxynucleotide chain-termination sequencing reactions were electrophoresed alongside the primer extension reaction product, using the same primer. The position of the primer extension band at −447 and the sequence coordinates are indicated to the right.
The two BAC clones containing LAG1Hs were used to cytogenetically map the gene by fluorescence in situ hybridization (FISH). Both clones of LAG1Hs DNA hybridized to chromosome 19 band p12 (Fig. 11). Both homologs of chromosome 19 were labeled in each case.

Cytogenetic mapping. BAC DNA is visualized by the light blue signal. The red signal is derived from the 19q13.1-specific probe. (Inset) Both chromosome 19 homologs enlarged.
DISCUSSION
Low nucleic acid sequence homology precluded the detection of clones of genes homologous to the yeast longevity-assurance gene LAG1in genomic and in cDNA libraries from a phylogenetically diverse array of organisms. Routine BLAST searches of the databases for a long time revealed only two potential homologs, which showed low and unconvincing levels of homology at the protein level. Nevertheless, cloning through the databases became the most expedient means to obtain a human homolog. Two developments proved instrumental in this regard. First, the Yeast Genome Project discovered a putative ORF (LAC1) with high sequence identity to LAG1. Furthermore, the C. elegans Genome Project released, in rapid succession, two putative ORFs (LAG1Ce-1 and LAG1Ce-2), with some, though low, homology to yeast LAG1. All of these genes, and the human geneUOG-1 (LAG1Hs) present previously in the databases, encode proteins that appear to contain several domains that are hydrophobic. This prompted the second important development. A computer-generated assessment of potential transmembrane domains revealed a large extent of overall structural similarity among all of these proteins.
The five LAG1 genes described above all appear to be related phylogenetically. This assessment comes from a comparison of the protein sequences based on their evolutionary conservation. Another potential homolog identified in the databases TRAM, which is found in both human and dog, seems to be on a separate branch from the remaining genes. TRAM protein at first blush appears almost as similar in terms of BLAST score to the yeast Lag1 protein as the other nonyeast gene products are. Furthermore, TRAM possesses several putative transmembrane domains. All of the LAG1 gene products possess a sequence of 52 amino acids that shows a higher degree of sequence identity and similarity than any other region. We call this the Lag1p motif. Interestingly, this motif is not conserved in TRAM protein. The Lag1p motif may be an important conserved functional element in the family of Lag1 proteins. A BLAST search of GenBank with this motif has not identified any other proteins containing the Lag1p motif. This analysis raises the possibility that the Lag1p function may depend on this motif and that the transmembrane domains alone may not be sufficient.
A deletion of both the LAG1 and LAC1 genes conveniently renders yeasts inviable. This set the stage for examining the functional interchangeability of the LAG1 genes fromC. elegans and human with the yeast genes. LAG1Ce-1and LAG1Hs were tested. Both genes were able to complement the yeast genes for viability, demonstrating that they are not only structurally related and evolutionarily conserved, but that they are also analogous to yeast LAG1 functionally. In contrast, humanTRAM did not complement the yeast genes. This coincided nicely with the phylogenetic comparison of the genes as depicted in the dendrogram. Clearly, human TRAM is a divergent gene, and the functional and evolutionary comparisons of the genes are in agreement. Furthermore, this constitutes strong evidence that we have indeed cloned the human (and C. elegans) homologs of the first yeast longevity gene described.
The phenotype of viability is measured by colony-forming ability. An exponentially growing colony can be established if each cell divides at least twice. We have shown that the LAG1Hs gene supports much more than that, providing the yeast with a near normal life span. This result shows that the human homolog has longevity gene function in yeast. In fact, this is the first human homolog of a longevity gene that has been verified functionally in this way. It will be of interest to determine whether the LAG1Hs gene is associated with human longevity.
LAG1Hs maps cytogenetically to 19p12. This gene is expressed in a limited number of tissues, including brain, skeletal muscle, and testis. It is not clear at this point what the common denominator connecting the expression in the three tissues is, but this may provide a clue to the function of LAG1 protein in humans. Clearly, brain neurons and skeletal muscle are postmitotic. Transcripts were detected in tissue culture cells of both glial and neural origin, suggesting that both brain cell types may express this gene in vivo. Furthermore, expression was also found in fetal brain, indicating that it begins during embryonal development. Analysis of potential transcription-factor binding sites in the promoter of the gene may provide some clues as to its function. The presence of a myogenin site is not surprising, given the expression in skeletal muscle. The GR and NF-κB sites might suggest some involvement in stress responses, such as the inflammatory response. The Zeste binding sites suggest a possible role for LAG1Hs during development and/or in modulation of heterochromatin structure.
Triplet repeat genes have been shown to be involved in several human neurodegenerative diseases. The repeats expand in successive generations (Aslanidis et al. 1992). These genes are expressed in brain and testis frequently (Bulle et al. 1997). A search for novel disease genes in human testis and infant brain cDNA libraries has localized 95 genes that contain (CAG/CTG)n repeats, with the highest density on chromosome 19 (Bulle et al. 1997). LAG1Hscontains a repeat of four CTGs in its coding region and similarity in tissue expression profile to these genes. In the myotonic dystrophy gene, the repeat length has been reported as short as five. Repeat lengths varying between 3 and 11 have been reported in various genes (Richards and Sutherland 1992). It may be worth investigating a possible link between LAG1Hs and human neurodegenerative diseases. It will also be of interest to determine the expression profiles of LAG1Hs during aging.
The LAG1Hs gene spans some 30 kb of genomic DNA. This includes two very large introns. The bicistronic structure of the mRNA also suggests that this gene may have unusual properties. It remains to be seen how these structural features affect expression at the protein level. The gene does code for a functional product, as evidenced by the complementation in yeast. We did not detect a TATA box in the upstream region of LAG1Hs. This is often the case for housekeeping genes (Sakai et al. 1998). The sequence does not show a Kozak consensus sequence characteristic of efficiently translated genes (Kozak 1996).
An interesting feature of the LAG1Hs genomic region is that it is rich in Alu repeats. There are 56 Alu repeats in a 40-kb region comprising upstream sequences and coding regions for exon 1 to exon 7 of LAG1Hs. Alu repeats are present in all large introns (>3 kb) of LAG1Hs but not in the region coding forGDF-1. The expansion of Alu repeats may be an explanation for the origin of these large introns.
Many human disease genes possess yeast homologs. Some of these are very similar, and it has been found that the human gene can complement the yeast homolog. A compendium of these genes has been prepared in the XREF database (Bassett et al. 1997). The cutoff point for functional complementation in this database, starting with genes displaying low BLAST P-values and moving in the direction of higher scores, is the NF1 gene that complements IRA2 (Ballester et al. 1990). LAG1Hs is clearly an outlier in this case, because its P-value is many orders of magnitude higher than that ofNF1. Indeed, this is the lowest sequence similarity that has been shown to support functional complementation. This raises the point that for certain types of proteins the degree of overall similarity may not be important. In the case of Lag1p, a short motif surrounded by appropriate transmembrane domains may be sufficient. It will be necessary to define the structural features that are necessary to preserve Lag1p function. This may provide insights into the function of the protein in humans and its role in longevity.
METHODS
Sequence Comparisons
Protein sequences were analyzed using BLAST, PILEUP, and PAUP. The GenBank database at NCBI was searched using BLAST. Multiple sequence alignments were carried out using the PILEUP program from the UWGCG package. The alignments were used in a search for the phylogenetic tree. The phylogenetic tree reconstruction, using the parsimony method and bootstrap analysis (50% majority rule), was performed using the UWGCG PAUP program. Transmembrane domain prediction was performed using the TMpred program (Prediction of transmembrane regions and orientation in protein sequences, ISREC, Switzerland). Putative Cap signals were identified using the PC/GENE program (IntelliGenetics). Potential transcription factor binding sites were identified in DNA sequences using the Signal Database file (TSSG program, Baylor College of Medicine, Houston, TX). Only sites that are an exact match to the consensus were considered. DNA sequences were searched for Alurepeats using the SMPL program (Milosavljevic and Jurka 1993) with an updated version of a reference collection of human repetitive elements (Jurka et al. 1992).
Construction of Strains and Functional Complementation
Standard yeast genetic techniques have been employed throughout (Guthrie and Fink 1991). YPK9 is a MAT a derivative of YPK4.7, obtained by sporulation. The life spans of bothMATα and MAT a haploids derived from YPK4.7 are identical, and they are not affected by centromere-containing plasmids. YPK4.7 was derived by self-mating of a MAT asegregant obtained from YPH501 (Sikorski and Hieter 1989) by sporulation. The construction of the plasmid containing theTRP1 marker used to delete LAG1 has been described previously (D’mello et al. 1994). The diploid strain YPK4.7 (MAT a/MATα,ade2–101ochre/ade2–101ochre, his3-Δ200/his3–Δ200,leu2–Δ1/leu2–Δ1,lys2–801amber /lys2–801amber ,trp1–Δ63/trp1–Δ63,ura3-52/ura3–52) was transformed with this plasmid and selected for tryptophan prototrophy. Deletion of one copy ofLAG1 was confirmed by Southern blot analysis. This strain was then transformed by the lithium acetate method with pLAC405 (containing the LEU2 marker), linearized with XbaI, and selected for leucine prototrophy to delete LAC1. pLAC405 was constructed by cloning the regions flanking the LAC1 gene, separated by an XbaI site, into the vector pRS405 to perform γ deletion of the gene (Sikorski and Hieter 1989). Deletion of a single copy of LAC1 was confirmed by Southern blot analysis. This diploid containing a single deletion of both LAG1 andLAC1 is called YPK100. To construct the haploid containing chromosomal deletions of both LAG1 and LAC1, YPK100 was transformed with plasmid pLAG416, which contains the yeastLAG1 with its own promoter on pRS416 (Sikorski and Hieter 1989) and the URA3 marker. The diploid was sporulated and haploid segregants were selected for tryptophan, leucine, and uracil prototrophy. The genotype of the haplois that were Leu+Trp+ Ura+ was verified for the absence of the chromosomal LAG1Sc and LAC1Sc genes by PCR amplification. For transformants containing LAG1Hs (see below), the presence of LAG1Hs was also verified by the PCR. In all cases, the transformants were deleted for chromosomal copies of both LAG1Sc and LAC1Sc.
LAG1Ce-1, LAG1Hs, and human TRAM cDNAs were each subcloned into the plasmid pBM150 behind the GAL1promoter (Johnston and Davis 1984), followed by the ADH2transcription terminator, so that the ATG start codon was within 20 bp of the GAL1 transcrption start site. This vector contains theARS1–CEN4 sequence for stable maintenance in low copy number in yeast and the selectable yeast marker URA3. YPK100 was transformed with the three clones to generate strains containingLAG1Ce-1, LAG1Hs, or human TRAM. As control, this strain was also transformed with pBM150 alone or with pBM150 containing LAG1Hs in the antisense orientation.LAG1Hs was also cloned behind the MET3 promoter in the HIS3-containing plasmid pJR1588, allowing induction ofLAG1Hs expression on medium lacking methionine. pJR1588 was constructed by inserting the SalI–EcoRV fragment of the MET3 promoter into pRS313 (J. Rine and C. Trueblood, cited in Fox et al. 1997). Finally, LAG1Hs was cloned into pBM150 behind the GAL1 promoter and in front of the ADH2transcriptional terminator. The GAL1 promoter was then replaced by the LAG1Sc promoter to yield plasmid pMZ207. TheLAG1Hs in all of the expression constructs contained only the cDNA for LAG1Hs, without the downstream GDF-1sequences that are found in the bicistronic mRNA. All of the expression constructs described in this report were sequenced, to verify that they possessed the correct structure.
YPK100, transformed by pBM150 carrying one of the homologs, was sporulated, and the resulting tetrads were dissected. This dissection and germination of the spores was carried out on agar plates lacking uracil and in the presence of 2% galactose and 1% raffinose to induce expression of the LAG1 homolog. Expression ofLAG1Ce-1, LAG1Hs, and human TRAM was verified by determining mRNA levels on Northern Blots. Each of the homologs was expressed significantly in inducing medium but expression in glucose, which represses transcription from the promoter, was reduced by 97–98%. After the spores had grown to form colonies, the plates were replicated onto agar plates containing the same medium except that in one case the replica plates lacked leucine and in the other they lacked tryptophan. The replica plates were incubated to allow colonies to form.
Life-Span Determination
Life-span determination has been described (Kim et al. 1998). Briefly, a Nikon Labophot-2 microscope with a 20× longworking-distance objective and a micromanipulator attachment was used. Yeast cells were deposited onto a YPD (2% peptone, 1% yeast extract, 2% glucose, 2% agar) plate and incubated at 30°C for life-span determination. Individual cells were pulled aside with the micromanipulator, and were allowed to grow until buds emerged. Thirty-five buds were removed, and were referred to as virgins (i.e., cells that have never budded). After they underwent their first cell division, buds were removed from these cells, and the virgin cells, now mothers, were recorded as one generation old. This process was continued until budding ceased. At the end of their life span, cells lysed. The number of buds produced prior to lysis is the individual yeast’s life span, which is expressed as age in generations. During the course of the experiment, cells were transferred to 12°C during the night to slow division. This treatment does not alter the replicative life span (Muller et al. 1980). The nonparametric Mann-Whitney test was used to compare survival curves. Life spans were considered to be different if P < 0.05 (two-tailed).
PCR Amplification
For making probes and library screening, the PCR kit from Promega was used. SuperScript II RNase H/Reverse Transcriptase (Life Technologies) was used for first strand cDNA synthesis. The Advantage cDNA PCR kit and GC Melt (Clontech) were employed for amplification of cDNA. A pair of primers specific for LAG1Ce-1 was designed using a predicted exon sequence from the GenBank database. With these specific primers, RT–PCR was performed using RNA from several C. elegans strains. This RT–PCR produced the predicted DNA fragment from all three strains. This fragment was sequenced and its identity was confirmed. The fragment was used to screen a C. elegansmale-specific cDNA library (Clontech). The human homolog,LAG1Hs was cloned by RT–PCR of total RNA extracted from human fetal brain. Primers were designed on the basis of the cDNA sequence in the database to amplify the entire ORF. This PCR routinely resulted in the amplification of products missing the same 235 bp close to the 5′ end, as determined by DNA sequencing. This was judged to be the result of the high GC content (80%) of this region that resulted in the generation of a stem–loop structure that was not replicated in the PCR. A full-length cDNA was amplified when GC melt was used to eliminate secondary structure in the DNA. The cloning procedure for human TRAM was similar to the cloning of LAG1Hs. Poly A-containing RNA from HeLa cells was used as a template to synthesize cDNA . Then PCR was performed and the amplified DNA was cloned. For colony PCR screening, bacterial or yeast cells were picked by toothpick and boiled for 5 min in water. Released DNA was used as template for PCR. The human BAC library was screened by the PCR method (Research Genetics).
Northern and Southern Blots
Multiple human tissue Northern blots were purchased from Clontech. Other Northern blots were made following electrophoresis of RNA in formaldehyde-containing agarose gels (Ausubel et al. 1993). The preparation of labeled probe and hybridization conditions were according to the manufacturer or standard published protocols (Ausubel et al. 1993). Probes were made using the Rediprime kit (Amersham) and purified on Nick columns (Pharmacia Biotech). Following prehybridization, hybridization was performed at 42°C in 50% formamide solution containing 5×SSC (1× = 0.15 mNaCl, 15 mm sodium citrate at pH 7.0), 50 mm sodium phosphate buffer (pH 7.0), 0.1% Na2P4O7, 0.5% heparin, 0.5% SDS, and 1 mg/ml salmon sperm DNA. Membranes were washed at room temperature in 2×SSC, 0.5% SDS for 30 min, and twice at 60–65°C in 0.1×SSC, 0.5% SDS for 30 min.
DNA was electrophoresed on a 1% agarose gel in TAE (0.04 mTris-acetate, 0.001 m EDTA at pH 8.5). The DNA was transferred to nitrocellulose (Schleicher & Schuell) by capillary action. Following prehybridization, hybridizations were performed at 42°C, in 2× PIPES buffer (1× = 0.4 m NaCl, 0.01m PIPES at pH 6.5), 50% formamide, 0.5% SDS, and 0.5 mg/ml salmon sperm DNA. Membrane washing was the same as described for Northern blot analysis.
Primer Extension
Primer extension was performed by using 10 μg of total RNA from human brain tissue. The primer was 5′ end-labeled with [γ-32 P]ATP and reverse transcription was performed as for RT–PCR, except specific reverse primers downstream from the transcription start site were used instead of oligo(dT) and random primers. The extended, single-stranded DNA was subjected to electrophoresis on a sequencing gel alongside the sequencing reactions with the same primers used to determine the transcription initiation site.
Fluorescence in Situ Hybridization
For fluorescence in situ hybridization (FISH) human peripheral blood lymphocytes were arrested in metaphase using the TC Chromosome Microtest kit (Difco Laboratories). The cells were exposed to 0.075m KCl for 20 min at 37°C, fixed three times in 3:1 methanol:acetic acid at −20°C for 30 min, and dropped onto microscope slides. DNA from BAC clone 421I13 or 157D6 (Research Genetics) containing human DNA was labeled with biotin–16-dUTP (Boehringer Mannheim) using the Large Fragment DNA labeling kit (Oncor) as instructed by the manufacturer with a 45-min incubation time. Biotin-labeled BAC DNA (300 ng) was combined with digoxygenin-labeled 19q–13.1-specific DNA (Oncor) and blocked with 60 μg of human Cot-1 DNA (Boehringer Mannheim). Hybridization of labeled DNA was done for 16 hr in 50% formamide, 2× SSC, in a humidified 37°C incubator. The slides were washed in 2×SSPE (1× = 0.18m NaCl, 10 mm sodium phosphate at pH 7.7, 1 mm EDTA) at 73°C for 5 min prior to detection of labeled DNA. The signal was detected using FITC–avidin and rhodamine–anti-digoxygenin, with DAPI counterstain. The signal was observed on a Zeiss Axioskop epifluorescence microscope using 63× Apo-Chromat (NA = 1.4) or 100× Apo-chromat (NA = 1.4) objectives. The images were captured using a Cohu CCD camera and MacProbe (v. 3.4.1) software (PSI Scientific Systems).
Acknowledgments
We thank Dr. Mark A. Batzer for help with the analysis of theAlu sequences. This work was supported by grants from the National Institute on Aging (NIA) of the National Institutes of Health. P.A.K. was the recipient of a postdoctoral fellowship from the NIA. The publication costs of this article were defrayed in part by payment of page charges.
This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL sjazwi{at}lsumc.edu; FAX (504) 568-4725.
-
- Received June 11, 1998.
- Accepted November 9, 1998.
- Cold Spring Harbor Laboratory Press








