
Strategies for Mutational Analysis of the Large Multiexon ATM Gene Using High-Density Oligonucleotide Arrays
Abstract
Mutational analysis of large genes with complex genomic structures plays an important role in medical genetics. Technical limitations associated with current mutation screening protocols have placed increased emphasis on the development of new technologies to simplify these procedures. High-density arrays of >90,000oligonucleotide probes, 25 nucleotides in length, were designed to screen for all possible heterozygous germ-line mutations in the 9.17-kb coding region of the ATM gene. A strategy for rapidly developing multiexon PCR amplification protocols in DNA chip-based hybridization analysis was devised and implemented in preparing target for the 62 ATMcoding exons. Improved algorithms for interpreting data from two-color experiments, where reference and test samples are cohybridized to the arrays, were developed. In a blinded study, 17 of 18 distinct heterozygous and 8 of 8 distinct homozygous sequence variants in the assayed region were detected accurately along with five false-positive calls while scanning >200 kb in 22 genomic DNA samples. Of eight heterozygous sequence changes found in more than one sample, six were detected in all cases. Five previously unreported sequence changes, not found by other mutational scanning methodologies on these same samples, were detected that led to either amino acid changes or premature truncation of the ATM protein. DNA chip-based assays should play a valuable role in high throughput sequence analysis of complex genes.
The Human Genome Project will soon provide the scientific community with the complete sequence of all human genes. A major challenge for medical genetics will be in using this information to elucidate genetic contributions to single gene and multifactorial diseases. Exhaustive mutational screens of candidate genes, identified through linkage analysis and proposed function, must be undertaken and sequence changes must be correlated with disease states to address these problems. Many technical obstacles must be overcome to make this approach both economical and time efficient for analyzing large genes with complex genomic structures.
A prime example of a challenging system for mutational analysis is theATM gene (Savitsky et al. 1995, 1997), responsible for ataxia telangiectasia (AT). AT is an autosomal recessive disorder characterized by cerebellar and progressive neuromotor degeneration, immune deficiency, and the appearance of dilated blood vessels in the eyes and face. Patients also manifest growth retardation, premature aging of skin and hair, chromosomal instability, lymphoreticular malignancies, and acute sensitivity to ionizing radiation. The protein-coding region of the ATM gene contains 9168 bp in 62 coding exons spread over 146 kb of genomic DNA (Platzer et al. 1997). It is a member of a family of proteins containing carboxy-terminal regions with homology to the catalytic domain of phosphatidylinositol 3-kinase (PI 3-kinase), which have been implicated in diverse activities such as telomere maintenance, cell-cycle arrest, and DNA repair (Platzer et al. 1997).
The ATM gene displays a complex mutational spectrum. Thus far, >100 different somatic and germ-line mutations have been identified, the majority of which cause premature protein truncation (Gilad et al. 1996b; Wright et al. 1997). Very few common alleles, most notably the 103 C → T allele among North African Jews (Gilad et al. 1996a), have been found (Telatar et al. 1998). The large size and genomic structure of the ATM gene greatly complicates the process of screening genomic DNA samples for all possible sequence variations. Single-strand conformation polymorphism (SSCP) assay (Vorechovsky et al. 1996; Stilgenbauer et al. 1997), restriction endonuclease fingerprinting (Gilad et al. 1998a,b), heteroduplex analysis (Telatar et al. 1998), and protein truncation assay (Telatar et al. 1996) protocols have been used in mutational analysis. These begin with separate amplification of individual ATM exons from genomic DNA or (when possible) transcript by PCR or RT–PCR protocols, respectively. The above methods are not amenable for complete analysis of the entire ATM genomic coding and splice junction sequences in a single reaction, and most require gel electrophoresis, which complicates scale-up and automation. The RT–PCR methods carry a further risk of missing mutations that result in unstable RNA.
Significant evidence exists that heterozygous ATM carriers have an elevated lifetime risk of developing breast cancer (Swift et al. 1987; Athma et al. 1996; Larson et al. 1998); however, there is disagreement about the magnitude of this effect (Easton 1994; Bishop and Hopper 1997; Fitzgerald et al. 1997). This debate is especially relevant owing to the large frequency of ATM mutation carriers, estimated to comprise ∼1.4% of the general population, who could account for up to 6.5% of all breast cancer cases (Athma et al. 1996). Large-scale mutational analysis of carefully selected test and control groups will be necessary to resolve this controversy but current mutational analysis tools preclude such an undertaking.
Hybridization-based methodologies for high throughput mutational analysis using high-density oligonucleotide arrays (DNA chips) have developed recently (Hacia et al. 1998a; Ramsey 1998). Light-directed combinatorial chemical synthesis approaches enable the manufacture of high-density oligonucleotide arrays of >105 distinct species, typically 25 nucleotides in length, on 1.2 × 1.2-cm2 glass surfaces (Fodor et al. 1991; McGall et al 1997). Oligonucleotide arrays have been used to screen for sequence variations in the CFTR gene (Cronin et al. 1996), the human immunodeficiency virus-1 (HIV-1) reverse transcriptase and protease genes (Kozal et al. 1996), the β-globin gene (Yershov et al. 1996), the mitochondrial genome (Chee et al. 1996), and theBRCA1 gene (Hacia et al. 1996, 1998a). Furthermore, they have been used to identify and genotype single nucleotide polymorphisms (Wang et al. 1998), monitor gene expression (Lockhart et al. 1996), analyze genetic screens (Shoemaker et al. 1996), design antisense oligonucleotides (Milner et al. 1997), identify bacterial species (Gingeras et al. 1998), and acquire information from orthologous genes in related species (Hacia et al. 1998b).
In this study we describe the design of a high-density oligonucleotide array-based assay to screen the ATM gene for all possible sequence variations in the heterozygous state. This represents one of the most ambitious application of DNA chips to mutation detection yet described. A streamlined strategy to analyze efficiently all 62ATM coding exons is described that should be applicable toward virtually any DNA chip-based mutation screen. Furthermore, improved algorithms for heterozygous mutation detection were developed to analyze the results of blinded studies conducted to determine the sensitivity and specificity of two-color DNA chip-based assays.
RESULTS
Oligonucleotide Array Design
Extending previous oligonucleotide array-based mutational analysis of the 3.45-kb BRCA1 exon 11 sequence (Hacia et al. 1996), a pair of DNA chips (interrogating sense and antisense strands) containing >95,000 oligonucleotides were designed to detect all possible sequence variations in the ATM coding region including the 3′ GT donor and 5′ AG acceptor splice junction sequences of each coding exon. Four 25-mer sequencing probes, substituted with one of the four nucleotides in the central position, interrogate the identity of each target nucleotide (Fig.1). For every perfect match probe (fully complementary to the target sequence) in each set of sequencing probes, another identical perfect match probe is present elsewhere in the array. This redundancy increases assay sensitivity and specificity by compensating for nonspecific signals that may arise from localized surface imperfections such as microscopic scratches in the array surface. Although the BRCA1 exon 11 chip design included probes complementary to every (1–5) base pair deletion as well as single base pair insertions, the large size of the ATM gene (>2.5× larger than BRCA1 exon 11) precluded the inclusion of additional mutation-specific probes owing to space limitations on the array surface.
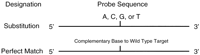
Classes of arrayed oligonucleotides. Each position is interrogated with 10 separate 25-mer oligonucleotides, 5 (two wild-type and 3-base substitution) each for sense and antisense strands. Substitution probes contain each of the four-nucleotide substitutions 13 bases from the 3′ end of the oligonucleotide (one of these will represent the wild-type sequence). A redundant set of wild-type perfect match probes are tiled in the lower portion of the array.
Iterative Strategy for ATM Target Preparation
A time-limiting and labor-intensive step commonly encountered in screening for sequence variations in large genes is the nonparallel analysis of individual exons. Because gel-based methodologies are dependent on mutation-based electrophoretic mobility shifts, analysis of multiple exons in a single gel lane has confounding effects. In addition to problems associated with separating multiple exons of similar size in one lane, mutant species may comigrate with other wild-type exons and thus mask their presence. Furthermore, it is generally not feasible to deduce the general identity of a mutant species (i.e., from which exon it is derived).
Hybridization-based mutational analysis methodologies allow parallel analysis of multiple nucleic acid species independent of their individual sizes (Wang et al. 1998). Therefore, it is possible to amplify separately every coding region of interest, pool them, produce RNA target, and hybridize to oligonucleotide arrays. Although this strategy works well when analyzing small numbers of exons, as pool sizes increase the most robust PCR/transcription reactions tend to dominate, and weaker amplifications are lost.
To address this issue, we pursued an empirical iterative hybridization-based strategy to evaluate rapidly and develop multiexon PCR reactions involving exons of similar sizes that allow for robust production of ATM test target samples (Fig.2). First, primer sets containing T3 and T7 RNA polymerase promoter sequences, based on those used in previous studies (Vorechovsky et al. 1996), were developed to amplify all individual coding exons using a single PCR protocol (Table 1). Second, all 62 primer pairs were pooled into a single multiplex PCR reaction. Reaction products were used as templates in T3 and T7 RNA polymerase-mediated in vitro transcription reactions to produce sense and antisense RNA targets. Target was hybridized to ATM arrays and sense and antisense strand data were averaged to produce a composite data set. Primer pairs that produced target giving >90% base-calling accuracy, based on the perfect match probe for a nucleotide position having a 1.2-fold greater hybridization signal intensity than that of the next highest single nucleotide substitution probe in each interrogation probe set (Chee et al. 1996; Hacia et al. 1996), were placed into a separate ATM target pool A. The remaining primer pairs were subject to another round of multiexon in vitro transcription template preparation. Second round and pool A multiexon PCR reaction products were combined and in vitro transcribed to produce target. Amplicons giving >90% base-calling specificity from the second round pool were placed into a separate pool B. The primers for the remaining exons underwent 17 additional rounds of multiexon target preparation analysis to yield 13 pools (A–M) ofATM exon primer pairs (Table 2). To further optimize these multiexon pools, five PCR primer sites were redesigned to produce increased hybridization signals for their respective exons. In the finished system, separate multiexon PCR reactions were carried out, products pooled, and in vitro transcribed in a single reaction. Several different hybridization conditions were then tested to determine which one produced optimal signal-to-noise ratios based on the aforementioned base-calling algorithm.
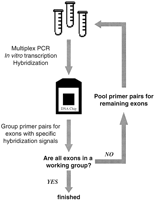
Iterative multiexon PCR and in vitro transcription optimization strategy based upon DNA chip hybridization.
ATMAmplification Primers
ATM Coding Exon Multiplex PCR groups
Two-Color Loss of Hybridization Signal Analysis
Two-color cohybridization experiments were used previously in scanning exon 11 of the BRCA1 gene for all possible heterozygous sequence changes (Hacia et al. 1996). Wild-type fluorescein-labeled (green) reference and biotinylated [stained with phycoerythrin–streptavidin (red) conjugate after array hybridization] test targets were competitively cohybridized to arrays and the relative binding of both targets to all probes was quantitated. The ratio of reference and test target occupancy to each perfect match oligonucleotide probe was used to detect sequence variations between samples (Chee et al. 1996; Hacia et al. 1996).
Test sample targets containing nonrepetitive sequence variations should have reduced affinity (relative to reference wild-type target) toward the family of perfect match probes designed to hybridize to the nucleotide tract now containing these variations (Chee et al. 1996;Hacia et al. 1996). This results in relative localized losses of a hybridization signal that can be displayed by plotting the ratio of reference to test sample hybridization signals for all overlapping perfect match oligonucleotide probes. A peak of perfect match probe hybridization signal intensity ratios should be centered nearby mutant sequences as a result of the localized relative decrease in mutant test sample hybridization. In practice, the baseline reference/test perfect match probe signal ratio values (which we will refer to as “loss of hybridization signal” baselines) obtained by comparing hybridization intensities on a single chip generally fluctuate between 0.5 and 2.5. However, fluctuations in the reference/test hybridization signal ratios for each perfect match probe can be normalized against data obtained from similar experiments because of reproducible two-color cohybridization results and probe redundancy (Hacia et al. 1996).
For convenience in visualizing relative losses of hybridization signal in test samples, it is useful for the average baseline of the reference/test perfect match probe hybridization signals for each of the 62 individual ATM coding exons to have a value of one. However, this is not always the case with unprocessed data, because of subtle variations in target concentration (in part due to a variability in multiexon PCR product yields), hybridization conditions, or in the arrays themselves. Therefore, it is necessary to calculate and apply a specific correction factor tailored for each of the individual coding exons to produce an average baseline value of one in each instance.
After using this correction factor we still observed that certain perfect match probe sets tended to have larger deviations from loss of hybridization signal baseline values (above or below the ideal value of one) than the majority of the data set. We reasoned that in some circumstances target hybridization could be significantly more sensitive to probe sequence composition and structure than others. Although reference sample concentration is equivalent in each hybridization reaction (as it is produced in a single large batch and aliquoted into each reaction), test sample concentrations may vary slightly because they are individually prepared. Concentration differences are likely attributable to subtle changes in the multiexon PCR reaction conditions that may cause specific exons within each multiexon PCR reaction pool to be amplified at different efficiency. Based on these considerations, we searched for and analyzed patterns in probe sequence composition and potential secondary structures to predict the relative likelihood and extent of any given probe to provide a lower signal-to-noise ratio. Using these predictions we formulated multiplicative correction factors to help minimize hybridization signal baseline fluctuations (see Methods).
A third general type of correction was used to minimize the effects of nonreproducible phenomena, such as microscopic scratches on the chip surface. Two steps were taken toward this end. The first was to truncate the corrected ratios to 1.50 and 0.67 for values >1.5 and <0.67, respectively. The second was to discard potential outlier values. This was effected by taking a moving window average of the corrected and truncated loss of hybridization signal ratio values over nine consecutive overlapping probe positions. The highest and lowest values within a window are discarded before the average within the window is calculated from data from each strand.
When incorporating these correction factors, stable baseline values generally between 1.0 ± 0.15 can be obtained for ratios of reference and test perfect match probe hybridization signals interrogating nucleotide tracts that are identical between reference and test samples. Ideal heterozygous mutations produce peaks with a maximum magnitude of 1.5, reflecting the maximum value allowed by the signal-processing algorithm. Cross-hybridization of the mutant allele to the wild-type probe will reduce this ratio closer to 1.0. The width and shape of these peaks are primarily a function of the sequence change and probe length. In arrays consisting of probes 25 nucleotides in length, point mutations should produce a peak width of 25 nucleotides, whereas deletions and insertions of x bp should produce peaks ∼(25 + x) bp in width. Although these theoretical properties are not always consistent with empirical observations, because of sequence context effects, peaks of widths <21 bp are considered to result from background noise rather than the presence of a test sample sequence variation.
Two-Color Loss of Signal Analysis
Two-color loss of signal analysis displaying the 5932 G → T ATM nonsense mutation in sample GMO8388 is given in Figure 3. The corrected ratio of reference (green) to test (red) perfect match probe hybridization signals averaged for sense and antisense strand target data are plotted for allATM-coding nucleotide positions. Only a single peak is of sufficient magnitude and peak width in this analysis to indicate a sequence change. The beginning of an intronic loss of signal peak several nucleotides preceding exon 35 is not scored, as the only intronic sequences we are considering in these experiments are the immediate 3′ AG acceptor and 5′ GT donor dinucleotides. Perfect match probe tilings extend 10 bp into adjacent intronic sequence to allow accurate loss of signal analysis for these splice junction dinucleotides. Analyzing sequences further into intronic regions is more challenging because of the repetitive polypyrimidine and polypurine tracts that accompany splice junctions and may confound hybridization-based sequence analysis (Hacia et al. 1996). The increased level of polymorphism in these noncoding regions also increases the difficulty of hybridization-based sequence analysis and data interpretation.
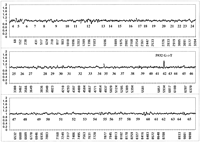
Two-color loss of signal assay for a nonsense mutation. Fluorescein-labeled (green) reference and biotinylated (red) test GMO8388 targets were cohybridized to the array. To correct for reproducible differences in the hybridization efficiencies of reference and test targets, the ratio of fluorescein to phycoerythrin signal at each wild-type position was normalized against ratios derived from 10 separate chip cohybridization experiments as described in Methods. Averaged sense and antisense strand ratios from GMO8388 are shown with the identity of each exon listed below the appropriate data points. The labeled peak at the mutated 5932 G/T position (a nonsense mutation) is present on both strands.
Blinded Analysis of ATM Samples
To ascertain the specificity and sensitivity of two-color hybridization analysis, blinded ATM mutational analysis was performed on genomic DNA samples. Twenty-three samples were screened for sequence variations in the ATM-coding region (including the 3′ AG and 5′ GT acceptor and donor sites for each exon). These included ATM homozygotes, compound heterozygotes, and heterozygous carriers of AT mutations.
Sequence variations were scored relying solely on loss of hybridization signal data. Averaged sense and antisense strand (composite) data were first considered. Composite loss of hybridization peaks of maximum height 1.3 or greater were flagged immediately for further dideoxy sequencing analysis, unless found in an exon of unacceptable baseline fluctuation. Individual sense and antisense strand loss of signal data were considered for averaged composite peaks of magnitudes between 1.2 and 1.3. Only if the sense and antisense strand loss of signal peaks were of similar shape (not necessarily of the same magnitude) and in phase with respect to one another were these sequences flagged for further analysis.
Of 18 distinct heterozygous changes confirmed to occur in theATM coding regions, 17 were detected using the ATMchip assay (Table 3). The 6015insC mutation was the only known heterozygous change not detected at least once in the assay. In addition, all eight known homozygous sequence changes were detected. Of the 18 distinct heterozygous sequence changes confirmed to occur in the assayed ATM sequence, 8 were present in the heterozygous state in more than one sample because of the relatedness of the individuals from which the genomic DNA was obtained. In two of these cases (8266 A → T and 1141insGACA) the mutation was identified correctly in one sample but not in another. Interestingly in both these cases, changes were missed owing to a failure of the chip designed to interrogate sense strand sequence to detect the change. This was mainly due to baseline noise masking weak two-color loss of hybridization peaks. For this ATM chip design it may be necessary to repeat hybridization experiments for samples with marginal loss of signal peaks occurring in regions of baseline fluctuation to increase assay sensitivity. Five false-positive sequence tracts were flagged for confirmatory dideoxy sequencing analysis based solely on the loss of signal assay. Of 1240 exons interrogated, seven (exons 5 and 12 twice, as well as exons 19, 26, and 27 each one time only) provided baseline values too variable for accurate loss of signal analysis, corresponding to >99.4% PCR amplification success rate.
Blinded Mutational Analysis of Genomic DNA from AT Patients and Carriers
Gain of Signal Analysis
In this ATM array design, gain of signal analysis is limited to the detection of single nucleotide substitutions. The simple base-calling algorithm described previously (Chee et al. 1996; Hacia et al. 1996) provides a useful metric by which to evaluate the specificity of these probes. When combining sense and antisense data derived from biotinylated reference target, 98.5% of the ATM-coding nucleotides are called correctly. Many of the miscalls occurred in regions of high A/T content where weak hybridization signals were presumably the result of the decreased stability of probe/target interactions. To increase the base-calling efficiency in these regions, target was transcribed in the presence of 5-methyluridine triphosphate in place of uridine triphosphate. Incorporation of 5-methyluridine into target has been shown previously to increase hybridization of A/U-rich regions of BRCA1 (Hacia et al. 1998c). In theory this is due to the increased thermodynamic stability of A/T relative to A/U base pairs (Saenger 1984). ATM targets containing 5-methyluridine substitutions gave a composite base-calling efficiency of 99.1%. Although not used in loss of signal analysis in this current study, it appears likely that the modified targets will prove useful in mutational analysis.
Nucleotide substitution probes showed promising sensitivity in detecting heterozygous single nucleotide substitutions on retrospective analysis of the blinded ATM sample study. Figure 4 displays a false colored image of the entire ATM chip fluorescent hybridization patterns produced from antisense test GMO8388 target. Many of the darker regions represent array probes interrogating several bases further into intronic regions of high A/T-content, and could be rescued partially by 5-methyluridine incorporation. The region of the array interrogating nucleotide position 5932 in samples GM11261 (5932 G/G) and GMO8388 (5932 G/T) is shown in Figure 4, b and c. The only significant difference between the hybridization patterns is the increased fluorescent signal at the 5932 T probe in sample GMO8388, indicating the presence of the 5932 T allele. The wild-type 5932 G allele is also detected in GMO8388. Therefore, chip hybridization analysis successfully genotypes GMO8388 as a 5932 G/T heterozygote.
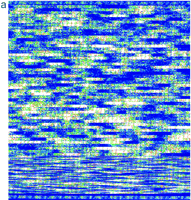
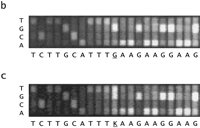
Chip image comparisons for a heterozygous nucleotide substitution. (a) False colored image of GMO8388 target hybridization to anATM sense array (1.2 × 1.2 cm) is given. Probes with greatest hybridization signal are given in white and red; those of lowest signal are given in blue and black. A magnified view of the probe sets (50 μm feature size) interrogating nucleotide position 5932 in samples GM11261 (5932 G/G) and GMO8388 (5932 G/T) is shown inb and c, respectively. The identity of the interrogated ATM cDNA nucleotide positions 5922–5942 is given with IUPAC ambiguity codes and position 5932 is underlined.
Although gain of signal analysis was not used directly in mutational analysis because of the absence of probes interrogating for insertions and deletions, single nucleotide substitution probes were evaluated for their sensitivity toward detecting likely mutations (Table 3) or neutral variants that do not affect the function of the ATMgene (Table 4). Applying the simple base-calling algorithm described for the iterative multiexon PCR strategy, we assayed whether the gain of signal probes could be used to detect single nucleotide changes. In 5 of 17 heterozygous single nucleotide substitutions, the gain of signal probes for the mutant allele did not show hybridization signal for either strand within 1.2-fold intensity of the wild-type probe. The fact that these base substitutions could not be detected demonstrates that single nucleotide substitution probes lack the predictable hybridization properties necessary for high sensitivity mutation screening purposes. On the other hand, the loss of hybridization signal assay clearly identified these substitutions missed by the gain of hybridization signal assay. Nevertheless, in one case the gain of hybridization signal probes could be used to supplement the loss of signal analysis. This involved the identification of the 8266 A → T base substitution in GM03188A that was missed by the loss of signal assay attributable to unstable baseline values in that particular analysis. Interestingly this mutation was detected in sample GMO3189C using the loss of hybridization signal assay.
Confirmed Newly Identified Probable Neutral Sequence Variations Detected by DNA Chip Analysis
DISCUSSION
Of the 7 carriers and 15 affecteds in the blinded ATMsequence analysis, there is a theoretical total of 37 mutantATM alleles, assuming one mutant allele per carrier and two per patient sample (Table 3). However, sample GMO2782B is reported to contain three separate cDNA mutations (Wright et al. 1996), which could be the result of three separate mutations (which would bring the total up to 38 mutant alleles) or complex alternative splicing events. We choose to use the value of 38 mutant alleles of which 28 (found either in the homozygous or heterozygous states) were previously known for these samples (Gilad et al. 1996a,b; Wright et al. 1996). An additional seven alleles likely to cause the ATM phenotype were discovered using the ATM DNA chip assay (Tables 3 and 4). Three of these involved heterozygous base substitutions, 8585TG → AA and 9022 C → T (found twice), leading to nonconservative amino acid changes. In fact, the 8585TG → AA sequence variation alters two resides in the highly conserved PI 3-kinase domain. Three other nucleotide substitutions, 4852 C → T and 5932 G → T (found twice), involved nonsense mutations. Two other changes, 1141insGACA (found twice), involved a 4-bp insertion that was detected in sample GMO3189C but not in GMO3187A, the father of GMO3189C. The mutant allele in sample GMO3187A was not known previously; however, because sample GMO3189C was found to contain the 1141insGACA allele, GMO3187A was subject to dideoxy sequencing analysis and found to be a carrier.
The DNA chip-based hybridization analysis helped to elucidate the genomic basis for two distinct mutations previously described in patient cDNAs. Loss of hybridization signal analysis indicated a possible 3′ splice junction sequence change in sample GMO2782B reported to contain the 2251del19 cDNA mutation (Wright et al. 1996). Dideoxy sequencing analysis of subcloned PCR products indicated the presence of the heterozygous 2251-1 G → A genomic DNA base substitution, which alters the 3′ acceptor splice site. Furthermore, the genomic DNA change responsible for the complete lack of exon 32 in sample AT29RM ATM cDNA has not been reported (Gilad et al. 1996b). Exon 32 from sample AT29RM failed to amplify using standard primer sets, as first indicated by lack of hybridization signal at exon 32 specific probes and confirmed by the absence of PCR product when this exon was amplified and analyzed individually by agarose gel electrophoresis. Another intronic primer pair (31A 5′-TTTCAGAGTAATTTTCCAGAAC-3′ and 31B 5′-CACTCAAATCCTTCTAACAATACT-3′) was used to amplify exon 32 from sample AT29RM. The PCR product was subcloned and individual colonies sequenced to find that it contained a homozygous 10-bp deletion (4611delG + 9) that extends 9 bases into the intronic region and alters the 5′ donor splice junction sequence.
In addition to finding these new mutations and sequence variations, the DNA chip assay helped to correct other genotyping assignments as well. For example, the 7517del4 frameshift mutation found in AT patient sample AT22RM, previously reported as being in the homozygous state (Gilad et al. 1996b), was found to be in the heterozygous state by dideoxy sequencing analysis. The initial assignment was questioned because the previously unreported 4852C → T nonsense mutation was found in the same sample. The researchers indicated that the homozygote genotype could be a misassignment as it was based on RNA sequence analysis where message derived from one mutant allele in a compound heterozygote could be unstable and thus not scored (Gilad et al. 1996b). This highlights an inherent drawback in using RNA for mutational analysis as sequence changes can be missed if they decrease sufficiently the lifetime of the mutant RNA.
Currently 36 of 38 possible mutant alleles in these samples are known with the remaining 2 alleles (one from the carrier GMO2781B, with D1853V being a probable polymorphism as it is not found in the offspring AT patient GMO2782B, and one from the affected GM11254) as yet undiscovered. These mutations could be the result of the lack of sensitivity of both the DNA chip and alternative assays toward detecting these mutations. However, it is also possible that otherATM mutations may reside further in intronic or the promoter region. In addition, large-scale genomic deletions could be present that would confound any PCR-based assay. Of the 36 known ATMmutant alleles, 34 occur within the genomic regions interrogated by the DNA chip-based assay. A total of 31 of 34 of these mutant alleles, counting each as many times as it is found in the sample set, were detected using the ATM chip to give an overall 91% sensitivity.
A number of factors may confound hybridization analysis and lead to false-positive and false-negative mutation detection assignments. Length differences among reference and test RNA species as well as internal dye or hapten incorporation may affect assay sensitivity and specificity. Hybridization will be affected by intra- or intermolecular structures caused by test target sequence variations in addition to the thermodynamic properties of mismatched target/probe duplex formation. Repetitive nucleotide tracts and duplications have been recognized as posing significant challenges to hybridization based assays (Hacia et al. 1996). The potential for cross-hybridization attributable to the formation of stable partial target/probe duplexes caused by probe slippage is increased in these sequence contexts. In these cases, the variant target sequence may still bind to wild-type probes and minimize the sensitivity of the loss of signal assay. Increasingly sophisticated hybridization data analysis algorithms will have to be developed to compensate for potential losses of mutation detection specificity in these sequence contexts.
The false-positive mutation detection rate (five identified in >200 kb scanned) in the blinded ATM mutational analysis study was higher than that found in a previous nonblinded two-color DNA chip-based analysis of BRCA1 exon 11 samples (none identified in >120 kb scanned). This is primarily attributable to the lower number of mutation-specific probes per target nucleotide in theATM relative to BRCA1 exon 11 arrays, where single base-pair insertion as well as (1–5)-bp deletion gain of signal probes were represented. It was not possible in the absence of insertion/deletion probes to eliminate ATM loss of signal peaks with marginal widths from consideration, because of a lack of confirmatory evidence from gain of signal probes specifically interrogating these sequence changes. Nevertheless, such false positives carry relative little risk, as their number was relatively few and confirmatory dideoxy sequencing was always done before calling a mutation.
We have demonstrated the efficient use of oligonucleotide arrays to screen for heterozygous and homozygous sequence variations in the large multiexon ATM gene. The ability of hybridization-based assays to analyze amplicons of identical sizes simplifies multiexon PCR amplification reactions. Although this limits the resolution of independent measurement of target quality, such as simple agarose gel electrophoresis analysis of PCR reactions, highly robust multiexon PCR reactions can, nonetheless, be developed in a rapid time frame (Wang et al. 1998). Encouraging results obtained from blinded ATMmutational analysis and advances in high-density oligonucleotide array manufacturing has now prompted the design of a second generation pair of ATM chips, designed to interrogate sense and antisense strand sequence, collectively containing >260,000 probes. FutureATM array designs with greater perfect match probe redundancy and additional mutation-specific oligonucleotide probes, such as those representing every possible single nucleotide insertion and small deletions should enhance the reproducibility, sensitivity, and specificity of the DNA chip-based assay. Furthermore, including additional wild-type probes based on common ATM polymorphic allele sequences will optimize mutation detection in regions surrounding these nucleotide changes. These chips could be used to help resolve the controversy surrounding cancer risks to ATMcarriers, as well as in screening for sequence variations in theATM gene for several AT- like diseases (Gilad et al. 1998a). Because DNA chip technology has the potential to analyze virtually any gene for heterozygous sequence variations, the hybridization-based strategies used in this study should be applicable to mutational analysis in many other systems.
METHODS
PCR From Genomic DNA and RNA Target Preparation
PCR reactions were performed on genomic DNA isolated from human AT fibroblast or lymphoblast cell lines, obtained from the Coriell Institute or as a gift from Yossi Shiloh (Tel Aviv University, Israel), using the AmpliTaq Gold (Perkin-Elmer) PCR kit and the manufacturer’s recommended protocols. The primer pairs used are listed in Table 1 and contain T3 (5′-ATTAACCCTACTAAAGGA-3′) and T7 (5′-TAATACGACTCACTATAGGGA-3′) promoter sequences for forward and reverse primers, respectively. Genomic DNA from reference and test samples were subject to the multiexon PCR reactions listed in Table 2. Reference and test sample multiexon PCR reaction pools were combined separately and subjected to in vitro transcription reactions. These reactions were performed in 10-μl reaction volumes using T3 RNA polymerase transcription buffer (Promega), 0.7 mm of ATP, CTP, GTP, and UTP [or 5-methyl-UTP (Hacia et al. 1998c)], 10 mm DTT, 0.7 mm fluorescein–12-UTP or 0.15 mm biotin-16-UTP (Boehringer Mannheim) for reference and test samples, respectively, and 10 units of T3 or T7 RNA polymerase as indicated. To generate and optimize multiexon PCR reactions, an iterative hybridization-based strategy was used (Fig. 3). Target was generated from multiexon PCR reactions through in vitro transcription reactions using biotin–UTP.
Reference and test sample in vitro transcription products were diluted into a 25-μl solution of 30 mm MgCl2 and incubated at 94°C for 15 min to fragment targets (Hacia et al. 1996). Cofragmented targets were diluted 1:100 into a 300-μl volume of hybridization buffer [3 m TMAC-Cl (tetramethylammonium chloride), 1× TE (pH 7.4), 0.001% Triton X-100, 1 nm 5′-fluorescein-labeled control oligonucleotide 5′-CGGTAGCATCTTGAC-3′ (designed to hybridize to specific array probes to aid in image alignment)] and passed through a 0.22-μm syringe filter.
Chip Hybridization and Data Analysis
Target was hybridized to either the sense or antisense ATMarrays in a 250-μl volume for 4 hr at 42°C. The chip surface was washed with 10 ml of wash buffer (6× SSPE, 0.001% Triton X-100) and stained with phycoerythrin-streptavidin conjugate (Molecular Probes) (2 μg/ml in wash buffer) for 5 min at room temperature. The chip was washed with 5 ml of wash buffer and data were accumulated using a scanning confocal microscope equipped with a 488-nm argon laser [GeneChip Scanner (Affymetrix)]. After passing through 515- to 545-nm (green) band-pass and 560-nm (red) long-pass emission filters, fluorescent hybridization signals were detected using a photomultiplier tube.
GeneChip Software (Affymetrix) was used to produce digitized images of fluorescent target hybridized to arrays by converting photomultiplier tube output into spatially addressed pixel values. Probe signal intensities are calculated from the mean of the non-outlier photon counts for each feature (i.e., per probe). The contributions of reference and test targets to each probe hybridization signal were extracted from each set of green reference and red test images using custom software.
Sequence Normalization Algorithms
To correct for reproducible fluctuations in the ratio of test and reference perfect match probe hybridization signals present after normalization against data generated from separate two-color cohybridization experiments, we calculated 79 quantities for each perfect match probe that form the basis for a first round of signal normalization. Of these, 62 are specific for probes in a given exon (representing individual multiplicative correction factors for each of the 62 ATM coding exons) and are set to a value of one for the exon containing a probe and to zero for all other exons. Two additional quantities reflect multiplicative intra- and intermolecular probe structure normalization scores. These attempt to normalize for inter- and intramolecular array probe hybridization and secondary structures that may also contribute to fluctuations in perfect match probe signal ratios.
We developed an algorithm to predict the potential for duplex formation between adjacent probes (intermolecular structure) within a feature in the array (each feature contains ∼107 probes of identical sequence) as well as within a given probe (intramolecular structure). The inter- and intramolecular probe structure predictions differ in that complementary nucleotides less than five positions apart are only considered in intramolecular probe structure analysis. This reflects the need for a loop of at least four or more residues to be present in a stable intramolecular hairpin structure (Zucker 1994).
The intermolecular probe structure normalization score is written as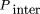
 where the sum is over the positions in the probe sequence (positions 1 to 25 for a 25-mer probe);F(Si, Sj
) was set to 3 if theith and jth positions form a GC base pair, 2 if they form an AT base pair, and 0 otherwise. These arbitrary values only serve as rough
estimates of base-pair stability; more sophisticated algorithms using more detailed thermodynamic relationships in theory
could be used to further refine this correction factor (Zucker 1994).
where the sum is over the positions in the probe sequence (positions 1 to 25 for a 25-mer probe);F(Si, Sj
) was set to 3 if theith and jth positions form a GC base pair, 2 if they form an AT base pair, and 0 otherwise. These arbitrary values only serve as rough
estimates of base-pair stability; more sophisticated algorithms using more detailed thermodynamic relationships in theory
could be used to further refine this correction factor (Zucker 1994).
The intramolecular probe structure normalization score is similar: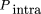
 The 15 quantities related to perfect match probe ratio correction are derived from the probe sequence composition. We address
this issue by systematically searching for patterns in baseline fluctuations that appear to act as a function of nucleotide
identity at a particular position. For example, consider a hypothetical situation where the average of the perfect match probe
ratios is one for all probes except those having an A in the fifth position where the average ratio is three. By dividing
the perfect match probe signal ratios generated these probes by three, one can correct for systematic variation in these intensity
ratios. A more complete way of formulating the multiplicative correction factor is to take one type of base (e.g., T) as a
reference, and have a correction factor for each of the other three bases at each probe position. For a 25-nucleotide probe,
this results in a scheme that involves 75 variables (three correction factor for each of the 25 nucleotides in the probe).
To simplify this process, we have chosen a function with five terms (a fourth degree polynomial function of nucleotide position
within a probe sequence fitted to estimate the function of 75 variables) to describe and correct for probe sequence composition
effects. For example, if T is (arbitrarily) chosen to be the reference base type, we calculate CX, j = Σi = 1,Nδ[X,seq(i)]L
(j − 1) (i/N), where X is A, C, or G; j is 1, 2, 3, 4, or 5;N is the number of bases in the probe sequence; δ(X,Y) is one if Y is the same asX and zero otherwise; seq(i) is the identity of theith base in the probe; and Lk
(x) is the kth Legendre polynomial. The first five Legendre polynomials are given by L0(x) = 1;L1(x) = x;L2(x) = 3/2x2 − 1/2;L3(x) = 5/2x3 − 3/2x; andL4(x) = 3/8x4 + 35/8x2 − 15/4.
The 15 quantities related to perfect match probe ratio correction are derived from the probe sequence composition. We address
this issue by systematically searching for patterns in baseline fluctuations that appear to act as a function of nucleotide
identity at a particular position. For example, consider a hypothetical situation where the average of the perfect match probe
ratios is one for all probes except those having an A in the fifth position where the average ratio is three. By dividing
the perfect match probe signal ratios generated these probes by three, one can correct for systematic variation in these intensity
ratios. A more complete way of formulating the multiplicative correction factor is to take one type of base (e.g., T) as a
reference, and have a correction factor for each of the other three bases at each probe position. For a 25-nucleotide probe,
this results in a scheme that involves 75 variables (three correction factor for each of the 25 nucleotides in the probe).
To simplify this process, we have chosen a function with five terms (a fourth degree polynomial function of nucleotide position
within a probe sequence fitted to estimate the function of 75 variables) to describe and correct for probe sequence composition
effects. For example, if T is (arbitrarily) chosen to be the reference base type, we calculate CX, j = Σi = 1,Nδ[X,seq(i)]L
(j − 1) (i/N), where X is A, C, or G; j is 1, 2, 3, 4, or 5;N is the number of bases in the probe sequence; δ(X,Y) is one if Y is the same asX and zero otherwise; seq(i) is the identity of theith base in the probe; and Lk
(x) is the kth Legendre polynomial. The first five Legendre polynomials are given by L0(x) = 1;L1(x) = x;L2(x) = 3/2x2 − 1/2;L3(x) = 5/2x3 − 3/2x; andL4(x) = 3/8x4 + 35/8x2 − 15/4.
If we group these 79 quantities together into a 79-dimensional vector, denoted Ci (where i runs from 1 to 79), then our correction scheme starts with finding the best least-squares fit to the logs of the measured perfect match intensity ratios. We need to find the values of the 79 parameters Xj that minimize the quantity Σprobes (logR measured − logR fit)2 = Σprobes (logR measured − {Σj = 1, 79 CjXj })2, where the sum over probes is taken over all perfect match probes;R measured is the measured value of the intensity ratio of ratios; and R fit is a fit to the measured ratios. The Cj values are different for each probe sequence; the Xj values are the same for all probes. The least-squares parameters Xj are found by solving the normal equations by Cholesky decomposition (Branham 1990). Then the corrected ratio for each probe, R corrected, is given by the residual to the fit: log R corrected = logR measured − log R fit.
After this signal normalization process, reference/test sample perfect match probe hybridization signal ratios are truncated to values between 1.5 and 0.67, as described in the text. Finally, corrected test/reference perfect match probe hybridization signal ratios were averaged against those generated from individual comparisons to data sets generated from 10 separate ATM chip hybridization experiments. This provides a final set of multiplicative correction factors to minimize systematic fluctuations in hybridization signal ratios. Afterward the reference/test perfect match probe signal ratios are plotted against their respective nucleotide positions using Microsoft Excel 7.0. Loss of hybridization peaks are scored based on peak height and width as described in the text.
Dideoxy Sequencing Analysis
PCR primer pairs (Table 1) containing M13 forward (5′-GTTTTCCCAGTCACGACG-3′) and reverse (5′-AGGAAACAGCTATGACCAT-3′) sequences were designed to amplify each of the 62 ATM coding exons. Individual exons were amplified using the AmpliTaq Gold System (Perkin Elmer) using the manufacturer’s recommended protocol. Dye primer dideoxy sequencing reactions were performed using the DYEnamic Energy Transfer Dye Primer Sequencing Kit (Amersham Life Science) with the suggested protocol and supplied M13 forward or M13 reverse primers.
In cases where the dye primer sequencing strategy did not definitively indicate the presence of a heterozygous sequence variation in a test sample, a subcloning strategy was used. PCR product of interest was subcloned using Zero Blunt Cloning Kit (Invitrogen) and inserts from individual colonies were sequenced using dye terminator chemistry.
Acknowledgments
We thank Yossi Shiloh (Tel Aviv University, Israel) for the generous gift of human AT fibroblast and lymphoblast cell lines. We thank Larry Brody at the National Institutes of Health and Gil Chu, Yvonne Thorstenson, and Virginia Goss at Stanford University for comments and suggestions on the manuscript. Partial support for this work was provided by 5POLHGO1323-03 (S.P.A.F.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL fc23a{at}nih.gov; FAX (301) 402-0837.
-
- Received August 26, 1998.
- Accepted November 9, 1998.
- Cold Spring Harbor Laboratory Press








