
A Comparative Analysis of ABC Transporters in Complete Microbial Genomes
Abstract
The ABC transporter is a major class of cellular translocation machinery in all bacterial species encoded in the largest set of paralogous genes. The operon structure is frequently found for the genes of three molecular components: the ATP-binding protein, the membrane protein, and the substrate-binding protein. Here, we developed an “ortholog group table” by comparison and classification of known and putative ABC transporters in the complete genomes of seven microorganisms. Our procedure was to first search and classify the most conserved ATP-binding protein components by the sequence similarity and then to classify the entire transporter units by examining the similarity of the other components and the conservation of the operon structure. The resulting 25 ortholog groups of ABC transporters were well correlated with known functions. Through the analysis, we could assign substrate specificity to hypothetical transporters, predict additional transporter operons, and identify novel types of putative transporters. The ortholog group table was also used as a reference data set for functional assignment in four additional genomes. In general, the ABC transporter operons were strongly conserved despite the extensive shuffling of gene locations in bacterial evolution. InSynechocystis, however, the tendency of forming operons was clearly diminished. Our result suggests that the ancestral ABC transporter operons may have arisen early in evolution before the speciation of bacteria and archaea.
In recent years the complete genome sequences of various organisms from the three domains of life are rapidly accumulating. It is now possible to attempt to reconstruct and analyze a complete set of biochemical reaction pathways that an organism adopts (Bono et al. 1998), especially on the transport, synthesis, and degradation of specific chemical compounds. Through comparative studies, the organisms’ transport and metabolic capabilities can be correlated to the evolutionary relationships of different organisms and to the environments in which they inhabit. This would eventually lead to both a genetic description of life and a chemical description of life. However, a major difficulty in attempting such a synthetic approach is the lack of appropriate function assignments in the gene catalogs produced by the genome sequencing projects. Different investigators use various criteria for interpretation of sequence similarity, which causes widely varying assignments and leaves many genes unassigned. In the Kyoto Encyclopedia of Genes and Genomes (KEGG) project (Kanehisa 1997a,b), we wish to provide a standard set of assignments for all known complete genomes, which can then be used as a reference for assignment in newly sequenced genomes. Here, we report our results on developing such a reference set for ABC transporters.
The ABC (ATP binding cassette) transporter is one of the active transport systems of the cell, which is widespread in archaea, eubacteria, and eukaryotes (Higgins 1992). It is also known as the periplasmic binding protein-dependent transport system in Gram-negative bacteria and the binding-lipoprotein-dependent transport system in Gram-positive bacteria. The transporter shows a common global organization with three types of molecular components. Typically, it consists of two integral membrane proteins (permeases) each having six transmembrane segments, two peripheral membrane proteins that bind and hydrolyze ATP, and a periplasmic (or lipoprotein) substrate-binding protein. The ATP-binding protein component is the most conserved, the membrane protein component is somewhat less conserved, and the substrate-binding protein component is most divergent (Tam and Saier 1993; Saurin and Dassa 1994) in terms of the sequence similarity. The ABC transporters form the largest group of paralogous genes in bacterial and archaeal genomes (Tatusov et al. 1996), and the genes for the three components frequently form an operon (Higgins 1992).
In the present analysis, we first search and compare ABC transporters in the complete genomes of seven microorganisms: three from Gram-negative bacteria, two from Gram-positive bacteria, one from cyanobacteria, and one from archaea. Experimentally determined transporters and putative ones deduced by similarity are classified into groups, both as orthologs and paralogs, according to the sequence similarity and the conservation of operon structure. Based on the ortholog/paralog relationship, we assign substrate specificity of hypothetical transporters and predict novel transporters in the seven organisms. The assignment is summarized in the “ortholog group table,” which is then used as reference data set to make functional calling in the four additional genomes. The up-to-date version of the ortholog group table of ABC transporters is made available publicly through KEGG (http://www.genome.ad.jp/kegg/).
RESULTS
Clustering of ATP-Binding Proteins
Because the ATP-binding protein is the most conserved component in the ABC transporter unit, our strategy was to first identify ortholog/paralog relations of the ATP-binding proteins among the seven organisms, which was followed by the assignments of the membrane protein permeases and the substrate-binding proteins around the ATP-binding protein gene in the genome (see Methods). The number of ATP-binding protein paralogs in the seven organisms is summarized in the top block of Table 1. It is ∼2% of the entire proteins, forming the largest paralog group in each genome.
Summary of ABC-Type ATP-Binding Proteins Identified in the Seven Organisms
Figure 1 shows the result of clustering the ATP-binding proteins in Escherichia coli, where only the basal clusters that satisfy the clustering criteria for both the single- and complete-linkage analysis are shown. The number in the box represents the ortholog group as described below. The results by the two clustering methods coincided well except group 4-1 (peptide transporters), in which multiple clusters were placed according to the complete linkage cluster analysis. The clustering was performed independently for each organism, and the number of basal clusters is summarized in the middle block of Table 1.
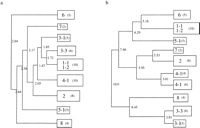
A schematic representation of the results of the hierarchical cluster analysis for the ATP-binding proteins in E. coli. For convenience, only the basal clusters are depicted according to (a) the single-linkage cluster analysis and (b) the complete-linkage cluster analysis. The distances are denoted at their branchpoint. Each box represents a group with the group number (Table2) and the number of sequences in parentheses.
Ortholog Groups
After identifying pairs of orthologous proteins in all possible pairwise comparisons among the seven organisms (see Methods), we used this information, together with the information of paralogous protein clusters in each organism, to define ortholog groups of ATP-binding proteins. The results are summarized in Table 2, in which the orthologs were classified into 13 groups and, when the similarity of membrane proteins was also considered, into 25 groups.
The Summary of the Ortholog Groups Showing the Number of ATP-Binding Proteins, the Operon Structure, and Known Functions
This grouping was generally in agreement with the grouping of known substrate specificity (see Table 2), as well as with the conservation of operon structures. Figure 2 illustrates some of the operon structures in group 1-1, and Table 3 is the corresponding ortholog group table. Thus, Table 2 represents not only the ortholog grouping of ATP-binding proteins but also the ortholog grouping of ABC transporters. Note, however, that the number of ATP-binding proteins shown in Table 2 is generally greater than the number of ABC transporters because two different genes often code for the two ATP-binding protein components. Table 2 also contains ATP-binding proteins that are not related to the standard ABC transport system (see below), which are indicated by the numbers in parentheses.
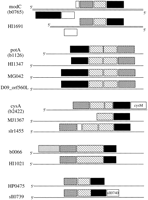
The gene organization in some of the operons of group 1-1 showing conserved patterns among the ortholog group. The ATP-binding proteins are represented by solid boxes, the integral membrane proteins by hatched boxes, and the substrate-binding proteins by dotted boxes. The last example is sll0739 in Synechocystis, is a fused gene for the permease and the ATP-binding protein.
An Example of the Ortholog Group Table for the ABC Transporter
Operon Structure
The shading in Table 2 represents the tendency that the transporter is formed by a set of genes in an operon. There is a striking feature in Synechocystis; namely, it significantly lacks the operon structure in comparison to other bacterial genomes. This is also shown by the unusually high number of orphan ATP-binding proteins in the bottom block of Table 1. Whenever the operon is observed, the gene order, as well as the number of genes, in the operon tends to be conserved in each group. For example, the operon of group 1-1 often consists of one substrate-binding protein, two permeases (sometimes fused), and one ATP-binding protein with a single domain (probably forming a homodimer), which are ordered in the 5′ → 3′ direction (Fig. 2). In contrast, a common gene arrangement in group 2 is a substrate-binding protein, an ATP-binding protein that is a double-sized protein with two domains, and a permease in the 5′ → 3′ direction.
The operons of groups 3-5 (branched-chain amino acids) and 4-1 (peptides) tend to contain a full set of five genes, that is, duplicated genes for both the permeases and the ATP-binding proteins. In contrast, the operons of group 3-3 (polar amino acids) usually contains a single gene for the single-domain ATP-binding protein. It is interesting to note that in these three groups, as well as in group 1-1, there are cases of duplicated (extra) substrate-binding proteins, for example, HisJ (b2309) and ArgT (b2310) in group 3-3 and LivK (b3458) and LivJ (b3460) in group 3-5 of E. coli. Thus, a general tendency is observed where the increased diversity and specificity of transporter functions are apparently correlated with the increased numbers of both paralogous operons in the genome and paralogous genes in the operon.
Substrate Specificity in Known Groups
Group 1-1, which is widespread in all organisms, is a functionally divergent set of transporters despite its conserved sequence similarity extending not only to membrane proteins but also to substrate-binding proteins. A case in point is the significant sequence similarity (∼50% identity) and functional exchangeability of the ATP-binding proteins MalK (encoded in b4035) and UgpC (b3450) in E. coli(Hekstra and Tommassen 1993) that are involved in different transports of maltose/maltodextrin and sn-glycerol-3-phosphate, respectively. Group 1-2 is somewhat less conserved, lacking the similarity of substrate-binding proteins. Two members (b0366 and b0933) of this group in E. coli are involved in sulfate starvation response (van der Ploeg et al. 1996).
Group 2 is a relatively uniform set of simple-sugar transporters. It is not observed in the autotrophs of Synechocystis andMethanococcus jannaschii (Table 2), but there are eight paralogs in E. coli including the four experimentally determined operons, rbs, mgl, xyl, andara (one of the other four operons contained two ATP-binding protein genes). In the Gram-positive bacteria of Mycoplasma genitalium and Mycoplasma pneumoniae, the operon does not contain a substrate-binding protein but highly diverged and duplicated permeases. The carboxy-terminal domain of one of them does not exhibit similarity with any protein, and it may be involved in substrate binding.
Group 3-3 corresponds to the polar amino acid transporters. In E. coli, there are four experimentally identified operons,gln, glt, art, and his, but we could identify two more putative operons in this group. Group 3-4 is only found in E. coli for the alkyl-phosphonate transport. Group 3-5 is the branched-chain amino acid transporter, for whichE. coli apparently has only one operon with duplicated substrate-binding proteins. Group 3-7 is the phosphate transport system that is observed in most organisms with conserved sequence similarity and operon structure.
Group 4-1 is the peptide transport system that is also highly duplicated in E. coli; there are three experimentally identified operons, dpp, opp, and sap, but we could identify three more. The nik operon of E. coli that encodes the transporter for nickel uptake can also be classified into group 4-1. It is intriguing that only the nickel transporter does not belong to the metal transporter of group 5, but a similar result was also obtained by Kuan et al. (1995).
Groups 5-1, 5-2, and 5-3 are involved in metal uptakes: iron, manganese, and cobalt, respectively. Group 5-1 contains the E. coli operon for vitamin B12 transport. E. coliapparently lacks the cobalt transporter (group 5-3) and the ability to synthesize major intermediates in cobalamin (vitamin B12) biosynthesis (Raux et al. 1996), which must be complemented by the vitamin B12 transporter.
Group 6 contains the family of ATP-dependent translocators that includes E. coli hemolysin B and its mammalian homolog, the mammalian multidrug resistance (MDR) proteins. Most of the ATP-binding proteins of this group have the length of >550 amino acids, and all contain membrane protein components that are fused at the amino-terminal region. However, the membrane protein portions do not exhibit any significant sequence similarity with other groups. There is no substrate-binding protein in this group.
Group 7, as well as probably, group 11, corresponds to the ABC-2 type transporters that include the capsular polysaccharide export system in Gram-negative bacteria (Reizer et al. 1992) and NatB for sodium export in Bacillus subtilis (Cheng et al. 1997). There is no substrate-binding protein in this group.
Functional Predictions in Other Groups
The function for the rest of the groups cannot be well established, but some predictions can be made. Group 3-1 contains the largest number of ATP-binding proteins among the unknowns and is found in all the seven organisms. This group entirely lacks the substrate-binding protein component. Most of the membrane proteins in this group possess conserved regions as shown in Figure 3, although the similarity is somewhat weak. The membrane protein appears to contain four transmembrane segments, rather than six. Conserved sequence patterns are observed in the first transmembrane segment and in the putative loop between the second and third transmembrane segments and at the carboxy-terminal end. An E. coli membrane protein, FtsX (b3462), exhibits a weak similarity with the other membrane proteins in group 3-1 and shares the putative motif G-X[9]-F-X[10]-G in the loop. FtsX and FtsE (b3463) are the members of a cell division operon (Gill et al. 1986). Thus, we suspect the involvement of group 3-1 with cell division.
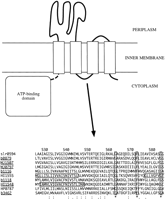
The predicted architecture of an E. coli protein in group 3-1, b0879, which consists of the ATP-binding domain and the membrane protein domain. The G-X[9]-F-X[10]-G motif at a putative loop region has been identified by the multiple alignment of group 3-1 membrane proteins; the protein names in this group are underlined, and the boxes in the sequence data represent putative transmembrane regions derived from the SWISS-PROT entries, P44250 (HI1548) and P44252(HI1555). The alignment was obtained by CLUSTAL W (Thompson et al. 1994). The numbers above the alignment represent the residue positions of b0879.
Groups 1-3, 1-4, and 3-2 seem to conform to the gene organization of ABC transporters, but we could not find any clue to their functions. Group 3-6 may not be an ABC transporter for the ATP-binding protein appears to belong to the operon containing RNA polymerase σ54 factor and/or phosphotransferase system (PTS) system-related proteins. Group 4-2 is only found in E. coliand consists of two ATP-binding proteins that are among thephn operon. Because there is already the phosphonate transport system of group 3-4 in this operon, these ATP-binding proteins may be alternatives for the phosphonate transport or have different functions.
In group 10, the putative operon in E. coli contains eight genes that are involved in cytochrome c maturation (Thony-Meyer et al. 1995). Among them, the ATP-binding protein, CcmA (b2201), and the membrane proteins, CcmB (b2200), CcmC (b2199), CcmD (b2198), may act as a heme exporter. Groups 8, 9, 12, and 13 are not likely to belong to the ABC transport system.
Prediction of Putative Flagellar Operon in E. coli
In addition to these predictions, we have made the following observation: There is a report of fliAZY operon (b1922–b1920) that contains the σ-factor fliA (b1922) involved in the transcription of the class III flagella genes in E. coli(Mytelka and Chamberlin 1996). We predict that the adjacent ATP-binding protein (b1917) in group 3-3 would constitute an ABC transporter, together with the membrane protein (b1918) and the substrate-binding protein FliY (b1920). The three genes may form a cluster with another gene in between that is a possible ACC deaminase gene (b1919) but whose role on the transcription regulation is unclear. The three orthologous transporter genes are conserved in Haemophilus influenzae(HI1080, HI1079, and HI1078). In Helicobacter pylori, two orthologous genes without the ATP-binding protein are also conserved as a gene string (HP0940 and HP0939). Therefore, we suggest thatfliAZY operon is in fact longer, containing six genes (b1922 to b1917), and the interacting partners of fliY (b1920) would be the membrane protein (b1918) and the ATP-binding protein (b1917).
Functional Assignment in Additional Genomes
After completion of the initial grouping of the seven organisms, the complete genomes of four additional organisms became available. Among them, we repeated the same procedure for B. subtilis, performing the clustering of ATP-binding proteins and identifying orthologs of ATP-binding proteins. The result was in good agreement with the initial grouping. For the other three organisms,Methanobacterium thermoautotrophicum, Archaeoglobus fulgidus, and Borrelia burgdorferi, a systematic search of ATP-binding proteins was not performed, but only the putative operons of ABC transporters were analyzed by comparing with the ortholog/paralog group table that includes B. subtilis, using the tool that is made available in KEGG (http://www.genome.ad.jp/kegg-bin/srch_orth_html).
Table 4 is a summary of predicted ABC transporters in the 11 organisms, which was taken from the KEGG ortholog group table as of June 17, 1998. The KEGG table is represented in a form shown in Table 3, and it is continuously updated as more predictions are made and new genomes are added. Table 4 indicates general tendencies of different repertoires of ABC transporters in different genomes. For example, E. coli, B. subtilis, and H. influenzae have similar sets of transporters, butSynechocystis has a different set that seems somewhat like that of archaea. The parasitic bacteria, M. genitalium,M. pneumoniae, B. burgdorferi, and H. pylori, apparently lack many transporters, which is consistent with our previous observation that they lack entirely or partially the biosynthetic pathways for the 20 amino acids (Bono et al. 1998).
The Number of Predicted ABC Transporters in the Complete Genomes
DISCUSSION
Unique Features in Synechocystis
Synechocystis has the lowest degree of operon structures among the complete genomes thus far sequenced, not only for the ABC transporters but also for the regulation of metabolic pathways (H. Ogata, W. Fujibuchi, and M. Kanehisa, in prep.). There are two possibilities for the implication of disrupted ABC transporter operons in Synechocystis. One is that the orphan ATP-binding proteins may form a novel type of operon containing different components, although we could not identify any striking conservation among gene strings except the unique protein mentioned below. The other possibility is that separately encoded components of an ABC transporter may form a regulon or are under a novel type of gene expression regulation. However, we have not been able to detect any regularities between the ATP-binding proteins and the putative partners identified by searching the entire genome.
When we searched conserved genes around the orphan ATP-binding proteins in Synechocystis, we found four proteins that were conserved in the putative operons of different groups: sll0740 in group 1-1, slr0981 in group 11, sll0488 in group 7, andsll0913 in group 8. There were 16 additional proteins with sequence similarity to these 4 proteins in the entire genome. However, we do not yet know the function of this uniquely conserved set of proteins.
Another characteristic feature of the ABC transporter operons inSynechocystis is that there are more fused proteins compared with other organisms (Fig. 2; Table 3). It was reported that no bacterial substrate-binding proteins were found to be fused to the other components of the ABC transporter (Tam and Saier 1993), but inSynechocystis, there are substrate-binding proteins that are apparently fused with permease (sll1270 in group 3-3) or with ATP-binding proteins (sll1452 and slr0043 in group 1-2).
Comparison with Other Classifications
We used the most conserved ATP-binding protein components for classifying ABC transporters. Although our method is based on simple clustering using similarity scores, our result correlated well with the phylogenetic tree analysis of ATP-binding proteins by Kuan et al. (1995). There are previous reports on other classifications, two based on the substrate-binding protein component and one based on the membrane protein component. Tam and Saier (1993) classified substrate-binding proteins according to the sequence similarity and obtained eight clusters. Saurin and Dassa (1994) classified 70 membrane proteins into eight groups according to the sequence similarity, which correlated well with the clusters that Tam and Saier had reported. The Structural Classification of Proteins (SCOP) database (Murzin et al. 1995) also provides the classification of periplasmic-binding proteins based on the tertiary structures, where two folds named periplasmic-binding protein-like I and II are currently identified.
The result of our grouping is generally consistent with the three previous classifications as shown in Table 5. Among the new groups we identified, group 5-2 is highly related to group 5-1. Groups 5-3, 6, 7, 11, and 3-1 are not typical ABC transporters because they lack entirely the periplasmic-binding protein components. According to the SCOP classification, known tertiary structures of periplasmic-binding proteins in groups 1-1, 3-3, 3-7, and 4-1 take the same fold of periplasmic-binding protein-like II, whereas those in groups 2 and 3-5 take the periplasmic-binding protein-like I fold. Thus, it will be interesting to see whether periplasmic proteins of group 5-1 will have a distinctive third fold.
Comparison of the Four Classification Schemes for the ABC Transporter System
Tatusov et al. (1997) defined clusters of orthologous groups (COGs) in the seven complete genomes by identifying orthologous relations among multiple species. According to their classification, most of the ABC-type ATP-binding proteins fall in a single group (COG044), for their method is aimed at classifying the entire set of proteins in the genome rather than subclassifying a large set of paralogous proteins. In addition, there were four minor groups that corresponded to our grouping: COG0410 and COG0411 to the branched-chain amino acid transporter (group 3-5) and COG0488 and COG0396 to unknown tranporters of groups 8 and 9, respectively.
Evolution of ABC Transporters
There have been surveys on the positional relationship of orthologous genes between bacterial genomes (Mushegian and Koonin 1996;Watanabe et al. 1997). The general observation is an extensive shuffling of orthologous genes even between closely related species such as E. coli and H. influenzae. The present study has confirmed for the majority of species in bacteria and archaea that the unit of shuffling in the ABC transporters is a group of genes in the operon (transcription unit) rather than a single gene (translation unit). In Synechocystis, there are less cases of conserved operons, but there are more cases of multiple components of a transporter unit being fused into a single gene. These observations indicate that there is a positive selection of the genome organization for clustering of related genes forming a functional unit or under a common regulatory mechanism.
By examining Tables 4 and 5, groups 1-1/1-2 (sulfate/nitrate), 3-7 (phosphate), and 5-1 (iron complex) are most conserved among the 11 organisms. We speculate that the original form of these ABC transporter operons could have arisen early in the evolution, that is, before the branch point of bacteria and archaea. In terms of the number of paralogs in bacteria, groups 2 (simple sugars), 3-3 (polar amino acids), and 4-1 (peptides), as well as group 1-1/1-2, seem to stand out. These ABC transporters could provide clues to the diversification of biological species.
METHODS
Complete Genomes
For construction of the ortholog group table, we analyzed the complete genomes of seven organisms: E. coli (Blattner et al. 1997), H. influenzae (Fleischmann et al. 1995), and H. pylori (Tomb et al. 1997) from Gram-negative bacteria, M. genitalium (Fraser et al. 1995) and M. pneumoniae(Himmelreich et al. 1996) from Gram-positive bacteria,Synechocystis PCC6803 (Kaneko et al. 1996) from cyanobacteria, and M. jannaschii (Bult et al. 1996) from archaea. Using the ortholog group table derived, we performed additional analysis for the complete genomes of four organisms: B. subtilis (Kunst et al. 1977) from Gram-negative bacteria, B. burgdorferi (Fraser et al. 1977) from spirochetes, and M. thermoautotrophicum (Smith et al. 1977) and A. fulgidus (Klenk et al. 1977) from archaea. The amino acid sequence data and the information of gene locations were taken from the complete genomes section of GenBank (ftp://ncbi.nlm.nih.gov/genbank/genomes/bacteria/), which is incorporated into KEGG (http://www.genome.ad.jp/kegg/), together with the annotations made by the original authors, SWISS-PROT, and KEGG itself.
Overall Analysis Procedure
The procedure of our analysis consisted of the following six steps. (1) Starting with experimentally known ABC transporters in E. coli, we extracted paralogs of ABC-type ATP-binding proteins in each organism by the Smith–Waterman similarity searches. (2)Then, we performed the single- and complete-linkage cluster analysis to identify groups of paralogous ATP-binding proteins within each organism. (3) We then performed all possible (7 × 6) organism-by-organism comparisons to catalog ortholog pairs of ATP-binding proteins among the seven organisms. (4) Using the paralogous groups of proteins in each organism and the orthologous relations of proteins between two organisms, we constructed a table of ortholog groups for the ATP-binding proteins. (5) By examining five (occasionally more) genes on both sides of the ATP-binding protein gene in the genome, we identified additional components of the transporter, where the functions of permeases and substrate-binding proteins were assigned according to experimental evidence in some cases but mostly by similarity searches against SWISS-PROT. (6) Based on the similarity of membrane proteins, we subdivided the ortholog grouping by ATP-binding proteins and constructed the ortholog group table of ABC transporters among the seven organisms.
Sequence Similarity Search
For amino acid sequence comparisons of ATP-binding proteins, we used the SSEARCH program (Pearson 1991) that uses the Smith–Waterman algorithm (Smith and Waterman 1981) with the default parameters, BLOSUM50 matrix (Henikoff and Henikoff 1992) and the gap penalties of −12 and −2 for opening and extension, respectively. We used thee (expectation) value of <0.001 for the similarity criterion. Because the e value is dependent on the database to be searched, the value is not identical when a pair of proteins is compared between two complete genomes depending on which genome is taken as a database. We simply used the larger e value (lower similarity) for the similarity score of the pair.
In the search of ATP-binding protein paralogs, the computed similarities were manually verified. For example, in the initial search we identified 83 sequences that were probable ATP-binding proteins among the E. coli gene products. However, we excluded five of them: UvrA (b4058) that was the subunit A of ABC excision nuclease, SbmA (b0377) that was the receptor protein sensitive to microcin B17, and three hypothetical proteins of ∼40 kD (b0792, b0793, b3485) that were apparently artifacts caused by the multidomain ATP-binding proteins fused to membrane proteins.
Cluster Analysis
The clustering of paralogous ATP-binding proteins was done by both the single- and complete-linkage cluster analysis, in which we used the value of 1000 divided by the Smith–Waterman alignment score as the distance. For the pairs that did not satisfy the similarity criterion, we gave 10.0 as their distances. To best represent the functional groups of known transporters in E. coli, we determined the threshold values of 1000/590 for the single-linkage and 1000/470 for the complete-linkage cluster analysis. Note that the single linkage that combines two clusters according to the minimum distance among all possible pairs of members tends to produce larger clusters with varying members, whereas the complete linkage that combines two clusters according to the maximum distance tends to produce smaller clusters with uniform members. The clusters that satisfied both clustering criteria were called the basal clusters (Fig. 1). For the other organisms the threshold values were adjusted around the ones mentioned above.
Definition of Orthologous Relation
An orthologous gene pair is defined operationally by the following procedure (Bono et al. 1998): When two genes between two organisms satisfy the similarity criterion mentioned above and when both exhibit the highest similarity according to the e value and the Smith–Waterman alignment score in the homology search against the counterpart genome, the gene pair is defined as an ortholog. When combining the pairwise relations into an ortholog group, conflicts of relations can happen. Basically, we gave the priority of the relation in order of the alignment score. When constructing an ortholog group table, separate groups in one organism can sometimes be merged into a single group because of the grouping in another organism. We used the threshold Smith–Waterman score of 350 for this merging process. Although most of our procedure was computerized, such global conflicts as well as others were eliminated manually.
Acknowledgments
This work was supported in part by a grant-in-aid for scientific research on the priority area “Genome Science” from the Ministry of Education, Science, Sports and Culture of Japan. The computational time was provided by the Supercomputer Laboratory, Institute for Chemical Research, Kyoto University. K.T. was supported by the Research Fellowship for Young Scientists from the Japan Society for Promotion of Science.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL kanehisa{at}kuicr.kyoto-u.ac.jp; FAX 81-774-38-3269.
-
- Received June 22, 1998.
- Accepted August 28, 1998.
- Cold Spring Harbor Laboratory Press








