
Zebrafish: Genomics on the Fast Track
The zebrafish (Danio rerio) has proven its worth as an excellent model system for the study of vertebrate development. In 1996, in an issue of the journalDevelopment, the results of the characterization of almost 2000 mutations derived from two screens of ENU-mutagenized zebrafish (Driever et al. 1996; Haffter et al. 1996) were reported in 36 articles (Vol. 123, Dec. 1996). The same issue, however, included only one article describing the generation of reagents for genomic analysis that will be required for the positional cloning and characterization of these newly identified developmental anomalies (Knapik et al. 1996). Since then, remarkable progress has been made in zebrafish genomics, based largely on technology that has been developed for mammalian genetic analysis. The rapid implementation of these techniques, which will enable the characterization of the large set of zebrafish mutations, speaks to the utility of research that aims to develop resources for a community of investigators rather than to test a specific hypothesis.
The use of saturation mutagenesis in zebrafish as a means to study vertebrate development has evolved from a convergence of diverse experimental approaches. Saturation mutagenesis of Drosophilahas been used with extraordinary success to identify abnormalities in early embryonic patterning. The fact that mutations in mammalian homologs of these genes often have profound developmental consequences demonstrates that many of these basic pathways have been maintained in highly divergent organisms. A similar analysis in vertebrates was proposed as a means to uncover additional loci of developmental importance, especially those affecting formation of internal organs, which are not effectively scored in Drosophila embryonic screens (Nüsslein-Volhard 1994). Zebrafish are particularly well-suited for this purpose, as they generate large numbers of transparent embryos that develop synchronously to a free-swimming hatchling in a period of 3 days (for review, see Driever et al. 1994). Zebrafish have an additional experimental advantage that has been exploited successfully for the purpose of genetic analysis. Based largely on the studies of George Streisinger, a variety of techniques can be used to manipulate the ploidy and parental origin of genes in zebrafish (Streisinger et al. 1981, 1986). For example, gynogenetic haploid embryos can be generated by fertilization of secondary oocytes with UV-irradiated sperm; these embryos develop to hatching, albeit with specific developmental abnormalities. It is also possible to generate both fully homozygous diploid adults, and “half-tetrad” diploid adults, which are partially homozygous (for review, seePostlethwait and Talbot 1997). The uses of these various genetic “tricks” for genetic mapping are outlined in Box 1.
Genetic Markers
The generation of a detailed genetic map for a largely uncharacterized organism represents a formidable task, but one that has been simplified dramatically by the application of technologies employed in other systems. A variety of PCR-based techniques have been employed successfully in zebrafish to detect genome-wide polymorphism. The first systematic analysis utilized the characterization of random amplified polymorphic DNA (RAPD) markers in haploid offspring of a heterozygous female obtained from a cross between two strains of zebrafish (Postlethwait et al. 1994). A total of 134 decamer primers identified 401 loci, which fell into 29 linkage groups [4 more than the expected haploid complement of 25 chromosomes suggested by karyotype analysis (Schreeb et al. 1993; Daga et al. 1996)]. Further characterization of this cross using additional primers increased the number of loci to 652 and consolidated the number of linkage groups to 25, with an estimated total genetic size of 2900 cM (Johnson et al. 1996).
Despite the limitations of RAPD analysis (described below), the strategy used can serve as a paradigm for the establishment of a genetic map for an otherwise genetically uncharacterized organism. With a relatively modest amount of analysis, one can obtain a wealth of information regarding many loci. Thus, framework maps can be generated in systems for which genome-wide sequenced tagged site (STS) markers have not been individually characterized.
The RAPD map has proven extremely useful as a framework map for the localization of PCR-based markers employing either simple sequence repeat length polymorphisms (SSLP) or single-strand conformational (SSC) polymorphisms. However, the routine application of RAPD analysis for genetic mapping has significant limitations. First, the efficiency of amplification of fragments using decamers is very sensitive to reaction conditions and may be inconsistently reproducible in different experiments. This is particularly problematic if one is interested in integrating mapping data from different crosses by comparison to common reference markers. Second, because many loci are not truly codominant (i.e., both alleles are not equally detectable in heterozygotes), the robustness of this analysis is more limited in an intercross or backcross as heterozygous and homozygous loci may not be distinguishable. (The zebrafish RAPD map was generated using a haploid mapping panel.) Finally, although the amplified fragments can be cloned, sequenced, and converted to an STS marker (Johnson et al. 1996), the primer binding site is the source of strain specificity, and the sequence obtained between the primers may not be polymorphic.
AFLP (amplified fragmentlength polymorphism) is a recently described strategy of DNA fingerprinting that has been shown to be useful for genetic mapping in zebrafish (Vos et al. 1995; L. Zon, pers. comm.). This technique maintains many of the advantages of RAPD analysis but is more amenable to characterization of convential crosses of diploid organisms. In this technique genomic DNA is digested with restriction enzymes, ligated to oligonucleotide adapters, and selectively amplified (Fig. 1). Restriction sites that are polymorphic between strains will result in a generation of strain-specific bands, and segregation of these bands with a mutation can be readily scored. This approach has the significant advantage over RAPD analysis in that stringent primer annealing temperatures can be utilized, increasing reproducibility. However, it is still necessary to position these markers with respect to a reference genetic map, and the complexity of band patterns obtained in this analysis may make it difficult. Again, it is possible to convert an AFLP marker to an STS by reamplification and subcloning; however, again the polymorphism is at the ends of the fragment and the sequences internal to the primers may not be themselves polymorphic.
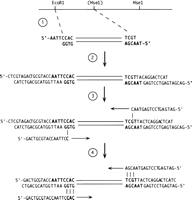
AFLP analysis (adapted from Vos et al. 1995). (1) Genomic DNA is digested with a combination of restriction enzymes. The products of this digestion will include some that are strain-specific due to sequence variation at the enzyme recognition sites (e.g., theMseI site shown in parenthesis). (2) The restriction products are ligated to adapters that carry cohesive ends for the respective enzymes and additional sequences to facilitate adapter-specific amplification. (3) A “preamplification” step is performed using primers corresponding to the adapters and each carrying an additional unique 3′-terminal nucleotide. This serves to limit the number of amplified products, as the template for this base will be derived from genomic sequence and will vary. Amplification of fragments carrying the same adapters (e.g., derived fromMseI–MseI restriction fragments) is less favorable than amplification of fragments with different ends, presumably because of the formation of stem–loop structures. (4) The population of products is further limited by amplification using a primer with three unique 3′-terminal nucleotides. These products can be separated on a denaturing gel and examined to identify the subset that are strain-specific. A kit containing reagents for AFLP analysis is commercially available from Life Technologies, Inc.
In mammals, the identification of large sets of primers that can detect SSLPs at loci distributed across the genome has proven to be an extraordinarily efficient means to pursue genetic mapping. SSLPs were shown to be present in zebrafish (Goff et al. 1992), and the characterization of 102 SSLPs that segregate in 25 linkage groups in an analysis of 520 intercross progeny (1040 meioses) has been reported (Knapik et al. 1996). The generation of a mapping panel containing this large number of progeny will provide the means to perform high-resolution genetic mapping in a similar fashion to that which can be pursued in the mouse using the European Community Interspecific Backcross (EUCIB) panel (Breen et al. 1994). A present limitation of these characterized markers is a significant amount of allele sharing found between zebrafish strains (reported to be 30%), so that not all markers will be informative in a cross. The more recent characterization of additional markers, for a total of 703, should ensure that there is sufficient map density such that any mutation can be localized with reasonable resolution (E. Knapik, pers. comm.). This map covers 2350 cM of the zebrafish genome, with an average resolution of 3.3 cM. (The smaller genetic distance obtained in this analysis may represent the difference between the sex-averaged meiotic map, and the female meiotic map, which was determined using the haploid mapping panel.) Given this estimated total size of the zebrafish genetic map and a haploid DNA content of 1.7 × 109 bp (Hinegardner and Rosen 1972), the present distribution of SSLP markers averages approximately one every 2400 kb.
The goal for this project is to generate a set of 3000 SSLP markers; however, experience gained in mammalian efforts suggests that even this number of markers may not be sufficient for efficient positional cloning. In crosses between commonly used laboratory mouse strains, up to 50% of the 6000 microsatellite markers available for murine genetic mapping will be polymorphic. Yet it is rare that a positional cloning analysis will not employ a cross to a feral mouse strain (such asMus castaneaus or Mus spretus), in which nearly all of the markers will be informative, to maximize the number of characterized markers in a recombinant interval. The availability of a very dense marker distribution is necessary to minimize the number of physical clones that must be examined in the effort to identify candidate transcribed units.
The existence of the mermaid repetitive element in the zebrafish genome suggests another possible strategy based on PCR amplification (Shimoda et al. 1996). Interspersed repetitive sequence PCR (IRS PCR) is a technique in which naturally occurring variation in the distribution of repetitive sequences is exploited to generate strain-specific markers (McCarthy et al. 1995). In this approach, PCR is performed using combinations of primers derived from repetitive elements under conditions of stringency such that unique bands are detected. The amplification of these bands is contingent on the specific position and orientation of the repeat sequences in the genome, and as these may vary with genetic background, subsets of these products may be strain-specific. A strategy has been described in which these polymorphic regions are cloned, mapped, and arrayed such that their representation in a selected population carrying a mutation can be scored by hybridization using the pooled products of IRS PCR as a probe (Elango et al. 1996). Although this strategy has not seen extensive utilization in analysis of mammalian mutations, it may prove particularly useful for the analysis of the zebrafish genome.
Mutation Mapping
Widely distributed polymorphic markers have two important functions in a positional cloning analysis (1) as a means to localize a mutation to a specific recombinant interval, and (2) as an entry point for identifying large-insert genomic clones (such as YACs, PACs, or BACs) that can be used for chromosome walking. Two YAC libraries, a PAC library, and a BAC library have been generated in zebrafish. One YAC library comprises 36,000 clones with an average insert size of 280 kb (L. Zon and G. Silverman, pers. comm.) and is available for screening from Genome Systems, Inc. Another YAC library, which comprises 17,000 clones with an average insert size of 470 kb (Zhong et al. 1997), has been generated and is available from Research Genetics, Inc. The PAC library contains 100,000 clones with an average insert size of 115 kb and corresponds to five-fold coverage of the zebrafish genome (C. Amemiya, http://med-humgen14.bu.edu). The BAC library contains 75,600 clones, with an average insert size of 120 kb. Filters containing BAC and PAC clones are available from Genome Systems, Inc.
Although marker density is presently less than that necessary for efficiently generating large-insert clone contigs, it is sufficient for mutation mapping. Three strategies have emerged as useful for this purpose: (1) RAPD (or AFLP) analysis on pooled samples (Postlethwait et al. 1994); (2) SSLP analysis on pooled samples; and (3) half-tetrad analysis (see Box). The first two strategies take advantage of the fact that a selected population of intercross or backcross progeny will have in common the region linked to the mutant locus and will be randomly segregating parental alleles at other loci. Thus, in a test of genetic markers in a pooled population, a marker showing bias toward representation of the allele corresponding to the mutant parent should prove to be linked to the mutation (Fig. 2). To maximize the efficency of this strategy, Haffter and colleagues are performing mapping crosses using the WIK strain of zebrafish, which was isolated from a wild catch in India and found to be polymorphic at 43% of 314 SSLP markers tested (Rauch et al. 1997;http://www.eb.tuebingen.mpg.de/Abt.3/1997.html Rauch). The third strategy takes advantage of the fact that it is possible to use early pressure parthenogenesis to generate partially homozygous diploid fish. Because these diploid fish are isogenic at most loci, the linkage group carrying the mutation can be identified by testing only centromere-linked markers (Johnson et al. 1995; for review, seePostlethwait and Talbot 1997). This linkage group can then be analyzed in detail with additional markers to localize the mutation more precisely. Although this strategy is less necessary as marker density increases, as mapping can be performed using genome-wide screens using conventional crosses, it still provides a means to localize a mutation with a minimum of molecular analysis.
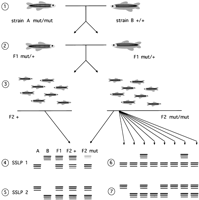
Linkage analysis using SSLP genotyping of pooled samples. (1) Fish of strain A homozygous for the recessive mutation mut are crossed to wild-type fish of strain B to generate F1 fish that are heterozygous at all polymorphic loci. (If homozygous fish cannot be bred, then F1 fish heterozygous for the mutation can be generated by mating a heterozygous mut/+ A parent with a wild-type B parent and identifying mutation carriers by progeny testing.) (2) F1 mut/+ fish are intercrossed and DNA is prepared from (3) phenotypically normal F2 progeny (which will be either wild type or heterozygous at mut) and mutant F2 progeny (which will be homozygous mut/mut). Samples from these two classes are pooled separately and tested using SSLP markers distributed across the genome. (4) For a marker linked to the mutation (SSLP 1), the PCR products corresponding to the A allele of the SSLP will predominate when the mutant pool is tested. (5) For unlinked markers (e.g., SSLP2), the abundance of PCR products of the two alleles amplified from the mutant population will be equal, and the distribution will appear to be the same as those from the F1 parents and the wild-type pool. (6) Linked markers are then tested in individual mutant progeny to determine their map positions with respect to the mutation. Two out of eight mutant progeny are heterozygous here. Because these are intercross progeny, this corresponds to 2 recombinants in 16 meioses, which is 12.5 ± 8.3 cM. (7) The distribution of SSLP 2 markers in individualmut/mut progeny is seen to be random.
A milestone in zebrafish genomics is the recent cloning of the first mutant locus, one-eyed pinhead (oep), using a purely positional cloning strategy. RAPD analysis was used to identify closely linked markers to oep, and 400 kb of contiguous genomic DNA sequences in BACs and PACs were assembled by genomic walking (Zhang et al. 1998). An illustration of one of the unique advantages of the zebrafish system is that transcription units within the minimal recombinant interval were characterized by whole-mount in situ hybridization. This identified a previously unknown protein as a strong candidate for oep, which was confirmed by the sequencing of mutant alleles as well as phenotype rescue.
Gene Mapping
Given the demonstrated power of candidate gene analysis in the characterization of vertebrate mutations, it would be extremely desirable to generate a dense gene map in zebrafish. Compared to mammalian systems, only a modest number of genes have thus far been localized relative to either RAPD or SSLP markers (Postlethwait et al. 1994). This has been accomplished mainly by identification of length variation in noncoding regions (which are relatively uncommon) or by identifying RFLPs in PCR-amplified fragments (which can be inefficient). However, the compelling need to localize on the zebrafish genetic map genes known to affect developmental processes in other organisms has led to rapid progress in this area.
SSC polymorphism analysis is an efficient PCR-based means to detect sequence variation; it has been shown previously that polymorphisms are abundant in noncoding regions of mouse genes and can be used for gene mapping in an appropriate mapping resource (Beier et al. 1992; Beier 1993; Brady et al. 1997). To test whether SSC polymorphism analysis could be used for mapping genes in zebrafish, >100 primer pairs derived from 3′ UTRs of zebrafish genes were analyzed (D. Foernzler and D. Beier, unpubl.). The polymorphism frequency between zebrafish strains represented in existing mapping crosses was ∼40%–50%, and these polymorphisms were useful for mapping in both the haploid and diploid mapping panels described by Johnson et al. (1996) and Knapik et al. (1996), respectively (Fig. 3). Approximately one-third of the tested loci were derived from the presumptive 3′ ends of ESTs, indicating that this strategy can be useful for localizing cDNAs for which little information about gene structure is known.

SSCP analysis of expressed sequences. Genotype distributions of two different primer pairs corresponding to 3′ UTR of expressed sequences are shown in progeny of two reference crosses. (Top): Reference cross described by Knapik et al. (1996). Lanes correspond to diploid progeny of AB × India intercross. (A): AB homozygote; (H): heterozygote; (I): India homozygote. (Bottom): Reference cross described by Postlethwait et al. (1994). Lanes correspond to haploid progeny of SJD × C32 intercross. (S): SJD; (C): C32.
Zebrafish “Primer”
BiologyGeneration time: 3–4 months Number of offspring per cross: ∼100 Size: 3–4 cm Time to develop to hatchling stage: 3 days Life span: 2 years
Genome and GeneticsHaploid genome size: 1.7 × 109 bp Number of chromosomes: 25 (metacentric) Linkage map: 2900 cM Number of characterized mutations: ∼2000 Strains: Wild-type strains in most common use are AB, TubAB, TupLF, WIK, and SJD. Interstrain polymorphism is variable; AB/SJD are ∼50% polymorphic for SSLPs Current no. of mapped markers: 652 RAPDs, 703 SSLPs Current no. of mapped mutations: ∼15 Current no. of mapped cloned genes: ∼150 ESTs: >3000
Genetic analysis—Special Tricks Gynogenetic haploids: Eggs fertilized by UV-inactivated sperm develop as haploids to the end of the embryonic stage. Polymorphic DNA markers can be mapped by following their segregation in the haploid offspring of a heterozygous female resulting from a cross between two different zebrafish strains.
Gynogenetic half-tetrad diploids: Pressure treatment of an egg fertilized by inactivated sperm inhibits the second meiotic division, leading to the development of diploid embryos. In these embryos, which mostly survive to adulthood, chromosomes that have recombined (there is usually only one crossover per chromosome in zebrafish) are homozygous at loci centromere-proximal to the crossover site, and heterozygous for loci distal to the crossover. Thus, any embryos homozygous for a recessive mutation will be homozygous for the centromeric region linked to the mutation. Pools of such embryos will harbor only one allele of the relevant centromere, whereas they will contain mixtures of centromere-linked alleles for each of the other chromosomes. Thus, in principle, 25 PCR reactions (1 for each centromere) performed on each of two DNA pools, one derived from gynogenetic mutant embryos and the other derived from gynogenetic wild-type embryos, will indicate the chromosome that carries the mutation. In addition, the frequency of homozygous mutants observed among these progeny will indicate the approximate genetic distance between the mutation and its centromere, which can be readily confirmed by subsequent marker analysis.
Gynogenetic diploids: Heat treatment of haploid embryos at the one-cell stage generates diploid embryos homozygous at all loci, which can be used to obtain clonal lines of fish.
Another strategy proven to work is RFLP analysis of progeny derived from a cross between two divergent zebrafish strains, such as Tübingen and WIK (P. Haffter, pers. comm.). Although this has some constraints as a result of the relatively small amount of DNA obtainable from single zebrafish (0.5–1.0 mg), careful use of filters used for Southern analysis in this strategy can maximize its utility.
An alternative to the direct genetic mapping of the zebrafish genes would be to localize them on a high-resolution physical map with respect to more easily scored markers, allowing their position on a genetic map to be determined by inference. In humans, this is exemplified by the publication of a transcript map containing 16,000 STSs derived from cDNA sequences (Schuler et al. 1996). These STSs were characterized primarily in radiation hybrid (RH) somatic cell panels, where the presence or absence of a product (rather than a polymorphism) is scored. The STS products are positioned in the RH panels with respect to microsatellite markers, although it should be appreciated that this localization may be less precise than a direct genetic analysis because anomalies in the latter are easily recognized as double crossovers, which are most likely artifactual if found in a small genetic interval.
Somatic cell hybrids have been developed in which zebrafish chromosomal material from a cell line transfected with a selectable marker has been introduced into mouse melanoma cells by whole cell fusion (Driever and Rangini 1993; Ekker et al. 1996). Retention of zebrafish genomic sequence in the hybrids has been demonstrated by FISH and can be stably maintained when cells are grown under selection. Most of the characterized cell lines are whole chromosomal, however, and cannot be used for high-resolution analysis. RHs containing fragmented zebrafish genomes have been developed, and a set of 97 lines is presently being characterized (L. McCarthy, pers. comm.) These are being expanded by Research Genetics, Inc., and will likely be available as a community resource in the near future.
An alternative to the direct localization of genes on a genetic map would be to extrapolate their position with respect to other genes, assuming that gene order is conserved across species. This is a more stringent requirement than the simple conservation of synteny, which refers to genes being present in the same linkage group in different organisms, without consideration of gene order (Eppig and Nadeau 1995). Conservation of gene order between mouse and human has been well documented for subchromosomal regions, and by comparison of the dense gene maps that have been developed for these organisms, the boundaries of the conserved regions are reasonably well-defined (for review, seeCarver and Stubbs 1997). Given the limited information with respect to gene positions in the zebrafish genetic map, assessment of significant gene order conservation cannot yet be made. Conclusive characterization of this conservation must necessarily consider gene order and not simply colocalization in a linkage group and must be pursued in an unbiased fashion, as some linkage conservation may be functional. That said, it is likely that there will be some regions where extensive gene order conservation will be found, which will markedly increase the information density of the gene map, and specifically its utility for suggesting candidate loci for positional cloning analyses. Although the mean length of the conserved chromosomal regions in the mouse compared to humans is 8.8 cM (and this calculation does not necessarily include small local rearrangements that can be shown to be common when detailed comparisons are made), there are regions of up to 40 cM in which gene order appears to be conserved (Debry and Seldin 1996).
Expressed Sequences
Finally, because it would be desirable to use a PCR-based method for gene mapping, it is important to build up a resource of zebrafish-specific cDNA sequence. In this regard, recent efforts using single pass sequencing of randomly cloned human or mouse cDNAs as a means to generate expressed sequence tags (ESTs) can serve as a useful model. The variation in the 5′ termini of cDNAs has allowed the generation of large regions of overlapping sequence, and the 3′ sequences have served both as templates for STS generation (as the 3′-terminal sequence rarely contains introns) and as a means to identify true cross-species orthologous genes (Makalowski et al. 1996). Although no large-scale EST projects on the order of mammalian efforts have yet been initiated, several investigators have pursued pilot projects, and there are >3,000 sequences in the dbEST database. Information about zebrafish EST sequencing projects at Washington University is available athttp://genome.wustl.edu/est/zebrafish_esthmpg.html.
A complement to EST characterization is the generation of arrayed cDNA libraries that can be probed by hybridization to identify zebrafish homologs of genes cloned from other species. By hybridization of murineHox and Pax probes to filters containing a total of 54,000 arrayed zebrafish embryonic cDNAs generated by M. Clark and H. Lehrach (Max-Planck Institut für Molekulare Genetik, Berlin, Germany), 34 clones with significant homology have been identified (D. Foernzler and D.R. Beier, unpubl.). Because the library was oligo-(dT)-primed, these clones could be assessed for 3′ SSC polymorphisms that will facilitate their mapping. Hybridization to arrayed cDNA libraries could be an efficient mechanism for obtaining homologs of the many other genes thought to have roles in organogenesis. The importance of this is illustrated by the successful molecular identification of a number of mutations in zebrafish by the candidate gene approach, using inferences based on gene functions determined in other organisms. For example, zebrafish no-tail(ntl) mutant embryos resemble mouse T/T mutant embryos in that they lack a differentiated notochord and the caudal region of their bodies. This observation suggested theBrachyury gene as a promising candidate and the zebrafish gene was cloned by homology (Schulte-Merker et al. 1994). The addition of positional information for developmentally important genes will considerably enhance their utility in candidate gene analysis.
Insertional Mutagenesis
Mutagenic strategies in which the disrupted loci are tagged in a fashion that facilitates their rapid molecular characterization represent a means to bypass the inherent difficulties in positional cloning. It has been shown that retroviral provirus insertions can be engineered in the zebrafish germ line, that these insertions can disrupt genes required for development, and that the insertion sites can be cloned efficiently (Allende et al. 1996; Gaiano et al. 1996). In the inital studies of this approach, three embryonic lethal mutations were found in a screen of 217 proviral insertions. Although the efficiency of this technique is very respectable as a method to obtain a cohort of tagged mutations, it cannot readily be employed to identify a specific mutation; that is, it is less useful if the screen is more narrowly constrained to a particular phenotype. Thus, although insertional mutagenesis represents a potent method to “jump start” the molecular characterization of interesting mutations in zebrafish, it will not preclude the necessity of developing the more widely applicable genomic resources required for positional cloning.
One very powerful application of this transgenic technology, however, is the opportunity to introduce readily scored phenotypic markers distributed across the zebrafish genome. These transgenic lines can serve as “balancer chromosomes,” which will markedly facilitate the sophistication of genetic studies that can be performed.
Conclusions
In summary, the enthusiasm for a vertebrate system in which efficient mutagenesis could be exploited as a means to study development has resulted in remarkable progress in a short period. Technologies developed for genomic analysis in mammalian systems have been employed rapidly and successfully to develop resources that make positional cloning of these novel mutants feasible. What is also clear, however, is that for the full potential of zebrafish to be realized, there needs to be considerable additional effort put into development of tools for genomic analysis, in particularly in the areas of marker development, cDNA resources, gene mapping, and especially bioinformatics.
Because zebrafish is not officially sanctioned as a model organism for the Human Genome Project and the generation of tools for genomic analysis does not test any particular hypothesis, the mechanism for supporting the development of these resources is not yet defined. Given the competition for finite resources, the rationale for investing in this effort must be clearly justified.
The generation of thousands of novel mutants in a vertebrate organism in a short period is in itself an impressive achievement; and yet it only hints at the true power of zebrafish. It will be possible in this system to pursue complex genetic analysis, which will facilitate the investigation of the effects of multiple interacting loci. This will always be difficult in the mouse, simply because it will be logistically and financially prohibitive to breed sufficient numbers of mice to obtain the few in which multiple mutant loci are cosegregating. An additional attraction of the zebrafish system is that it should be feasible to use mutagenesis to identify second-site mutations (e.g., in a suppressor screen) that will potentially illuminate developmental pathways. Thus, one can readily envision pursuing the elegant genetic studies in a vertebrate that have been so extraordinarily fruitful inDrosophila, Caenorhabditis elegans, and Saccharomyces cerevesiae. It is this opportunity that should make the development of robust tools for the genetic analysis of zebrafish compelling.
Acknowledgments
I thank Dorothee Foernzler, Len Zon, and anonymous reviewers for comments on the manuscript. This work was supported in part by the Center for Molecular Developmental Hematopoiesis (DK49216) at Children’s Hospital, Boston, Massachusetts.
Footnotes
-
↵1 E-MAIL ; FAX (617) 264-5292.
- Cold Spring Harbor Laboratory Press








