
A Gene Map of the Best’s Vitelliform Macular Dystrophy Region in Chromosome 11q12–q13.1
Abstract
Best’s vitelliform macular dystrophy is an autosomal dominant disorder of unknown causes. To identify the underlying gene defect the disease locus has been mapped to an ∼1.4-Mb region on chromosome 11q12–q13.1. As a prerequisite for its positional cloning we have assembled a high coverage PAC contig of the candidate region. Here, we report the construction of a primary transcript map that places a total of 19 genes within the Best’s disease region. This includes 14 transcripts of as yet unknown function obtained by EST mapping and/or cDNA selection and five genes mapped previously to the interval (CD5, PGA, DDB1, FEN1, and FTH1). Northern blot analyses were performed to determine the expression profiles in various human tissues. At least three genes appear to be good candidates for Best’s disease based on their abundant expression in retina or retinal pigment epithelium. Additional information on the functional properties of these genes, as well as mutation analyses in Best’s disease patients, have to await their further characterization.
[The GenBank/EMBL accession numbers and details of the isolation, localization, and characterization of ESTs and selected cDNAs are available as online supplements in Online Tables 1–3 at http://www.genome.org.]
The macular dystrophies are a heterogenous group of distinct disorders characterized by morphological changes most profoundly in the central fundus and loss of central vision assumed to result preferentially from degeneration of cone photoreceptors. In recent years, almost 20 hereditary maculopathies have been localized to specific chromosomal regions (for review, see Sullivan and Daiger 1996), laying the groundwork for the identification of the underlying disease genes by applying the various strategies of positional cloning (Collins 1995). To date, five genes associated with macular dystrophy have been identified: (1) The peripherin/retinal degeneration slow (RDS) gene when mutated can result in typical macular as well as retinitis pigmentosa (RP) phenotypes (Dryja et al. 1990;Kajiwara et al. 1993; Nichols et al. 1993; Wells et al. 1993); (2) the tissue inhibitor of metalloproteinase-3 (TIMP3), causing autosomal dominant Sorsby’s fundus dystrophy (Weber et al. 1994); (3) the photoreceptor-specific ABC transporter (ABCR) implicated in autosomal recessive Stargardt’s macular dystrophy (Allikmets et al. 1997); (4) ataxin-7 (SCA7), which is associated with progressive cerebellar ataxia as well as pigmentary macular dystrophy (David et al. 1997); and (5) the RS1 gene underlying X-linked juvenile retinoschisis (Sauer et al. 1997).
Our current efforts are directed toward the isolation of the gene responsible for Best’s vitelliform macular dystrophy (Best’s disease) (Best 1905). This disorder is characterized by an autosomal dominant mode of inheritance, with symptoms generally manifesting in the first or second decade of life. A subretinal eggyolk-like lesion in the macular area represents the most prominent feature of the disease. Histopathologically, an unusual accumulation of yellowish material is found at the level of the retinal pigment epithelium (RPE), probably between the RPE and Bruch’s membrane. It is composed of viscous, lipofuscin-like granula likely constituting incompletely digested remnants of the outer segments of the photoreceptor cells (Weingeist et al. 1982; O’Gorman et al. 1988). Electrodiagnostic testing demonstrates a typically reduced electro-oculogram (EOG) in the affected individuals, even in those patients with otherwise normal fundus appearance. Electrophysiology, together with the histopathological findings, favors a generalized dysfunction of the RPE complex as the basic defect in Best’s disease.
As a first step toward the cloning of the disease gene, the Best’s disease locus was mapped to chromosome 11 by genetic linkage analysis (Forsman et al. 1992; Stone et al. 1992). Subsequently, a detailed characterization of recombinant chromosomes in large Best’s disease pedigrees has refined the disease locus to a 1- to 2-cM interval in 11q12–13.1 flanked by markers at D11S1765 proximally and uteroglobin (UGB) distally (Stöhr and Weber 1995; Wadelius et al. 1996). As a prerequisite to the construction of a comprehensive gene map of the disease region we have recently assembled a sequence-ready PAC contig providing full coverage of the minimal 1.4-Mb Best’s disease locus (Cooper et al. 1997).
The identification of coding sequences within large genomic regions is still a challenging and formidable task, although a variety of advanced technologies is available to date (for review, see Parimoo et al. 1995). Currently, several approaches are widely used and include the exon trapping/amplification technique (e.g., Buckler et al. 1991), direct cDNA selection (e.g., Morgan et al. 1992; Rommens et al. 1993), identification of CpG islands (Lindsay and Bird 1987), and computational-based methods for the prediction of protein-coding potentials in genomic sequences (Uberbacher and Mural 1991). Because no single approach appears to be efficient enough to identify all and, in particular, the rare transcripts within a specific genomic region, it is desirable to apply a combination of these techniques.
Five genes, CD5, PGA, DDB1, FEN1, and FTH1, as well as seven EST clones have been mapped previously to the Best’s disease region (Cooper et al. 1997). Here we have expanded the mapping of known ESTs and have performed a PCR-based cDNA selection of large parts of the critical region using cDNA pools derived from eye tissues that most likely express the gene underlying Best’s disease, namely the retina and the RPE. Based on Northern blot analyses the identified cDNA fragments have been assembled into 14 distinct transcripts, thus increasing the total number of known genes in the Best’s disease region to 19. This primary transcript map provides the starting point for further characterizations of potential candidates for the Best’s disease gene.
RESULTS
Refined Mapping of Known ESTs
To further refine the localization of 15 ESTs assigned previously to proximal 11q by radiation hybrid RH mapping (Schuler et al. 1996;Shows et al. 1996) a minimal set of overlapping PAC clones encompassing the minimal Best’s disease locus were used for EST content mapping by PCR and/or Southern blot analyses of EcoRI-digested cloned DNA. In addition, several YAC clones were analyzed, extending the PAC contig ∼1 Mb centromeric and 300 kb telomeric (Qin et al. 1996;Cooper et al. 1997) (Fig. 1A). Of the 15 ESTs, two mapped within the Best’s disease locus, whereas four EST clones were localized to CEPH Mega YAC 942_g_8 and one EST to YACs yRP6a5 and yRP16b1, thus positioning these transcripts outside the disease region proximal to D11S1765 [Fig. 1A and online supplements (Online Table 1, http://www.genome.org)]. The location of the remaining eight ESTs could not be determined within the genomic clones utilized in this study.
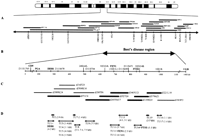
Schematic representation of the physical and transcript map of the Best’s disease locus. (A) Two nonoverlapping YAC contigs extending the Best’s disease region proximally and distally. The order and location of genes and anonymous DNA markers were determined by STS content mapping. (B) The minimal Best’s disease interval between DNA markers at D11S1765 and UGB. The physical size of the region and the position of known genes and STSs is shown. (C) A selected set of overlapping PAC clones (Cooper et al. 1997) covering ∼1 Mb of genomic DNA of the candidate region was used to localize 17 distinct transcription units (TUs). Bold bars indicate PAC clone DNAs used for cDNA selection. (D) Regional assignment of 17 distinct TUs. The transcript sizes obtained by Northern blot analysis are in parentheses.
Isolation and Physical Mapping of Clones Obtained by Direct cDNA Selection
To identify novel cDNAs from the Best’s disease locus we performed direct cDNA selection using seven PAC clone DNAs covering ∼800 kb of the core Best’s disease region (Fig. 1C). The primary cDNAs were generated from total RNA of retinal tissue and cell line ARPE-19 and were subsequently pooled for two rounds of hybridization-based selection with PAC insert DNAs. Three separate experiments were performed using PAC clone dJ71C4 (denoted C96), PAC clones dJ455M17, dJ527E4, and dJ465G21 (denoted C97A); and PAC clones dJ139E20, dJ379A21, and dJ221L18 (denoted C97B) [Fig. 1C and online supplements (Online Tables 2 and 3, http://www.genome.org)]. Initial overlap between dJ71C4 and dJ455M17 during PAC contig assembly could not be verified, leaving a small gap of ∼30 kb of genomic DNA between dJ71C4 and dJ455M17 not included in the cDNA selection (Fig.1C). A total of 182 clones were retrieved (C96, 41 clones; C97A, 95 clones; C97B, 46 clones) and arrayed into 96-well microtiter plates. The insert sizes were determined by PCR with oligonucleotide primers M13f/M13r flanking the EcoRI cloning site of the pBluescript vector and were found to range from 200 bp up to 2 kb.
Subsequently, the genomic origins of the selected cDNA fragments were verified by Southern hybridization to EcoRI-digested PAC clone membranes. In total, 29 (16%) cDNA fragments failed to hybridize specifically to the originating PAC clones. Based on sequential hybridizations of single cDNA clones to the membrane-spotted arrayed cDNAs, the remaining 153 retrieved cDNAs were assembled into 46 contigs. Twelve contigs were found to contain individual clones composed of >50% of interspersed repeat elements and were excluded from further analysis. The remaining 34 contigs were regionally assigned to dJ71C4 (9), dJ455M17, dJ527E4, and dJ465G21 (1), dJ527E4 and dJ465G21 (13), dJ465G21 (2), dJ139E20 and dJ379A21 (1), dJ379A21 and dJ221L18 (2), and dJ221L18 (6) (data not shown). DNA sequences of selected clones from each cDNA contig were determined and subjected to BLAST searches against nucleotide and protein sequence databases accessible through the National Center for Biotechnology Information (NCBI) via the internet (http://www.ncbi.nlm.nih.gov/BLAST/).
Identification of Sequences of Known Human Genes
Sequence alignments to the nonredundant GenBank/EMBL databases demonstrated significant similarities of five individual cDNA contigs to various regions of three human genes, namely to the damage-specific DNA-binding protein-1 (DDB1), the flap endonuclease-1 (FEN1) and the ferritin heavy-chain-1 (FTH1) (Online Table 2). The three genes have been localized independently to the chromosome 11q12–q13 region by FISH and RH mapping (Hentze et al. 1986; Dualan et al. 1995; Hiraoka et al. 1995) in excellent agreement with our PAC mapping data (Fig. 1B, D).
Clone contigs C96-1, C96-2, and C96-3 revealed sequence identity to distinct regions of the DDB1 cDNA (Dualan et al. 1995) (Online Table 2). Mapping of these contigs to PAC clone dJ71C4 or dJ398K24/dJ71C4 allowed us to determine the orientation of transcription of the DDB1 gene from telomere to centromere (Fig. 1C). Clone contig C97A-6 demonstrated identity to the 3′ UTR of FEN1 and was mapped to dJ527E4 and dJ465G21. In addition, the left end fragment from ICI–YAC 1EE1, 1EE1L (Fig. 1A), was sequenced and found to contain the 5′ end of FEN1, thus establishing the orientation of transcription of FEN1 from centromere to telomere. Finally, sequence alignment of the 439-bp fragment C97B-1 revealed 99% similarity to the published cDNA sequence of FTH1 (Boyd et al. 1984). A second ferritin heavy-chain gene, namely FTHL16, is known to map to chromosome 11q13 (Papadopoulos et al. 1992). However, sequence alignment with C97B-1 revealed 22 mismatches to the FTHL16 sequence, demonstrating that clone C97B-1, originating from the FTH1 locus, is the only known functional member of the FTH multigene family (Hentze et al. 1986).
Identification of Sequences of Unknown Function
Fifteen of the remaining 29 cDNA contigs revealed significant sequence matches in the dbEST database (Online Table 3). Six contigs identified EST clusters that were independently assigned to the proximal long arm of chromosome 11 (UniGene server athttp://www.ncbi.nlm.gov/UniGene/index.html) (Online Table 1). In addition, cDNA contig C96-7 and D11S2319E are part of a single EST contig that has been positioned within the Best’s disease region (Online Tables 1 and 3). Likewise, cDNA contig C96-8 partially overlaps EST173048, previously localized to the D11S1765–UGB interval by EST content mapping (Online Tables 1 and 3).
Expression Analysis
The expression profiles of ESTs and a representative clone of each of the 34 cDNA contigs mapping to the Best’s disease locus were analyzed by Northern blot analyses with emphasis on retina and retinal pigment epithelium. In addition, database searches were carried out to evaluate the representation of the cDNAs in tissue-specific cDNA libaries (Fig. 2; Table 1). Based on the localization to an identical genomic interval, a common transcript size, and an identical expression profile the cDNA fragments subsequently were assigned to distinct transcription units (TUs). Accordingly, we identified 17 TUs that were consecutively numbered from centromere to telomere (Fig. 1D; Table 1).
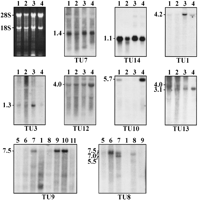
Northern blot analyses of cDNA fragments corresponding to the (TUs) as indicated (see Fig. 1). Examples are given to demonstrate the various expression profiles in lung (lane 1), cerebellum (lane2), retina (lane 3), retinal pigment epithelium cell line ARPE-19 (lane 4), and heart (lane 5), brain (lane 6), placenta (lane 7), liver (lane 8), skeletal muscle (lane 9), kidney (lane 10), and pancreas (lane 11). The transcript sizes are indicated atleft in kilobases.
Expression Analysis of TUs
TU 4–TU7, TU11 (FEN1), TU14 (FTH1), TU16, and TU17 appear to be expressed ubiquitously with a single transcript size present in all tissues tested (TU7 and TU14 are shown in Fig. 2). Using cDNA fragment IXD11 corresponding to FTH1 (Online Table 2) we detected a single transcript of 1.1 kb in all tissues tested including cerebellum (Fig. 2). An additional transcript of 1.4 kb recently identified in brain tissue (Dhar et al. 1993) could not be confirmed even after a prolonged exposure time.
TU1 (DDB1), TU3, and TU12, although widely expressed, show a higher abundance in some tissues. Interestingly, TU1 and TU3 are highly expressed in retina (Fig. 2). Similarly, the 4.0-kb mRNA of TU12 has a high abundance in cell line ARPE-19 (Fig. 2).
TU9 was originally isolated from a retina cDNA library (EST387646) (Online Table 1 and Table 1). Northern Blot analysis identified a 7.5-kb transcript in skeletal muscle, kidney, and more weakly in placenta (Fig. 2); however, no detectable signals were observed in retinal and ARPE-19 mRNA (data not shown). TU2, TU10, and TU15 appear to be specifically expressed in lung and ARPE-19, with TU10 highly abundant in the cell line RNA (Fig. 2).
TU8 displayed at least three species, a 7.0-kb mRNA in brain, liver, pancreas, and placenta, with single additional transcripts of 7.5 and 5.5 kb in brain and placenta, respectively (Fig. 2). TU13 is represented as 4-kb and 3.1-kb transcripts in cerebellum and retina. Only the larger transcript appeared in lung and the smaller species in the cell line ARPE-19 (Fig. 2), suggesting that this gene is subjected to alternative splicing that may be tissue specific.
DISCUSSION
Despite major achievements of the International Human Genome Project, to date, positional cloning of a disease gene is still a formidable task. In the case of Best’s disease extensive analyses of recombinant disease chromosomes have narrowed the locus harboring the disease gene to an interval encompassing ∼1.4 Mb of DNA (Stöhr and Weber 1995; Wadelius et al. 1996). Thus far, five genes have been localized within this interval (Cooper et al. 1997), encoding pepsinogen A (PGA) (Evers et al. 1989), a membrane glycoprotein of lymphocytes (CD5) (Jones et al. 1986), the p127 subunit of a damage-specific DNA-binding protein DDB1(Dualan et al. 1995), the ferritin heavy-chain subunit of the iron-storage protein ferritin (FTH1) (Hentze et al. 1986), and the structure-specific flap endonuclease (FEN1) (Hiraoka et al. 1995). Although a possible involvement of one of these genes in the pathogenesis of Best’s disease remains to be shown, the functional properties of the corresponding proteins appear not to provide supporting evidence for their candidacy.
To identify additional genes within the Best’s disease region and possibly those with functional relevance to the disease pathology, we have applied a combination of two approaches.
First, we have made use of the availability of an increasing number of ESTs that are localized to chromosome 11q12–q13 by RH mapping (Schuler et al. 1996). Of 22 EST clones assigned previously to the interval between markers at D11S1357 and D11S913 by RH mapping, a total of nine cDNAs (41%) were found to reside within the crucial region between D11S1765 and UGB (Cooper et al. 1997; Online Table 1). Northern blot analyses of these cDNA fragments revealed a minimum of six distinct transcripts. Another five EST clones were positioned outside the Best’s disease locus within an ∼1 Mb region proximal to D11S1765. From these results we conclude that refined EST mapping is a useful and reasonably efficient method to add novel transcripts to a genomic region of interest.
Second, we performed direct cDNA selection (Rommens et al. 1993) using cDNA pools from retinal tissue and the RPE cell line ARPE-19. Based on electrophysiological and histopathological data one can assume that the defective gene is expressed in one of these eye tissues. Overall, this approach has led to the identification of 13 transcripts with distinct signals on Northern blot analyses. It is of note that all three genes (DDB1, FEN1, and FTH1) and two EST clones (EST173048, D11S2319E) known to reside within the analyzed genomic region were represented in the retrieved cDNA clones, thus demonstrating the high sensitivity and efficiency of the method used.
We have mapped an additional 17 unrelated cDNA fragments to the disease interval but could not detect mRNA signals in Northern blot hybridizations of total RNA preparations. Although for practical purposes we have not included these cDNAs in our primary transcription map, low or cell-specific expression within a given tissue may account for the lack of Northern blot signals. Therefore, we intend to further characterize these possible low copy transcripts with respect to their presence in RT–PCR experiments and in retinal cDNA libraries. In contrast to the analysis of abundant transcripts, the identification of rare RNA species requires a disproportionally greater effort.
Sequence analyses of the retrieved cDNA clones have revealed a remarkably high percentage (40%) of fragments containing repetitive elements, with the great majority of repeats representing various members of the Alu repeat family (Schmid and Jelinek 1982). One can assume that these findings reflect the presence of unprocessed RNA fragments in the total RNA preparations used for first-strand cDNA synthesis. Accordingly, an illegitimate priming event of the RXGT12 oligonucleotide to stretches of adenine nucleotides present in Alu or L1 elements would then generate exon/intron-containing cDNA fragments. In support of this mechanism are our findings in at least two retrieved cDNA clone contigs [C96-1 (DDB1) and C97A-7] that unambiguously show the presence of coding sequences flanked by conserved splice acceptor and donor sites. Similar observations have been reported previously (Rommens et al. 1995) and have led to the suggestion that the unprocessed cDNAs may be favored in the selection process because of the enlarged target size that allows a more efficient annealing of cDNA/DNA molecules.
Thus far, of the 14 novel transcripts identified within the Best’s disease locus, no information regarding their possible function could be derived from DNA sequence or protein pattern database searches. This may be attributable primarily to a potential selection bias toward retrieved cDNA fragments originating from the 3′ UTRs of the respective genes, thus revealing little or no coding information. In the absence of this crucial piece of information the criteria for the assessment of candidate genes for Best’s disease has to be based purely on their expression profiles in various tissues. Considering the etiology of Best’s disease, several transcripts identified in this study are interesting candidates for the disease gene (e.g., TU3, TU10, or TU12). Their further characterization and mutational analysis in Best’s disease patients is currently in progress.
In summary, we have identified 14 novel transcripts by EST mapping and cDNA selection and have determined their expression patterns in various human tissues, including retina and retinal pigment epithelium. In addition, the precise location of these transcripts within the genomic locus harboring the Best’s disease gene was determined. Together with the five known genes mapped previously to this interval (CD5, PGA, DDB1, FEN1, and FTH1), this increases the number of distinct genes in the disease region to 19. As the physical size of the interval can be estimated to be ∼1.4 Mb, the primary transcription map contains, on average, one gene every 73 kb of genomic DNA. The gene density in this region may even be higher as additional genes with tissue-specific expression other than retina and RPE will likely be added to the primary transcript map. Therefore, we conclude that the Best’s disease region represents one of the more gene-rich regions of the human genome (Fields et al. 1994). Beyond the identification of the Best’s disease gene itself, the complete molecular characterization of this region will add to the efforts aimed at the construction of comprehensive gene and expression maps of the human genome.
METHODS
Genomic Clones
As part of the framework physical map of chromosome 11, YAC clones were isolated from the RPCI chromosome 11-specific YAC library, the CEPH Mega YAC library, and the London ICI–YAC library and assembled into 142 contigs (Qin et al. 1996). For the refined mapping of known ESTs several YAC clones were selected from two nonoverlapping YAC contigs extending the minimal Best’s disease interval in 11q12–q13.1 proximally and distally (Fig. 1A). Yeast cultures were grown in AHC complete medium [1.7 grams /liter of yeast nitrogen base without amino acids and (NH4)2SO4, 5.0 mg/liter of (NH4)2SO4, 10 mg/liter of casein, 20 mg/liter of adenine hemisulfate, adjusted to pH 5.8], and total chromosomal yeast DNA was isolated as described (Scherer and Tsui 1991).
For EST content mapping a minimal set of overlapping PAC clones were selected covering the entire Best’s disease region (Cooper et al. 1997). For cDNA selection seven PAC clones were used from the central region of the disease locus (Fig. 1C). The clones were grown in overnight cultures of LB broth containing 50 μg/ml of kanamycin followed by the isolation of PAC DNA using the standard alkaline lysis technique.
Fine Mapping of Known EST Clones
PCR-based EST mapping was performed using PAC or YAC DNAs as template and oligonucleotide primers designed according to the published sequences in the databases (GenBank accession nos. are given in Online Table 1). Reaction mixtures contained ∼50 ng of template DNA, 15 pmoles of each primer, 1.25 mm dNTPs, and 1× PCR buffer supplemented with 1–2 mm MgCl2 and 0.5 units of Taq DNA polymerase. The reaction mixture was denatured once at 94°C for 5 min and was subsequently subjected to 33 cycles of 30 sec at 94°C, 30 sec at an annealing temperature optimized for each primer pair (55°C–60°C), and 30 sec at 72°C, followed by a final extension of 5 min.
Alternatively, the mapping experiments included Southern blot analyses of EST clones obtained from the IMAGE Consortium clone distributors toEcoRI-digested PAC DNA. Restriction enzyme digestions were carried out as recommended by the manufacturer (GIBCO BRL) followed by electrophoresis and blotting to Hybond-N+ membranes (Amersham) using standard procedures. DNAs from individual cDNA clones were isolated, digested with specific restriction enzymes to excise the respective insert, and separated electrophoretically. Insert fragments were purified in low melting point agarose (Bio-Rad) and radiolabeled by random oligonucleotide priming. Hybridizations were carried out in 0.5m sodium phosphate buffer (pH 7.2), 7% SDS at 65°C (Church and Gilbert 1984).
Direct cDNA Selection
Total RNA was prepared either from frozen tissues or from cell line ARPE-19 derived from human RP epithelium (Dunn et al. 1996) using the RNA-Clean-LS system (Angewandte Gentechnologie Systeme). The cDNA selection was performed essentially as described (Rommens et al. 1993), with only minor modifications. Briefly, RNAs were reverse transcribed using the Superscript preamplification system for first-strand cDNA synthesis (GIBCO BRL) and the RXGT12 oligonucleotide primer (5′-CGGAATTCTCGAGATCTTTTTTTTTTTTT-3′). After poly(A) tailing with terminal transferase (U.S. Biochemical), a cDNA pool was generated by RXGT12-primed PCR at 94°C for 1 min; 2 cycles of 94°C for 30 sec; 37°C for 1 min, 72°C for 2 min, followed by 22 cycles of 94°C for 30 sec; 58°C for 30 sec, and 72°C for 2 min. Prior to hybridization the cDNA pools were preannealed to Cot-1 DNA (GIBCO BRL) enriched with sonicated LINE1 sequences (clones pA10 and pA12 containing 3.4 kb of the 3′ end and 4.1 kb of the 5′ end of the LINE/L1 fragment, respectively, were kindly provided by C. Collins, University of California, San Francisco). PAC clone inserts (∼1 μg) were isolated by NotI digestion, purified using QIAEXII agarose gel extraction beads (Qiagen), and immobilized on Hybond-N+ membrane filters with an average concentration of 60 ng/mm2. The insert filters were subjected to two consecutive rounds of hybridization with a starting mixture of 20 μg of retina and ARPE-19-derived cDNAs. Hybridization time was 4 days at 58°C in Church hybridization buffer (Church and Gilbert 1984). Filters were washed three times in 2× SSC/0.1% SDS at room temperature, once each in 0.5× SSC/0.1% SDS, 0.2× SSC/0.1% SDS, and 0.2× SSC/0.05% SDS (all at 58°C). A final wash was in 2× SSC. cDNAs were eluted in distilled H2O by incubating for 10 min at 98°C and reamplified by PCR using the RXGT12oligonucleotide primer. Four micrograms of the reamplified cDNAs were used for a second round of hybridization. After two rounds of selection the cDNAs were amplified using the RXGT12 oligonucleotide primer, digested with EcoRI and cloned into the EcoRI site of pBluescript (Stratagene).
Analysis of Selected cDNA Clones
Individual cDNA clones were archived in 96-well microtiter plates containing 100 μl of LB/ampicillin. To determine cDNA clone redundancy and to verify the genomic origin of the selected cDNA clones, insert fragments were individually PCR amplified with oligonucleotide primer RXGT12 and consecutively hybridized to nylon membranes containing the gridded cDNAs andEcoRI-digested PAC clone DNAs. Prior to hybridization, fragments were preannealed with sonicated human placental DNA (Sigma).
Sequencing of 300–400 bp of the 5′ and 3′ ends of selected cDNA clones was carried out by the dideoxy chain termination method using the Sequenase version 2.0 DNA sequencing kit (U.S. Biochemical). Sequences were analyzed using the MacVector program version 4.1.4 (Kodak) and publicly available nucleotide and protein databases at the NCBI (Gish and States 1993). GenBank accession numbers of retrieved cDNA sequences are given in Online Table 3.
Northern Blot Analysis
Total RNAs (12 μg) isolated from frozen lung, cerebellum, retinal tissue, and cell line ARPE-19 were separated electrophoretically in 1.2% agarose gels containing 0.6 mformaldehyde and vacuum blotted onto Hybond-N+ membranes. Repeated hybridizations with radiolabeled cDNA inserts were performed as described for the mapping of EST clones above. Final washing conditions were 0.2× SSC/0.1% SDS at 55°C. Multiple-tissue Northern blot membranes containing 2 μg of poly(A)+ RNA were purchased and used repeatedly according to the supplier’s recommendations (Clontech). Stripping of bound cDNA inserts from the membranes was done by incubating in boiled solution of 0.01× SSC/0.01% SDS.
Acknowledgments
We thank Dr. L.M. Hjelmeland (University of California, Davis) for generously providing the RPE cell line ARPE-19, Dr. A. Eckstein (University Eye Hospital, Essen, Germany) for the retinal tissues, and Dr. J. Rommens (Hospital for Sick Children, Toronto, Canada) for kindly introducing us to the direct cDNA selection technique. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (We 1259/2-3).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL bweb{at}biozentrum.uni-wuerzburg.de; FAX 49-931-888-4069.
-
- Received July 22, 1997.
- Accepted November 26, 1997.
- Cold Spring Harbor Laboratory Press








