
Comparative Sequence Analysis of a Gene-Rich Cluster at Human Chromosome 12p13 and its Syntenic Region in Mouse Chromosome 6
Abstract
The Human Genome Project has created a formidable challenge: the extraction of biological information from extensive amounts of raw sequence. With the increasing availability of genomic sequence from other species, one approach to extracting coding and regulatory element information is through cross-species sequence comparison. To assess the strengths and weaknesses of this methodology for large-scale sequence analysis, 227 kb of mouse sequence syntenic to a gene-rich cluster on human chromosome 12p13 was obtained. Primarily through percent identity plots (PIPs) of SIM comparative sequence alignments, the sequence of coding regions, putative alternative exons, conserved noncoding regions, and correlation in repetitive element insertions were easily determined. The analysis demonstrated that the number, order, and orientation of all 17 genes are conserved between the two species, whereas two human pseudogenes are absent in mouse. In addition, apart from MIRs, no direct correlation of distribution or position of the majority of repetitive elements between the two species is seen. Finally, in examining the synonymous and nonsynonymous substitution rates in the conserved genes, a large variation in nonsynonymous rates is observed indicating that the genes in this region are diverging at different rates. This study indicates the utility and strength of large-scale cross-species sequence comparisons in the extraction of biological information from raw sequence, especially when combined with other computational tools such as GRAIL and BLAST.
[The sequence data described in this paper have been submitted to the GenBank data library under accession nos. AC002393 and AC002397.]
While the human genomic sequence is being generated at an increasing rate, the extraction of biological information remains a formidable challenge. Together with exon prediction and gene modeling programs, data bases of ESTs (Hillier et al. 1996) have greatly improved the initial identification and characterization of the sequence. However, the complexity of the human genome requires additional approaches to efficiently determine complete gene structure and elucidate regulatory elements in a manner amenable to large-scale sequence analysis.
Large-scale cross-species DNA sequence comparisons are being used increasingly to address structural and evolutionary questions. Several studies have compared large segments (>30 kb) of human and rodent sequences, indicating that coding domains are generally well conserved, whereas noncoding (intronic and intergenic) domains exhibit highly variable levels of sequence conservation (Koop 1995; Hardison et al. 1997). Sequence comparison of the human and mouse T-cell receptor C-δ and C-α regions (Koop and Hood 1994), the human and hamster α- and β-myosin heavy chain genes (Epp et al. 1995), and the human and murine Bruton’s tyrosine kinase loci (Oeltjen et al. 1997) revealed a high level of sequence similarity at coding and noncoding regions. However, sequence comparison of the human and mouse β-globin gene cluster (Collins and Weissman 1984; Sheehee et al. 1989), the human and rat γ-crystallin genes (den Dunnen et al. 1989), the human and mouse XRRC1 DNA repair gene regions (Lamerdin et al. 1995), and the human, mouse, and hamsterERCC2 gene regions (Lamerdin et al. 1996) showed less conservation of noncoding sequence. A mixed pattern of sequence similarity has been observed in the human and mouse immunoglobulin C(Mu)–C(δ) heavy chain loci (Koop et al. 1996). These studies suggest that portions of human and rodent genomes evolve at different rates (Koop 1995; Hardison et al. 1997).
The importance of cross-species sequence comparison for detection of regulatory elements has been suggested in numerous studies (Koop and Hood 1994; Lamerdin et al. 1995; Oeltjen et al. 1997). Increased conservation of noncoding segments has been observed 5′ and 3′ of the first exon of the genes in the BTK region. The conserved region flanking the first exon of BTK regulates, at least in part, the lineage-specific expression pattern of BTK (Oeltjen et al. 1997). Some of the noncoding sequence conservation could be attributable to constraint by splicing, chromatin condensation, matrix association, and replication origins (Koop 1995). However, some of the conserved noncoding regions clearly represent promoter and transcriptional regulatory elements (Koop 1995; Hardison et al. 1997; Oeltjen et al. 1997).
Although a great deal of attention has been directed toward computer approaches for annotation and gene predictions in a genomic sequence, there is no agreement about the precise goals of large-scale comparative sequence analysis, the computer methods to attain them, or how to report and/or annotate the results. These issues need to be resolved before the benefits of comparative sequence analysis can be accurately assessed.
Previously, we identified a gene-rich cluster comprising 17 complete genes, one partial gene, two likely pseudogenes, and other transcribed regions, at the CD4 locus on human chromosome 12p13 (Ansari-Lari et al. 1996, 1997). The genes in this region possess diverse expression patterns and functions, ranging from signal transduction and glycolysis, to regulation of cell proliferation and ubiquitin-dependent proteolysis (Ansari-Lari et al. 1996, 1997). To gain insight into the evolution of coding and noncoding segments of this region and as a first step in elucidation of the regulatory elements of the genes, we isolated and sequenced the syntenic region in mouse chromosome 6. Several computational tools were utilized for human and mouse sequence comparison. This study indicates that genes closely clustered in one region have evolved at different rates.
RESULTS AND DISCUSSION
Organization of the Genes in the Mouse Sequence
Several mouse BAC clones corresponding to the gene-rich cluster at human chromosome 12p13 were isolated (Ansari-Lari et al. 1996, 1997). Based on PCR and hybridization analyses, one of the clones appeared to contain the majority of genes corresponding to the human cluster. The sequence of this clone was obtained using a modified shotgun M13 sequencing strategy (Richards et al. 1994). The mouse sequence is composed of two contigs totaling 226,708 bp, separated by a small gap of several hundred bases in a repeat-rich region. From the first exon of CD4 to the penultimate exon of C6, the GC content of the human sequence is 51.35%, whereas that of the mouse sequence is 49.68%. The number of CpG islands estimated by GRAIL is 22 and 14 for the human and mouse sequences, respectively. It has been estimated that 20% of the CpG islands in humans are absent in mouse (Antequera and Bird 1993).
Initial analysis of the mouse sequence with PowerBLAST (Zhang and Madden 1997) revealed the presence of all the genes in the human cluster, with the exception of the two putative human pseudogenes. The number, order, and orientation of the genes are conserved between human and mouse. RPL13-2 and Destrin-2, which appear to be pseudogenes in the human (Ansari-Lari et al. 1997), are absent in the mouse sequence, suggesting their formation subsequent to divergence of primates and rodents. The region encompassing RPL13-2and Destrin-2 contains an unusually large number of repeat elements. In mouse, this region is also repeat-rich and contains an ETn mouse early transposon (Sonigo et al. 1987), which is absent in the human sequence.
The boundaries of the coding exons could be determined accurately for all the genes in this region. The exact exon–intron organizations of the mouse CD4, TPI, ENO-2, DRPLA, U7 snRNA, PTPN6,and BAP genes were determined, based on the available published mouse cDNA sequences. In the absence of murine cDNA sequence information, the exact exon–intron organization of genes A, B, GNB3, ISOT, B7, C2f, and C3f were determined using homology to human cDNA sequences.
The nucleotide similarity for the coding domains of the genes in this region ranges from 70.2% for CD4 to 91.9% forISOT-1, whereas the percent amino acid identity is between 56.3% for CD4 and 100% for BAP (Table1). These ranges of values are consistent with earlier observations for other human and murine genes (Makalowski et al. 1996). The sizes of the majority of the coding exons are conserved between human and mouse. Of the 153 coding and noncoding exons in this region (excluding the C6 gene and the two putative pseudogenes), 141 are coding. Twelve of the coding exons in this region do not maintain the same number of nucleotides between the two species, although they differ by multiples of three nucleotides, conserving the reading frame. The sizes of 5′- and 3′-untranslated exons are not highly conserved between the two species. Sequence comparison may not allow the unequivocal identification of the 5′-untranslated boundaries of the genes; however, polyadenylation signals that are present in human are conserved in mouse. Similarly, the genes that are lacking recognizable polyadenylation signals are in common between the two species.
Nucleotide and Amino Acid Sequence Comparison Between the Coding Segments of the Genes
Several new EST matches that do not correspond to previously annotated exons of genes in this region have been observed. Some of these ESTs could be artifactual, whereas others could represent antisense transcripts, which have been hypothesized to play important roles in the regulation of gene expression (Nellen and Lichtenstein 1993). Alternatively, these ESTs could be differentially spliced exons or 5′-untranslated exons. An example of an alternatively spliced exon is seen in gene B7, where the human nucleotide positions 124740–125037 (see Fig. 2, below) show 76% sequence identity to the mouse sequence, and the ORF for this putative exon shows 87% amino acid similarity between the two species. GRAIL-2 (Xu et al. 1994) also predicts an exon corresponding to this region. An EST match (accession no. U25931) indicates splicing of this putative exon to the last exon of B7. This putative exon was not present in the original cDNA sequence for B7; however, human/mouse sequence comparison clearly directed us to look more closely at this region. An example of a putative antisense transcript for gene C3f is the IMAGE cDNA clone 40871 (accession no. U72507). The transcription of this clone, which contains a polyadenylation signal at its 3′ end, is opposite of gene C3f (Ansari-Lari et al. 1997).
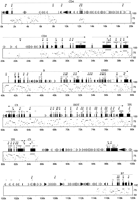
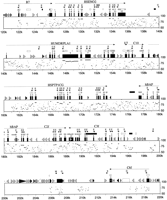
The PIP. The nucleotide position for the human sequence is shown in thex-axis, and the percent sequence identity (50%–100%) is shown in the y-axis. Exons (numbered black boxes), and repeats (SINEs other than MIR are light gray triangles pointing toward the A-rich 3′ end; LINE1s are open arrow boxes; MIR and LINE2 elements are black triangles and pointed boxes, respectively; other interspersed repeats are dark gray triangles) are indicated above the main boxes; BLAST hits in dbEST as shorter white (human), gray (mouse); or black (ESTs discussed in the text) boxes above that; and GRAIL-2 exon predictions (email to grail{at}ornl.gov) above EST matches. The GRAIL-2 hits show direction, extent, and quality [(E) excellent; (G) good; (M) marginal].
Human/Mouse Sequence Comparison
Initially, the human and mouse sequences were compared by dot plot analysis using DOTTER software (Fig. 1) (Sonnhammer and Durbin 1995). The diagonal line represents conserved coding and noncoding segments of the genes. Three major repeat-rich regions can be observed at the beginning, middle, and end of the sequence, interrupting the diagonal line of sequence conservation. In addition, repetitive elements throughout the sequence are represented by matches away from the diagonal. The dot plot indicated a substantial global sequence conservation across the entire segment and the potential for large-scale alignment within well-defined domains.
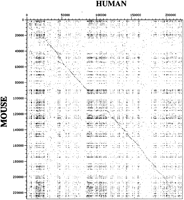
Human and mouse sequence comparison by DOTTER. The human and mouse nucleotide sequence positions are indicated on the axes.
A more detailed approach to cross-species sequence comparison is depicted in Figure 2. The repeat elements were masked by RepeatMasker [A.F.A. Smit and P. Green (1995–1997) http://ftp.genome.washington.edu/cgi-bin/RepeatMasker], and using a modified version of SIM (similar) (Huang et al. 1990), the local alignments scoring above 95% in approximate significance (Schwartz et al. 1991) were determined. The gap-free alignments were subsequently converted into segments of percent identity relative to positions of human sequence, and the resulting data drawn as a percent identity plot (PIP), using a modified version of the local alignment to postscript (LAPS) program (Schwartz et al. 1991). In addition, EST matches and exons predicted by GRAIL-2 were mapped on the PIP.
The majority of the exons could be identified by visual inspection of the PIP, even in the absence of EST and GRAIL-2 matches. In particular, with the exception of 5′- and 3′-untranslated regions, every exon of an active gene shows conservation between human and mouse that contrasts sharply with the typical noncoding region. Alternatively, the PIP could be used to improve predictions made by other means. For instance, of 146 exons predicted by GRAIL-2, the requirement that a predicted exon contain a gap-free aligned segment of at least 40 nucleotides eliminates 21, which are all false positives. Sequence conservation in intronic and intergenic regions is observed throughout the region. In particular, of 171 gap-free aligned regions of at least 100 bp in length, 34 do not overlap an identified exon. However, significantly more sequence conservation is observed upstream of the first exon and within the first intron of each gene. There are regions with strikingly high matches in other introns, such as intron 6 ofB7, and introns 4 and 12 of PTPN6. The PIP indicates that the genes in this region have diverged at different rates, with CD4 being the most divergent. Furthermore, interruption in sequence alignment is observed in highly repeat-rich regions in intron 3 of CD4, intergenic region betweenC9 and B7, and the region downstream ofC3f.
Based on sequence conservation identified by the PIP, three regions with potentially new genes were considered (Fig. 2). All three regions contain at least one EST match and exhibit 60%–86% sequence conservation, at least in part, between the two species. The corresponding human and mouse sequences were analyzed by GRAIL-2 (Xu et al. 1994), FGENEH (Solovyev et al. 1995), and Genie (Kulp et al. 1996) exon prediction/gene modeling programs (data not shown). None of the programs predicted a consistent gene model that was common to the two species in any of these three regions. However this analysis does not exclude the possibility of the presence of a gene in either of these regions. One of these is an intergenic segment between DRPLAand ENO-2, containing an EST match (accession no.M78236). This EST could represent the 5′-untranslated region ofDRPLA. The second region is located between the 3′ ends of PTPN6 and BAP genes. In this region there are two mouse overlapping EST matches (accession nos. AA423763 and AA272334). No continuous ORF is present in either EST. The third region is between the C3f and C6f genes, containing an EST (accession no. T83233) partially conserved between the two species. The segments of sequence conservation in these three regions could represent regulatory elements. Further experiments are required to resolve the significance of these three regions.
Extent of Coding and Noncoding Sequence Conservation
Using a modified version of CONSERVED (Oeltjen et al. 1997), the gap-free segments (⩾50 bp) showing sequence identity (⩾60%) were determined from the overall SIM alignment. Weighted percent identities were calculated for transcribed (coding and noncoding exons) and intronic noncoding regions between the first and last exon of each gene (Table 2). Using these CONSERVED parameters, 85.90% of the total weighted transcribed region, and 72.65% of the total weighted noncoding region are conserved. For CD4, the ratio of transcribed to noncoding sequence conservation has approached unity (Table 2). This is mainly due to extensive divergence at the coding region rather than higher conservation at the noncoding region.
Sequence Conservation of Transcribed and Noncoding Regions
Nucleotide Substitution Rate
The synonymous and nonsynonymous substitution rates of the coding regions were determined by method 1 of Ina (1995) (Table3). Similar results were obtained by Ina’s method 2 (data not shown). On average, the synonymous substitution rate was approximately six times higher than the nonsynonymous substitution rate. As expected, the synonymous substitution rate exhibits less variation (∼1.6-fold range of variation). The gene-specific differences in synonymous substitutions suggest either purifying selection or local differences in mutation rate (Wolfe et al. 1989). The rate of nonsynonymous substitution, on the other hand, is extremely variable among the genes, ranging from 0.01 × 10−9substitution per nonsynonymous site per year for BAP to 2.26 × 10−9 for CD4 (∼225-fold difference in variation). This high variation in nonsynonymous substitution rates could be due to adaptive selection and/or region-specific variation in mutation rate (Li and Graur 1991), although the contribution from the latter would likely be minimal, considering the close physical proximity of the genes in this region.
Synonymous and Nonsynonymous Substitution Rates
In mammals, synonymous substitution frequencies are gene-specific and they correlate with nonsynonymous substitution frequencies, implying that homologous mammalian genes evolve at similar rates (Mouchiroud et al. 1995). In addition, tissue-specific gene expression could determine the rate of evolution of genes, likely attributable to global functional constraints (Kuma et al. 1995). In vertebrates, proteins that are expressed in the immune system appear to evolve more rapidly than those expressed in nervous system, as has been suggested for members of protein kinase and immunoglobulin gene families (Kuma et al. 1995). The rate of evolution of C2 domains of members of the immunoglobulin superfamily, including CD4, have been compared for human and murine rodents (Hughes 1997). There is a strong positive association between expression in immune system and nonsynonymous substitution rate at C2 domains (Hughes 1997). The major histocompatibility complex (MHC) proteins and T-cell receptors, all lacking C2 domains, also exhibit high nonsynonymous substitution rates, which are believed to represent adaptive selection (Hughes 1997). Because CD4 interacts with class II MHC molecules, due to amino acid diversity in the class II MHC molecules, CD4 might be under weaker structural constraints and exhibit a faster rate of evolution (Hughes 1997).
Distribution of Repetitive Elements
From the first exon of CD4 to the putative penultimate exon of gene C6f, repetitive elements represent 33.36% and 26.39% of the human and mouse sequences, respectively. The number and the percentage of sequence occupied by different classes of repetitive elements is summarized in Table 4. These repeats can be mapped to the genomic sequence of the other species with some confidence.
Distribution of Repetitive Elements
Many of the mouse interspersed repeats could be accurately mapped onto human positions as follows. The alignment program was applied to the unmasked human and mouse sequences, and the four largest contiguous alignments, which covered human positions 1349–5688, 29117–85438, 116301–118786, and 124669–194806, totaling 59.8% of the human sequence and 60.9% of the mouse sequence, were extracted. The alignment of masked sequences, which was used to make the PIP, consists of 43 aligned segments totalling 64.8% of the human sequence. Thus, these four pieces contain the vast majority of the aligned positions. The distribution of repetitive elements in the unmasked aligned segments is summarized in Table 4.
As expected, there is a tendency for inserted elements to occur in unaligned regions, as their presence disrupts the alignment. In this respect, Alu repeats are more disruptive than common mouse SINEs, such as B1 elements, because of their greater length, a finding that may explain the lower frequency of Alu repeats in the aligned regions. MIRs (mammalian-wideinterspersed repeats) are highly abundant tRNA-derived SINEs (Smit and Riggs 1995). Detected MIRs in the human sequence did not favor either aligned or unaligned regions. In mouse, many fewer MIR relics were found by RepeatMasker, and they also all occurred in aligned regions. Because of the higher rate of mutation in rodents (Li et al. 1990), one would expect that the MIRs detectable in mice will tend to occur in well-conserved regions of the mouse genome, in accordance with what we observed. MIRs have been found at orthologous sites in different mammals and, hence, are believed to have amplified, at least in part, before the mammalian radiation (Jurka et al. 1995; Smit and Riggs 1995). Available evidence suggests that MIR activity stopped before the divergence of humans and mice (Jurka et al. 1995). Table 5 lists the potential orthologous repeat segments, as suggested by alignments and confirmed by inspection. Several other MIRs were detected by RepeatMasker in one of the sequences but either postdate the rodent–primate split or had mutated beyond recognition in the other species. A MIR fragment at human position 47150–47297 is 68% identical between human and mouse, which suggests that it may have been incorporated into a functional element.Makalowski et al. (1996) report a MIR insertion that retains 65.4% identity. Additionally, MIRs have been identified as parts of coding and 3′-untranslated segments of a few mammalian genes (Murnane and Morales 1995).
Orthologous Repeat Elements
More than half of all LINE-1 elements are believed to be inserted into the genome before the mammalian radiation, based on their presence at orthologous sites in different mammalian genomes (Smit et al. 1995). RepeatMasker identified nine human and nine mouse LINE1 fragments in the region studied here. Only two elements from human and three from mouse could be aligned with any confidence. Each of the human L1s was in roughly the same position as a mouse L1. Furthermore, the orientations, and in one case the LINE1 subclassification, were consistent with orthology. However, extensive divergence of these elements, particularly in mouse, and proximity to other repeat elements ruled out a definitive conclusion as to orthology.
In this study, ∼223 kb of human sequence on chromosome 12p13 was compared with 227 kb of syntenic mouse sequence on chromosome 6. Except for two pseudogenes that appear to be absent in mouse, the number, organization, and orientation of the genes are maintained between the two species. Conservation in the noncoding regions was observed more frequently in the areas flanking the first exon of each gene, representing the likely positions of regulatory elements. The genes in this region appear to be evolving at different rates, as inferred from great variation in their nonsynonymous substitution rates. Several orthologous MIRs were identified, supporting the insertion of MIRs before mammalian radiation. However, for the majority of the repeat elements, no direct correlation between their distributions and positions between the two species was found.
Cross-species sequence comparison permits variations in the rate of sequence conservation to be measured across a genomic region. That information is useful for identifying coding regions, regulatory signals, and other functional segments of the genome. Dynamic programming alignment methods permit precise identification of conserved segments, and recent algorithmic improvements allow 200-kb sequences to be aligned in a minute on a 200-MH workstation.
PIPs provide a useful visualization of sequence conservation. They could be added to interactive programs (Harris 1997; Zhang and Madden 1997) that view results from database searches and gene-prediction programs as an aid to annotating genomic sequence data. Alternatively, information from the alignment can be incorporated into an explicit gene-prediction algorithm, just as results from database searches are now incorporated (Xu and Uberbacher 1997). This will allow the benefits of genomic alignments as a source of information for gene prediction to be measured objectively.
METHODS
Isolation and Sequencing of the Mouse BAC
The initial screen of the mouse BAC library (Research Genetics, Inc.) was performed by PCR using four primer pairs specific for mouseCD4 (R2633, 5′-CCATCTCTCTTAGGCGCTTG-3′ and R2634; 5′-GAACTTCCAGGTGAAGACTG-3′), TPI (R2635, 5′-CTTGGTTTGCTCGAACACGAC-3′; and R2636, 5′-CTGGCATGATCAAAGACTTAG-3′), ENO-2 (R2673, 5′-GATCAATGGTGGCTCTCATG-3′; and R2674, 5′-CTGTTCTCCAGGATATTGGG-3′), and BAP (R2671, 5′-CCTGCACAATCTTCTGTCGC-3′; and R2672, 5′-CAAGGACTTCAGCCTCATCC-3′) genes. The five positive subpools were arrayed on filters and screened using radioactively labeled probes specific for mouse CD4, TPI, and BAP genes. Labeling and hybridization were performed as described (Ansari-Lari et al. 1996). A similar restriction pattern for all overlapping BACs was observed. Only the clone 284H12 was positive for all three probes. The shotgun M13 sequencing library and M13 sequencing templates were generated as described (Andersson et al. 1996a,b). Sequence assembly was performed by GAP (Bonfield et al. 1995) and Phrap software (Phil Green, http://genome.wustl.edu/gsc/finishing/PHRAP-INTRO.html).
Computer Software
The repeat elements were masked using the new release of RepeatMasker [A.F.A. Smit and P. Green (1995–1997)http://ftp.genome.washington.edu/cgi-bin/RepeatMasker]. Initial analysis of the sequence was performed by PowerBLAST (Zhang and Madden 1997) searching databases for nonredundant sequences, ESTs and STSs, using default parameters. CpG islands were found by GRAIL (http://avalon.epm.ornl.gov/Grail-1.3). Dot plot analysis was performed using DOTTER (Sonnhammer and Durbin 1995). cDNA and protein sequence comparison was performed by the GCG BestFit program. Synonymous and nonsynonymous substitution rates were determined using DISTS1 and DISTS2 methods from Ina (1995). GRAIL-2 (Xu et al. 1994), FGENEH (Solovyev et al. 1995), and Genie (Kulp et al. 1996) were employed for exon prediction/gene modeling programs.
Alignments of the genomic sequences were computed using the parameters specified in Oeltjen et al. (1997), though a new alignment program was written to give a 100-fold speed-up. Alignment analysis (by the CONSERVED program) and display (Fig. 2) were performed as described (Oeltjen et al. 1997). Programs were written to automatically process output from RepeatMasker, Grail-2, and BLAST (Altschul et al. 1990), where EST hits were required to have length of at least 60% and at least 90% identity.
Acknowledgments
We thank Dr. Arian Smit for helpful discussions and Michael Chiu for help in submission of the data. This work was supported in part by the grant RO1 HG01459 from the National Center for Human Genome Research. S.S., Z.Z., and W.M. were supported by grant LM05110 from the National Library of Medicine.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL agibbs{at}bcm.tmc.edu; FAX (713) 798-5741.
-
- Received September 8, 1997.
- Accepted December 2, 1997.
- Cold Spring Harbor Laboratory Press








