
Computational and Biological Analysis of 680 kb of DNA Sequence from the Human 5q31 Cytokine Gene Cluster Region
Abstract
With the human genome project advancing into what will be a 7- to 10-year DNA sequencing phase, we are presented with the challenge of developing strategies to convert genomic sequence data, as they become available, into biologically meaningful information. We have analyzed 680 kb of noncontiguous DNA sequence from a 1-Mb region of human chromosome 5q31, coupling computational analysis with gene expression studies of tissues isolated from humans as well as from mice containing human YAC transgenes. This genomic interval has been noted previously for containing the cytokine gene cluster and a quantitative trait locus associated with inflammatory diseases. Our analysis identified and verified expression of 16 new genes, as well as 7 previously known genes. Of the total of 23 genes in this region, 78% had similarity matches to sequences in protein databases and 83% had exact expressed sequence tag (EST) database matches. Comparative mapping studies of eight of the new human genes discovered in the 5q31 region revealed that all are located in the syntenic region of mouse chromosome 11q. Our analysis demonstrates an approach for examining human sequence as it is made available from large sequencing programs and has resulted in the discovery of several biomedically important genes, including a cyclin, a transcription factor that is homologous to an oncogene, a protein involved in DNA repair, and several new members of a family of transporter proteins.
[The sequence data described in this paper are available via the internet athttp://www-hgc.lbl.gov/sequencearchive.html.]
The Genome Project has shifted only recently to the sequencing phase for humans (Marshall and Pennisi 1996) while significant progress has already been made on the sequencing of selected model organisms. The genomic sequence of several organisms, including two eubacteria (Fleischmann et al. 1995; Fraser et al. 1995), an archaeon (Bult et al. 1996), and the extensively studied eukaryote, Saccharomyces cerevisiae (Walsh and Barrell 1996), have already been completed. The strategy employed to computationally identify and analyze putative genes in these model organisms has consisted of identifying protein-coding open reading frames (ORFs) followed by a search of the databases to determine whether these ORFs are homologs of previously characterized genes. Surprisingly, almost one-half of the protein-coding ORFs revealed during the analyses of these model organism genomes have shown no homology to previously characterized genes (Dujon 1996). In contrast to the genomes of model organisms, in which contiguous and annotated sequence data were released at defined intervals, human genomic sequence is being released to the public domain in noncontiguous minimally annotated fragments. Because the human genome is significantly larger and more complex, it is clear that the approaches employed to analyze it will have to vary from the approaches used previously for the genomes of model organisms.
The annotation of human genomic sequence is facilitated greatly by the availability of the large public expressed sequence tag database (dbEST) so that transcribed regions of the genome can be identified, whether or not homology to a previously characterized gene is present. Even in the absence of protein and EST similarity matches, gene prediction programs can provide important clues about the location of new genes in unannotated human genomic sequence. Although many putative new genes can thus be computationally predicted, ascertaining whether the putative new gene is transcribed and the tissues in which it is expressed can only be determined by experiment.
The 5q31 region is of particular interest for large-scale sequencing because of the presence of the cytokine gene family and the fact that a quantitative trait locus associated with inflammatory diseases has been mapped in this region (Marsh et al. 1994; Bleecker et al. 1995; Postma et al. 1995). The cytokines interleukin-3 (IL-3),IL-4, IL-5, IL-13, and granulocyte–macrophage colony-stimulating factor (GM-CSF) are clustered within a 1 Mb region of each other on chromosome 5q31 (Saltman et al. 1993;Nimer and Uchida 1995; Smirnov et al. 1995). Although the loci encoding these proteins are not homologous at the nucleotide or amino acid level, they are considered a gene family because of their localization, overlap in biological activities, and secondary and tertiary structural similarities. It is remarkable that because of the uniqueness of each cytokine, new ones have been discovered not based on homology to old ones but solely using classical methods of cloning genes based on biological activity. Because of the clustering of the cytokine gene family on chromosome 5q31, it has been hypothesized that other, as yet unidentified, interleukins may be located in this region.
In this report we have computationally and biologically analyzed the genomic organization of the cytokine gene cluster region on human chromosome 5q31 and compared it to the genomic organization of the syntenic region on mouse chromosome 11q. As a result of our efforts we have identified a large number of new genes, determined their expression patterns and, in many cases, predicted their possible functions.
RESULTS
Physical Map, Sequencing Strategy, and Contig Assembly
Overlapping P1 and P1-derived artificial chromosome (PAC) clones from the 1-Mb region on chromosome 5 containing the cytokine gene family were isolated by screening genomic libraries (J. Cheng 1996, Chromosome 5 Physical Mapping, http://www-hgc.lbl.gov/clone-info.html) by hybridization and PCR. The minimum tiling contig of the region, which is represented schematically in Figure 1, consists mainly of P1 clones, although one gap in the contig is filled with a PAC clone. The directed sequencing strategy used to generate the data has been described previously (Martin et al. 1995). The sequence of the entire 1-Mb region on chromosome 5 containing the cytokine gene family is being generated and released as assembled fragments ∼3 kb in size (LBNL 1996, LBNL/BDGP Sequence Archive,http://www-hgc.lbl.gov/sequence-archive.html). We used the BSPASS program (S. Pitluck 1996, Towards Automated Assembly for the Directed Sequencing Strategy, http://www-hgc.lbl.gov/inf/spass.html) to build the currently available overlapping sets of assembled 3-kb DNA sequence into 34 blocks of sequence, ranging in size from 1.6 to 95.8 kb, which together compose a total of 680 kb of sequence.
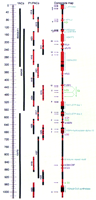
Physical map of the megabase region containing the cytokine gene cluster on human chromosome 5q31. The scale on the left is in kilobases. In the middle, YAC clones are represented by the medium-length bars, P1 and PAC clones are represented by the shorter bars. The long bar on the right represents a composite map of the overlapping P1 and PAC (H37) clones in the region. The regions depicted in red have been sequenced and analyzed in this study while the regions depicted in black have not been sequenced and/or analyzed. Known and putative genes are color coded according to the computational method by which they were identified as described in Fig. 2. (Dark blue) Genes that were sequenced previously. Except for Ril, all of the genes that were sequenced previously had also been localized to this region of 5q31. (Light blue) Putative genes identified by homology to known proteins. (Green) Putative genes identified by EST matches. (Purple) Putative genes identified by analysis of GRAIL-predicted exons and verified by expression studies. The genomic regions containing known and putative genes that have protein database matches are indicated by an increase in the width of the composite map bar. The direction of transcription for each gene is indicated by a vertical arrow. Locations of identical EST matches are indicated by the horizontal arrows on the left side of the composite map. The number next to the arrow indicates how many EST matches were found at each location. An arrow without a number represents a single EST match.
Computational Analysis
Seventeen new genes were discovered computationally in the 5q31 region and their expressions verified using the strategy diagrammed in Figure 2. The distribution of these 17 new genes as well as the 6 genes previously known to lie in 5q31 are illustrated in Figure 1.
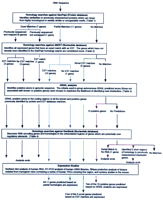
Approach used to computationally and biologically analyze the 5q31 sequence data. The homologies and expression patterns of the putative genes identified during this analysis are described in Tables 1–4.
Comparison of protein translations of the 5q31 sequence data with the proteins in GenPept identified 18 matches: 6 (33%) were exact matches to known genes mapped previously in this region, 1 (6%) was an exact match to a previously identified human gene whose location had not yet been determined, 4 (22%) were orthologs of characterized genes in the rat, mouse, and Drosophila organisms, and 7 (39%) were partial matches that ranged from being recognizable motifs to being moderately homologous with known genes (Table 1; Figs. 1 and3). Analysis of these GenPept database matches localized, determined the direction of transcription, and predicted the functions of the new 5q31 putative genes based on their similarities to known genes. In addition, comparison of human and mouse or rat orthologous genes frequently identified the splice sites in the human gene.
Expression Analysis of 12 New Putative Genes on 5q31 Discovered Based on Homology to Previously Characterized Genes


Summary of the computational analysis of the DNA sequence data in the 5q31 region. The scale is in kilobases. The locations and directionality of the putative genes are indicated by arrows and were determined based on database matches and/or GRAIL predictions. Similarity matches to known proteins, ESTs, and to the noncoding regions of known genes are color-coded as described in the key. If the conceptual translation of a putative gene was homologous to more than one protein then the locations of the similarity matches to those proteins with the highest and second highest BLASTX scores are indicated. Matches with human ESTs generated by the WashU–Merck Human EST project are distinguished from matches with ESTs generated by other groups. Only exact human EST matches and mouse EST matches with 85% or greater nucleotide identity are shown. If an EST contained gaps and matched the genomic sequence in more than one place, this was presumed to be attributable to the splicing of an intron and the EST segments were joined together by a line. GRAIL predicted exons in the forward strands are distinguished from those in the reverse strand by their color, as described in the key. If GRAIL predicted exons at the same location in both the forward and reverse DNA sequence strands, those that correspond to the coding strand (i.e., those GRAIL predictions having the same direction as the putative gene) are shown.
Similarity searches against dbEST localized 155 exact matching human and 19 highly similar (85% or greater identity at nucleotide level) mouse ESTs in this 1-Mb region of 5q31. To avoid using an arbitrary percent nucleotide identity cutoff, high scoring human ESTs were assessed to be an exact match or not based on individual inspection. Analysis of the human EST data revealed 19 genes that contain at least one exact match, of which 4 were known genes, 10 had been identified in the GenPept database searches based on similarities to known genes (Table 1), and 5 were new putative novel genes (Table2). We named these new putative novel genes E1–E5 to indicate that they had been identified by EST matches. Further analysis of the human and mouse EST matches localized and determined the direction of transcription of the E1–E5 genes, as well as yielded information about the spatial expression patterns of the putative genes in 5q31 and the relative abundance and splice sites of their transcripts (Figs. 1 and 3). For example, the Homo sapiens (HS) septin putative gene had 51 exact human EST matches, obtained from six different tissues, thus suggesting that it is a highly expressed ubiquitous gene. On the other hand,E1, E2, E3, E5, HSRil, cyclin-like, IL-4, and GM-CSFhad only one or two exact EST matches and IL-13, IL-5, andIL-3 had no exact EST matches, suggesting that these genes are either expressed at low levels or in a tissue-specific manner, as is known to be the case for the interleukin genes.
Expression and Structural Analysis of Five Putative Genes Discovered by EST Matches
The gene prediction program GRAIL was employed to locate exons in the 5q31 sequence. GRAIL predicted a total of 484 exons, of which 259 (54%) were associated with known genes or the putative genes identified through the GenPept and dbEST database searches, 67 (14%) were associated with repetitive elements, such as the LINE family called L1, which contain ORFs, and 158 (32%) were completely novel. These 158 novel GRAIL predicted exons were analyzed to identify new genes without either protein or EST database matches. The criteria used to group the GRAIL predicted exons into putative genes maximized the likelihood of identifying new interleukins. All the members of the 5q31 cytokine family, although lacking sequence homology, possess four exons spanning between 2 and 9 kb of genomic sequence. Thus, the novel GRAIL-predicted exons were analyzed to identify genomic regions that fit the following criteria: (1) contained four or more excellent or good GRAIL-predicted exons on the same strand of DNA sequence; (2) of the GRAIL predicted exons, at least four had to be located within 9 kb of one another; and (3) the opposite strand could not obviously code for a gene. Ten genomic regions were identified that fit the above criteria and were named G1–G10 to indicate that they had been identified by GRAIL analysis (Table 3; Fig. 3).
Expression and Structural Analysis of 10 Putative Genes Identified by Analysis of GRAIL-Predicted Exons
To assess the performance of GRAIL we examined the GRAIL predictions of five genes, IRF1, IL-4, IL-5, IL-13, and GM-CSF, in which the genomic structure, including the intron/exon boundaries, has been reported previously in detail. Of the 25 coding exons comprising these five genes, GRAIL Ia identified 18 (72%) with a false-positive rate of 10% (percent of predicted exons that are not real), whereas GRAIL II identified 21 (84%) of the exons and had a false-positive rate of 19%. If the GRAIL Ia and II exon predictions are combined, 23 (92%) of the known exons were identified with a false-positive rate of 15%. For the set of 21 exons recognized by GRAIL II, 11 (52%) had both splice junctions predicted correctly and 20 (95%) had at least one splice junction predicted correctly, whereas GRAIL Ia predicted correctly one of the two splice junctions for only 2 (11%) of the exons it recognized, and none of the exons had both splice junctions correctly predicted. It should be noted that GRAIL is a “learning” program, and the specific results will vary over time as the genes used in the training set are changed.
Comparison of the 5q31 DNA sequence data with the DNA sequences in GenBank identified a putative new RNA encoding gene and two regions, which are potential DNA regulatory sequences, homologous to sequences in the untranslated regions of known interleukin genes (Table 4; Fig.3). Both of these regions of homology were short but statistically significant; one was to a region 5′ of IL-13(86 bp) (91% identity), and the other was to the third intron ofIL-4 (52 bp) (78% identity); these regions were named IL-13SH and IL-4SH, respectively.
Analysis of 5q31 DNA Sequence Matches to the Noncoding Regions of Known Genes
Biological Analysis of Computationally Predicted Genes
Expression Studies Using Materials Developed from Human RNA
To biologically verify and determine the expression patterns of the computationally predicted genes, a variety of expression studies were performed (Tables 1–3). The putative genes were first examined for expression by RT–PCR analysis using four human cDNA libraries (infant brain, HeLa, placenta, and T-cell), and oligonucleotide primers were chosen based on homology matches and GRAIL predictions. RT–PCR analyses demonstrated that 10 of the 11 putative genes identified by similarities to known proteins, 4 of the 5 novel genes identified by EST matches, and 2 of the 10 putative genes predicted based on GRAIL analysis were expressed. Northern blot analyses were used to examine the transcript sizes and tissue distributions of the putative new genes identified by protein and EST database matches (Tables 1 and 2). Northern blot analyses were also used to decipher questions arising from the computational studies of the 5q31 sequence data. In several cases, it was unclear whether an EST corresponded to a particular gene. One reason was that the GenPept and dbEST database matches did not overlap, for example, the APXL2 putative gene appears to be located in the intron of EST (N48057). An alternative reason was that the gene sequence was incomplete, as in the case of HSacyl-CoA(coenzyme A) synthetase and the ESTs at its 3′ end (Fig. 3). Examination of transcript sizes and expression patterns by Northern blot analyses determined that APXL2 and EST (N48057) derive from same gene and similarly forHSacyl-CoA synthetase and the ESTs at its 3′ end.
Expression Studies Using Human YAC Transgenic Mice
To develop substrates for examining expression patterns and possible functions of the identified putative genes, we created a panel of transgenic mice containing three human yeast artificial chromosomes (YACs), 854G6-F1 (350 kb), A94G6 (450 kb), and 131F9 (500 kb), spanning 900 kb of the 5q31 region (Fig. 1). Previous studies of transgenic mice containing human genes on large insert vectors, such as YACs, bacterial artificial chromosomes (BACs), and P1s, have shown that the human transgenes are usually expressed in an appropriate spatial- and temporal-specific manner (Frazer et al. 1995; Smith et al. 1995). RNase protection assays, which are highly specific and detect a single-base-pair difference between the probe and the transcript being analyzed, were performed on numerous tissues isolated from the 5q31 YAC transgenic mice using probes for the HSseptin, HSKIF3, andHSRAD50 putative genes (Table 1). Expression of theHSseptin gene was detected in all tissues examined by RNase protection assays and Northern blot analysis of human RNA. RNase protection assays are more sensitive than Northern blots; therefore, expression of the HSKIF3 and HSRAD50 genes was detected in some tissues by the former method of analysis but not by the latter. Nevertheless, tissues in which expression by Northern blot analysis was detected typically had stronger signals in the RNase protection assays than other tissues.
The 5q31 YAC transgenic mice that we have developed will serve as valuable reagents to study the details of expression, genomic organization, and biological properties of genes in this region. This is exemplified by RNase protection assays of several GRAIL-predicted exons in the HSRAD50 gene, some of which are not homologous with the yeast RAD50 gene. These GRAIL predicted exons all displayed identical patterns of expression and thereby supported the hypothesis suggested by the 5′ and 3′ similarity matches of the human and yeast RAD50 genes, namely that HSRAD50 is a single gene >100 kb in length.
Comparative Mapping of the 1-Mb Region Containing the Interleukin Gene Family in the Mouse and Human Genomes
The mouse interleukin gene family is located on the long arm of chromosome 11 in a region syntenic with human chromosome 5q31 (DeBry and Seldin 1996). The genes known to lie in this segment include (from proximal to distal) IL-4, IL-13, IL-5, IRF1, GM-CSF, andIL-3. To ascertain whether the new human genes that we identified in 5q31 are also located in this syntenic region of the mouse genome, we isolated a series of mouse YACs by PCR using primers directed to the mouse IRF1 and GM-CSF genes. These mouse YACs were tested for the presence or absence of eight of the new genes (Fig. 4) by Southern analysis using hybridization probes generated from human genes. All eight of the human genes hybridized to the mouse YACs. Restriction analysis of the mouse YACs also indicated that the eight genes are in the same proximal-to-distal order in the mouse and human genomes (data not shown). These comparative mapping studies demonstrate that at least a significant fraction of the new human genes identified through our analysis of 5q31 sequence data is syntenic in the mouse and human genomes.

Content mapping of mouse YAC clones from the 11q region. The map at thetop is based on the relative order and spacing of the genes in the syntenic 5q31 region in the human genome.
DISCUSSION
The approach that we employed to identify genes in a large segment of the human genome relied first on searching for sequence similarities in protein and EST databases, second on coding potential predictions, and finally on a panel of biological confirmation studies. This is different from the approach used to analyze the genome of S. cerevisiae, as well as other model organisms, which first identified ORFs and then compared the ORFs with sequence databases to determine protein and nucleic acid homologies. This latter approach was successful for the analysis of the yeast genome because of both its compactness (intergenic regions are short and introns are rare), and its simplicity—only 6%–7% of ORFs do not correspond to real genes (Dujon 1996). Because the overwhelming majority of yeast ORFs are real genes, biological confirmation of expression is not necessary for initial purposes. In comparison, the human genome is very complex. It contains large intergenic and intronic sequences, numerous repetitive elements some of which contain protein coding ORFs, and genes that are typically composed of multiple exons whose boundaries are difficult to predict. Although the strength of human gene recognition programs lies in accurate prediction of coding regions (exons), their weakness is in splicing these exons together to correctly predict gene structure. This is especially problematic if a long genomic sequence contains exons from multiple genes or if it contains some gaps. For the foreseeable future it will be standard practice to analyze large segments of human genomic sequence that contains gaps and frequently will code for multiple genes. It is considerably easier therefore to annotate human sequence by first identifying the locations of putative genes based on protein and EST database matches and to then use gene recognition programs to identify those genes without database matches.
In this study we computationally and biologically annotated 680 kb of noncontiguous genomic sequence in a 1-Mb region of the 5q31 cytokine gene cluster region resulting in the identification and verification of 23 genes, 16 of which had not been reported previously. Approximately one-third of the computationally identified new 5q31 genes were not homologous to previously characterized genes. These results suggest that similar to S. cerevisiae and the other model organisms, a large proportion of the human genes remaining to be discovered will be novel and lack homology with any of the currently existing sequences in databases. Of the 11 new human 5q31 genes that were homologous to known genes, the quality of the sequence similarities ranged from recognizable motifs to highly conserved orthologs of mouse and rat genes. Naturally, the greater the level of sequence similarity between the new 5q31 gene and its homolog, the more confident we were in using that similarity as a form of sequence annotation and assigning a putative function.
Of the 23 genes identified in 5q31, 19 (83%) had at least one exact matching EST in dbEST. These data are congruent with the recent analysis of the ESTs generated by the WashU–Merck Human EST Project, which indicated that between 50% and 80% of all the human genes have at least one exact EST match (Hillier et al. 1996). Greater than 85% of the human ESTs in dbEST were deposited by this project, which for the most part generated ESTs from oligo(dT)-primed normalized libraries constructed from 17 different tissues. Therefore, it has been predicted that genes that are primarily expressed in cell types and tissues, such as T cells, thymus, testes, and others, not used to generate the WashU–Merck ESTs will be underrepresented in dbEST. This prediction was confirmed in our study, where three of the five interleukins, which are predominantly expressed in T cells, had no EST matches. Despite the fact that the libraries used to generate these ESTs were normalized, one-third of the ESTs in the 5q31 region matched the HSseptin gene. This demonstrates that even though normalization brings the frequency of all cDNA clones to within a narrow range, highly expressed transcripts will still be present at a greater frequency. Because the libraries used to generate the WashU–Merck ESTs were oligo(dT)-primed and normalized, which favors truncated clones over their longer counterparts, the majority of the ESTs in dbEST are in the 3′-untranslated regions of genes. These ESTs are extremely useful for gene-based mapping strategies and sequence annotation of the 3′-untranslated regions of genes but are less useful for sequence annotation of coding regions. Because the average aligned nucleotide and amino acid identity of mouse and human orthologous genes is 85% in the coding region (Makalowski et al. 1996), the 400,000 mouse ESTs currently being generated (Washington University and Howard Hughes Medical Institute Mouse EST Project;http://genome.wustl.edu/est/mouse/#est.mpg.html) are potentially a great resource to annotate coding regions of human genomic DNA, as the libraries used to generate these mouse ESTs should contain longer insert cDNA clones because of improved methods of normalization and subtraction (Bonaldo et al. 1996).
The performance of gene recognition programs in identifying genes lacking both protein and EST database matches depends on the criteria used for analysis. The criteria used in this study to group GRAIL predicted exons into putative genes were chosen in part to identify new interleukins in the 5q31 region. Of the 10 putative genes predicted based on GRAIL analysis, two, G4 and G5, were confirmed to be expressed by RT–PCR analysis. Because G4 and G5 may represent nonhomologous coding regions of the RATLSTP-like2, andHSpropyl4-hydroxylase genes, respectively, they are not counted as separate genes in this study. The GRAIL-predicted coding regions of several putative genes identified by this analysis, G1, G2, G3, and G8, share features with the GRAIL-predicted coding regions of the interleukin genes (Fig. 3); however, their expressions were not detected by RT–PCR analysis.
It is worthwhile to examine what types of genes would have been missed by our analysis. These omissions would include genes that both lack database matches and whose sequences would not be recognized by GRAIL, such as genes whose transcripts are not translated into proteins. Because of the criteria we used to group the novel GRAIL-predicted exons into putative genes, our analysis also would not have identified genes that lack a database match and either code for short proteins or have long introns.
To biologically verify the computationally predicted genes, we used RT–PCR analyses of human cDNA libraries to rapidly determine whether a predicted gene was expressed. We then relied on Northern blot analysis of human RNA and RNase protection assays of 5q31 human YAC transgenic mice to determine transcript sizes and expression patterns, and to clarify ambiguous situations such as whether two expressed sequences are part of the same gene. In general, these two methods of analyses as well as EST tissue-specific frequencies, indicated similar expression patterns of the new 5q31 genes. In several cases, however, one method indicated that a gene was expressed in a particular tissue while the other methods indicated that it was not. Because the substrates used for these analyses were generated from RNA isolated at different developmental stages, the differences observed may be attributable to temporal regulation of gene expression. EST data errors may be another explanation for some of the discrepancies observed. For example, ∼2% of the WashU–Merck cDNA clones are derived from intronic or intergenic sequences (Hillier et al. 1996); therefore, it is possible that the single EST match defining the E5 putative gene, which is not detected by the RT–PCR and Northern analyses, may be spurious.
In the future, whole genome expression studies are likely to be performed using automated high-throughput expression technologies (Lander 1996). These technologies, however, will not be suitable for exquisitely examining expression patterns spatially, temporally, and under variable stimuli, which for many genes may offer invaluable clues about function. YAC transgenic mice can provide much of this information and because of the large size of the human genomic insert it is possible to examine multiple human genes in a single line of animals. For example, in this study with three lines of transgenic mice each containing a single human YAC, we have the potential to examine in detail the in vivo properties of >900 kb of human DNA containing >20 genes.
In these studies we have employed a strategy that allows an in-depth computational and biological analysis of human genomic sequence data as they are produced. We have discovered a dense clustering of genes in the 5q31 cytokine gene cluster region and identified several genes that can be tested to determine whether they are the quantitative trait locus associated with inflammatory diseases that has been mapped to this region. Furthermore, we have demonstrated that the new genes identified in the 5q31 region are conserved in content and order in the syntenic region of the mouse genome.
METHODS
Computational Analysis
Alu and other human repetitive elements were identified and masked using a combination of BLASTN version 1.4 (Altschul et al. 1990) (default parameters) searches and XBLAST (Claverie and States 1993; Claverie 1996) filtering against the Alu.327.dna (National Center for Biotechnology Information; ftp://ncbi.nlm.nih.gov/pub/jmc/alu/) and humrep (National Center for Biotechnology Information;ftp://ncbi.nlm.nih.gov/repository/repbase/REF1/humrep.ref) databases, respectively. Protein translations of the masked 5q31 sequences were compared with the sequences in GenPept release 94 using BLASTX with default parameters and the SEG (Wootton and Federhen 1996) plus XNU (Claverie and States 1993; Claverie 1996) programs to remove low entropy similarities. The HSPcrunch program (Sonnhammer and Durbin 1994) was used to simplify the BLASTX output. The masked 5q31 sequences were compared with the dbEST release 072996 and GenBank release 93 using BLASTN with default parameters. The unmasked 5q31 sequences were analyzed for potential coding regions using XGRAIL (v. 1.3b; Uberbacher et al. 1996).
RT–PCR Analysis
cDNA libraries were purchased from Clontech Laboratories (catalog nos. HL3003a, HL5014a, HL5013a, and HL5007a) and amplified according to the manufacturer’s recommendations. PCR amplifications of known and putative genes were performed as follows: 50 ng of cDNA from each amplified library was mixed with 80 μm of each deoxyribonucleoside triphosphate, 20 ng of each oligonucleotide primer, 2 μl of 10× PCR buffer (Boehringer), and 1 unit of Taqpolymerase (Boehringer) in a 20-μl volume. The samples were amplified using standard PCR reaction conditions in an automated (Perkin Elmer Cetus) thermal cycler for a total of 35 cycles.
RT–PCR analyses were performed using the following primers chosen based on database matches and GRAIL predictions: LAF-4-like(120 bp)—forward, 5′-TGCTCTTTGGAAAGCTGTTCAGCTTGG-3′; reverse, 5′-TCTTCAGTGACCATTCCACAGAAGATC-3′; APXL2 (114 bp)—forward, 5′-CAGGAAAGCTGGATCGTGTGG-3′; reverse, 5′-CCGGATGCTGGAACTCCAAGG-3′; HSseptin2 (125 bp)—forward, 5′-GAACATTATTCCCATCATGCC-3′; reverse, 5′-CATCATCCGTGGCCAACTG-3′; cyclin-like (235 bp)—forward, 5′-AGCTTGGCCACAGGATGGGCATTAAGCCT-3′; reverse, 5′-AAGGATTCCTCTGAGGCAGC-3′; HSKIF3 (220 bp)—forward, 5′-CCTCAAGTTCTCTGCGAAGTTC-3′; reverse, 5′-CTTGAAACAAAGCTCGACATG-3′; HSRAD50 (91 bp)—forward, 5′-TTCACAATCTCTGAGCACTGATCG-3′; reverse, 5′-AAGATTTTGTGGAGCTTTTAGGACGT-3′; RATLSTP-like1 (118 bp)—forward, 5′-CACCACCAGAGTGCCACG-3′; reverse, 5′-TATTTTATGCATTTGGCTACATGG-3′; HSprolyl4-hydroxylase α(II) (153 bp)—forward, 5′-GAGCTCCAGGGCACGGTGCAG-3′; reverse, 5′-GAAGGGGACTATTATCATACG-3′; ankyrin repeat motif (125 bp)—forward, 5′-GCTGCAAAATGCAGAGGTGATT-3′; reverse, 5′-GGGTGTACTCCTCTACATTATGCATG-3′; HSacyl-CoA synthetase (124 bp)—forward, 5′-GATCCTGAGGATACTGCGACTG-3′; reverse, 5′-GTGAGTGAACCAGTAGGCAAG-3′; E1 (241 bp)—forward, 5′-AAGTCGAGCTCCTTCAGCAAG-3′; reverse, 5′-TGATTGGGCTGCAGTCTTG-3′; E2 (185 bp)—forward, 5′-AGGAACGTGGTTTAATTGTGCA-3′; reverse, 5′-GTCTGTTGCTGTATATGGCAGAGCT-3′; E3 (275 bp)—forward, 5′-AGCCACAGCACTTAGAGCTGAG-3′; reverse, 5′-AAGTGGATCAGTGAGCACAGCT-3′; E4 (238 bp)—forward, 5′-GGAATAGGTGTCCCTGGGAAC-3′; reverse, 5′-GCAAAGAGACCAAGCATGTCTG-3′; E5 (150 bp)—forward, 5′-AGATTCACATGAATTAGGAGCTACAC-3′; reverse, 5′-ATGAGATGCTGCTTTGAGCCCTTGG-3′; G1 (99 bp)—forward, 5′-TTGCACAGGCAAGTAACCAGGCCATC-3′; reverse, 5′-TAGGCTGCGGAATTCTTCTCTGATCAGTA-3′; G2 (90 bp)—forward, 5′-CATCACCACCACCATCATCATTAATAGCAC-3′; reverse, 5′-AGACCTGGGGCCACCTGCGACTTC-3′; G3 (73 bp)—forward, 5′-ACCACTCCACACCTCGCAGCT-3′; reverse, 5′-CCCTGGCTTGTTCTTGGTGCC-3′; G4 (120 bp)—forward, 5′-TGGCACGGCAGTATAAGGC-3′; reverse, 5′-CAGTGCCAGTGTACAGGGCC-3′; G5 (76 bp)—forward, 5′-GAGAGCTGGGCACCTCCAAGCAG-3′; reverse, 5′-TCCAGAGTACCCCTGCCAGAGATG-3′; G6 (164 bp)—forward, 5′-GTACCAGAAGCTCCCTGTCAACCC-3′; reverse, 5′-TCAGTCTCTCAGCCACAGAGTCCG-3′; G7 (161 bp)—forward, 5′-CCATGTCCATGTAAACCTTCGTTATGTG-3′; reverse, 5′-GGATGAGGCTGGGTCATCTGGTGC-3′; G8 (121 bp)—forward, 5′-AGCTTGGAACACCAGGACAGGGAAC-3′; reverse, 5′-CTGCTCTCCCAGGCTGATGGCCAG-3′; G10 (118 bp)—forward, 5′-GTGAATCCTTCTGACTGTGCTATAG-3′; reverse, 5′-ACTCAATCACACTCAAGTCAATCTC-3′.
Southern and Northern Hybridization Analyses
Mouse YAC DNA was separated on Bio-Rad CHEF Mapper with 120 sec initial pulse to 20 sec final pulse time at 6 V/cm for 22 hr. DNA was then transferred to a Nytran Plus membrane (Schleicher & Schuell). An adult human multiple tissue total RNA blot was purchased from Clontech Laboratories. Probe DNA fragments of known or putative genes were amplified by PCR and purified from agarose gels on DEAE membranes (Schleicher & Schuell) using standard protocols. Approximately 25 ng of purified DNA was labeled with [α-32P]dCTP using Megaprime DNA labeling systems (Amersham Life Science) according to the manufacturer’s protocol. Both Southern and Northern blots were prehybridized in 0.5 m sodium phosphate (pH 7.2), 1 mm EDTA, and 7% SDS for 1 hr at 65°C. Hybridization was then carried out in the same solution with addition of 100 μg/ml of sheared salmon sperm DNA and radiolabeled probe, and incubated overnight at 65°C. The filters were washed once with 2× SSC–0.1% SDS, twice with 0.5× SSC–0.1% SDS, and if necessary once with 0.1× SSC–0.1% SDS at 68°C, and then exposed to X-ray films with an intensifying screen at −80°C.
Hybridization probes were generated using the following primers:LAF-4-like (323 bp)—forward, 5′-AGCTACACTGATACAAGTGGACCTAA-3′; reverse, 5′-GGGGAAGACTTAGACTCCTTCTTT-3′; APXL2 (253 bp)—forward, 5′-TCCCTGCTGCAGCGACTCCGGCTCC-3′; reverse, 5′-AAGGTCGTCCCTGATGGCGTCCAGTTG-3′; HSseptin2 (202 bp)—forward, 5′-AGACCGGCATTGGCAAAT -3′; reverse, 5′-GCCTCTTGCCTCTCATCCTT-3′; cyclin-like (235 bp)—forward, 5′-AGCTTGGCCACAGGATGGGCATTAAGCCT-3′; reverse, 5′-AAGGATTCCTCTGAGGCAGC-3′; HSKIF3 (262 bp)—forward, 5′-CAGAGAAGCCAGAAAGCTGC-3′; reverse, 5′-TTGTAGCCTTCAAGTACAGAATCAAT-3′; HSRAD50 (225 bp)—forward, 5′-CACTTTCTGAGGACCTACATTTCTATG-3′; reverse, 5′-AGTCGCTCACAGCAGCGTA-3′; RATLSTP-like2 (172 bp)—forward, 5′-GAACAGAAATTCTTGGCAAGTCAGTT-3′; reverse, 5′-ACCACAGCGGGACACACAG-3′; HSprolyl4-hydroxylase α(II) (199 bp)—forward, 5′-TCTCCTTACCAAGGGAGAGCA-3′; reverse, 5′-CCGCTCGGCTACAATGAAG-3′; HSacyl-CoA synthetase (262 bp)—forward, 5′-CTTGTTTCAACAGAGTTTGTCCTC-3′; reverse, 5′-CCTGCTCACCTCTACTTCTTCTGA-3′; E1 (241 bp)—forward, 5′-AAGTCGAGCTCCTTCAGCAAG-3′; reverse, 5′-TGATTGGGCTGCAGTCTTG-3′; E3 (275 bp)—forward, 5′-AGCCACAGCACTTAGAGCTGAG-3′; reverse, 5′-AAGTGGATCAGTGAGCACAGCT-3′.
Production of Transgenic Mice
Because YAC (854G6) was too large to isolate intact, it was truncated using the acentric YAC deletion vector pBCL (Lewis et al. 1992). The derivative clones were phenotyped as Lys−, Trp−, and Ura+, and the sizes of the fragmented YACs were determined by pulse-field gel electrophoresis. In this study derivative clone 1 (350 kb) was used. YAC DNA (854G6 no. 1, A94G6, and 131F9) was isolated as described previously (Frazer et al. 1995), with the following modification: The DNA was dialyzed overnight on a 0.05-mm dialysis filter (Millipore) against injection buffer [10 mm Tris-HCl (pH 7.5), 0.1 mm EDTA, 100 mm NaCl]. The isolated DNA, at a final concentration of ∼1 ng/ml, was microinjected into fertilized FVB hybrid mouse eggs using standard procedures.
RNase Protection Assays
Total RNA was extracted from various tissues of 3- to 4-week-old mice using RNA STAT-60 (TelTestB) according to the manufacturer’s instructions. Radiolabeled antisense riboprobes were generated using the MAXIscript kit (Ambion, Inc., Austin, TX) from DNA templates generated by PCR using the following probes: HSseptin2 (125 bp)—forward, 5′-GAACATTATTCCCATCATGCC-3′; reverse, 5′-CATCATCCGTGGCCAACTG-3′; HSKIF3 (195 bp)—forward, 5′-TTTTTCTTCAATATCCAAGCGTT-3′; reverse, 5′-AGATCTATTTTGGTTATTTTATTTCCG-3′; HSRAD50 probe (140 bp)—forward, 5′-TTTACCTAACAGTGAACCTGTGACGTT-3′; reverse, 5′-CCAGAGCATGTGCAAGAGATACTTAC-3′. RNase protection assays were performed using the RPA II kit (Ambion, Inc., Austin, TX) according to the manufacturer’s recommendations. The protected fragments were separated on a 7.5% acrylamide denaturing gel, and the gel was exposed to X-ray film.
Acknowledgments
This work was supported by National Institutes of Health grants to E.M.R. (PPG HL18574); Human Genome Distinguished Postdoctoral Fellowship (K.A.F.) sponsored by the U.S. Department of Energy, Office of Health and Environmental Research, and administered by the Oak Ridge Institute for Science and Education. E.M.R. is an American Heart Association Established Investigator. Research was conducted at the Lawrence Berkeley National Laboratory (Department of Energy contract DE-AC0376SF00098), University of California, Berkeley.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL emrubin{at}lbl.gov; FAX (510) 486-6816.
-
- Received December 27, 1996.
- Accepted March 5, 1997.
- Cold Spring Harbor Laboratory Press








