
Fine Localization of the Torsion Dystonia Gene (DYT1) on Human Chromosome 9q34: YAC Map and Linkage Disequilibrium
- Laurie J. Ozelius1,
- Jeffrey Hewett1,
- Patricia Kramer4,
- Susan B. Bressman2,
- Christo Shalish1,
- Deborah de Leon2,
- Marc Rutter1,
- Neil Risch3,
- Mitchell F. Brin7,
- Elena D. Markova5,
- Svetlana A. Limborska6,
- Irina A. Ivanova-Smolenskaya5,
- Mary Kay McCormick1,
- Stanley Fahn2,
- Alan J. Buckler1,
- James F. Gusella1, and
- Xandra O. Breakefield1,8
- 1Molecular Neurogenetics Unit, Massachusetts General Hospital and Departments of Neurology and Genetics, Harvard Medical School, Boston, Massachusetts 02114; 2Dystonia Clinical Research Center, Department of Neurology, Columbia Presbyterian Medical Center, New York, New York 10032; 3Department of Genetics, Stanford University, Stanford, California 94305; 4Department of Neurology, Oregon Health Sciences University, Portland, Oregon 97201; 5Institute of Neurology, Moscow, 123367 Russia; 6Institute of Molecular Genetics, Moscow, 123367 Russia; 7Department of Neurology, Movement Disorders Center, Mt. Sinai Hospital, New York, New York 10029
Abstract
The DYT1 gene, which maps to chromosome 9q34, appears to be responsible for most cases of early-onset torsion dystonia in both Ashkenazic Jewish (AJ) and non-Jewish families. This disease is inherited in an autosomal dominant mode with reduced penetrance (30%–40%). The abnormal involuntary movements associated with this disease are believed to be caused by unbalanced neural transmission in the basal ganglia. Previous linkage disequilibrium studies in the AJ population placed the DYT1 gene in a 2-cM region between the loci D9S62a and ASS. A YAC contig has now been created spanning 600 kb of this region including D9S62a. The location of the DYT1 gene has been refined within this contig using several new polymorphic loci to expand the linkage disequilibrium analysis of the AJ founder mutation. The most likely location of theDYT1 gene is within a 150 kb region between the lociD9S2161 and D9S63.
Torsion dystonia is a movement disorder characterized by sustained muscle contractions, frequently causing twisting and repetitive movements or abnormal postures (Fahn 1988). Primary or idiopathic torsion dystonia (ITD) has unknown etiology and comprises a number of clinically and genetically distinct syndromes. At least six different autosomal genes can cause clinically distinct forms of ITD, all of which are inherited in a dominant fashion with reduced penetrance and variable expressivity (for review, see Gasser et al. 1992; Kramer et al. 1995). The gene DYT1, which underlies early-onset generalized dystonia, the most severe form of dystonia, maps to chromosome 9q34 (Ozelius et al. 1989; Kramer et al. 1990,1994). The gene GCH1, which is responsible for dopa-responsive dystonia (including the Segawa variant), has been mapped to chromosome 14q21–22 (Nygaard et al. 1993; Endo et al. 1995) and encodes the enzyme GTP cyclohydrolase I, which is the rate-limiting enzyme for synthesis of tetrahydrobiopterin, the cofactor for tyrosine hydroxylase that converts tyrosine to dopa (Ichinose et al. 1994). A gene for one form of paroxysmal dystonia has been mapped to chromosome 2q (Fink et al. 1996; Fouad et al. 1996), and some types of late-onset focal dystonia are caused by genes on chromosomes 18p (Leube et al. 1996) and 8 (L. Almasy, S.B. Bressman, D. deLeon, P.E. Greene, G. Heiman, B.A. Ford, D. Raymond, A.C. Jones, H. Shen, P.L. Kramer, unpubl.). Genes responsible for other hereditary types of dystonia, including rapid-onset (Dobyns et al. 1993), myoclonic (Kurlan et al. 1988;Kyllerman et al. 1990), and other late-onset focal dystonias (Forsgren et al. 1988; Bressman et al. 1994a) have not been mapped yet.
The DYT1 gene appears to be responsible for most cases of early-onset ITD in both Jewish and non-Jewish families (Kramer et al. 1990, 1994). The incidence of this form of dystonia has been estimated to be ∼5- to 10-fold greater in the Ashkenazi Jewish (AJ), as compared to non-Jewish populations or non-Ashkenazi population (Zeman and Dyken 1967; Eldridge 1970; Korczyn et al. 1980; Bressman et al. 1989). This increased incidence results from a founder mutation that was introduced into the AJ population some 20–30 generations ago and probably originated in Lithuania or Byelorussia; this mutation can be identified by a haplotype of alleles at polymorphic loci surrounding it (Risch et al. 1995). The frequency of this founder mutation is estimated between 1/6000 and 1/2000 in individuals of AJ descent with 30% penetrance of the disease phenotype (Bressman et al. 1989; Risch et al. 1990, 1995). Initial linkage disequilibrium studies in affected AJ individuals indicated that the DYT1 gene was located within a short distance of the argino-succinate synthetase (ASS) locus (Ozelius et al. 1992a). Using six polymorphic markers spanning a 3-cM region, including the ASS gene (centromere–D9S62a/b–D9S63–ASS–ABL–D9S64–telomere), the highest lod scores were obtained with D9S62a/b–D9S63 and the strongest disequilibrium with D9S63 in 54 AJ families with early-onset torsion dystonia (Risch et al. 1995), suggesting thatDYT1 is in between D9S62 and ASS, in close proximity to D9S63.
In this study we have assembled a YAC contig (∼600 kb), including a number of cosmids, which spans the DYT1 region fromD9S62a to a new marker, D9S2163, which is telomeric to D9S63. We have defined and mapped seven polymorphic marker loci in this region and used a set of 11 marker loci between and including lociD9S62a and ASS to define the most likely location of the DYT1 gene by linkage disequilibrium studies in AJ families. This high-resolution genetic and physical map of the chromosomal region bearing the DYT1 gene paves the way for identification of this and other genes in the region.
RESULTS
YAC Map
A yeast artificial chromosome (YAC) contig was constructed for the region implicated by initial linkage disequilibrium studies spanning loci D9S62a and D9S63 (Fig.1). Initial screening of YAC libraries with these genetic markers and D9S62b resulted in two YAC contigs around loci D9S62a/b and D9S63. Alignment of the clones and ordering of markers was based on the presence or absence of each marker in the clones as assessed by PCR or hybridization. To extend these contigs, Alu–PCR products and end clones were generated from these YACs and used to identify overlapping YACs, including 8H12 and 21B5. To link the two contigs, end clones were isolated from YACs 5H7 and 8H12. These clones did not identify any new YACs but did hybridize to the same cosmid, LL09NC0150H11, allowing joining of the contigs around D9S62a/b and D9S63. To get a more accurate estimate of the size of the contig, because of the discrepancy in the size of YACs 251H9 and 183D9 covering the same region, the end clones from YACs 22A4 and 21B5 were isolated and hybridized to both the YAC and cosmid libraries. Again, no new YACs were identified, but cosmid LL09NC01198B10 was found to link YACs 22A4 and 21B5 and overlap 251H9. The resultant YAC physical map has one gap that is covered by a cosmid (LL09NC0150H11), contains 10 simple sequence repeat (SSR) markers, and spans a physical distance of ∼600 kb on chromosome 9q34 based on YAC sizes estimated by pulsed-field gel electrophoresis, and assuming an average size of cosmids (40 kb).
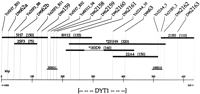
Physical map from markers D9S62a to D9S2163containing the DYT1 gene. Markers (SSR and end clones) used to screen YACs and cosmids are represented by a label at the topof the map and a vertical line. YAC and cosmid clones are drawn to scale under the loci that they contain. YACs are indicated by bold horizontal lines with a corresponding name and size. YAC names preceded by asterisks are CEPH coordinates, and the remaining YACs are from the flow-sorted chromosome 9 library. The thin line in YAC 183D9 indicates a region of deletion. YAC clone lengths were estimated by pulsed-field gel electrophoresis and are in brackets next to the YAC names. Cosmids are indicated by smaller horizontal lines. The cosmids were obtained from a chromosome 9-specific cosmid library generated at Lawrence Livermore National Laboratory and should be preceded by the designation LL09NC01. Cosmids were given the average size of 40 kb. Distances between markers are estimated based on their presence or absence in the clones and the clone lengths. A kilobase scale is shown at thebottom. The minimal region containing the DYT1 gene, as defined by haplotype analysis in the AJ population, is indicated by the bracketed region at the bottom.
Markers
Eleven SSR polymorphisms within the DYT1 region were used to align YAC contigs and to carry out linkage disequilibrium studies in affected AJ individuals. Four of these SSRs have been described and placed on a genetic map previously; in order from centromere to telomere they are: D9S62a, D9S62b, D9S63, and ASS(Kwiatkowski et al. 1992; Ozelius et al. 1992b). D9S62a andD9S62b lie within the same 40-kb cosmid (Henske et al. 1993). No recombinations have been observed between D9S62a/b andD9S63, in >800 meiotic events evaluated in the large Venezuelan reference pedigrees (VRPs) and our AJ dystonia families (Ozelius et al. 1992b; Risch et al. 1995). Results of marker-to-marker linkage analysis gives a maximum likelihood estimate for the recombination fraction between the D9S62a/b–D9S63 cluster andASS of ∼1.8 cM (Risch et al. 1995).
Six new polymorphic markers were generated from YACs and cosmids in theDYT1 region (Table 1). The other marker,D9S159, was described previously by Weissenbach et al. (1992)but repositioned compared to previous genetic maps as part of this study. These markers comprise four (GT)n repeats (D9S2158, D9S2159, D9S2163, and D9S159); two tetranucleotide repeats [D9S2160 (ATAG)nand D9S2161 (GATA)n]; and a trinucleotide repeat [D9S2162 (AAT)n]. The (GT)n polymorphism D9S2158 was isolated from a cosmid overlapping the end of YAC 183D9; another,D9S2159, from a cosmid that spans YACs 8H12 and 5H7 (Fig. 1); and the third, D9S2163, from a cosmid that hybridized toAlu–PCR products from YAC 21B5. D9S2160 andD9S2162 were isolated from YACs 183D9 and 21B5, respectively, by sequencing cloned Alu–PCR products from them.D9S2161 was identified in a cosmid that hybridized to anAlu–PCR product from YAC 8H12. Allele frequencies for these seven new markers were determined in the AJ population (Table 1). Allele frequencies for the four previous markers, D9S62a, D9S62b, D9S63, and ASS, in this population are provided in Risch et al. (1995).
New Polymorphic Markers on 9q34
Linkage Disequilibrium
Based on haplotype analysis in the immediate chromosomal region surrounding DYT1, it is clear that most cases of early-onset dystonia in the AJ population result from a founder mutation (Ozelius et al. 1992a; Bressman et al. 1994b; Risch et al. 1995). Here we have determined the haplotype associated with the founder mutation for 11 polymorphic markers, including eight (GT)n repeats (D9S62a/b, D9S63, ASS, D9S2159, D9S159, D9S2158, andD9S2163), two tetranucleotide repeats (D9S2160 andD9S2161), and a trinucleotide repeat (D9S2162). The allele associated with the DYT1 founder mutation at each of these markers, its percentage in affected and control AJ chromosomes, and the linkage disequilibrium parameter δ are given in Table 2 for all markers except D9S2161 andD9S2163. These markers were identified later and used to confirm recombination events. The 11 markers fromD9S62a to ASS define a haplotype, portions of which were observed in 94% (64/68) of unrelated AJ individuals with early-onset dystonia (Table 3 illustrates these 64 chromosomes with all or part of the founder haplotype). In families with more than one affected individual, a definite disease-bearing chromosome could be identified (38/64); however, when there was only one affected member in a family, only a probable disease-bearing chromosome could be assigned (26/64; Table 3). The full haplotype (all 11 markers) was not seen in any of 280 phased chromosomes from AJ controls (Table 4), although two of the control chromosomes do carry a portion of the founder haplotype from D9S62a to D9S63(7 markers). When these two chromosomes were typed for the two new loci (D9S2161 and D9S2163) neither carried the associated allele at these loci. Some marker combinations in controls appeared at an increased frequency, suggesting that there may be linkage disequilibrium between them, particularly for the 4-4-4-16 haplotype at markers D9S2158, D9S2159, D9S2160, andD9S63, which lie in the 150-kb region believed to contain theDYT1 gene (see below; total of 19 occurrences vs. 11.9 expected).
Linkage Disequilibrium Between 9q34 Markers and theDYT1 Gene in the AJ Population
Haplotype Analysis on AJ Disease Chromosomes
Frequency of Associated Haplotype in Control AJ Chromosomes
Analysis of the distribution of marker alleles in affected AJ individuals who bear only a portion of the founder haplotype allows finer assessment of the location of the DYT1 gene (Table 3). Although variations at any one allele could result from de novo mutations (“slippage” events of unknown mechanism) of the repeat element at that site, variations in a linear set of adjacent alleles indicate a recombination event, which could have occurred in any of the meiotic events from the time of the founder mutation to the generation of the affected individual under study. On the telomeric side, there are nine chromosomes in affected individuals that showed recombinations between D9S63 and ASS (chromosome categories 2–10; Table 3), four are directly observed recombination events that occurred in the generation under study (Table 3, footnote c), whereas the other five represent historic recombinations that occurred in a preceding generation. One of these historic recombinations was also seen withD9S2162 (Table 3, category 10), but five others (three historic, and two observed) were homozygous 4 at this locus. The new marker D9S2163 was used to type all of the telomeric recombinant chromosomes. Three of the chromosomes that were homozygous 4 at the D9S2162 locus had the associated allele atD9S2163, suggesting that the recombination occurred betweenD9S2163 and ASS. However, the other two crosses had different alleles at marker D9S2163, placing the DYT1gene proximal to this locus. The chromosome that recombined at theD9S2162 locus also crossed with the D9S2163 marker, consistent with placement of the disease gene proximal toD9S2162. One chromosome (Table 3, category 11) that displayed a historic cross between D9S2160 and D9S63 was homozygous 4 at D9S2162, suggesting the possibility of a slippage event at D9S63. This chromosome also recombined atD9S2163, however, thus confirming that a recombination event most likely occurred proximal to locus D9S2163 and probably proximal to D9S63 as well. The closest distal marker toDYT1 thus appears to be D9S63. On the centromeric side of DYT1 there are six putative historic recombination events between D9S62b and D9S63 (Table 3, chromosome categories 12–17). One of these chromosomes (Table 3, category 12) recombines with all six markers, thus placing the disease gene distal to the D9S2160 locus. Another chromosome (Table 3, category 13) has a historic recombination with five of the six markers, placing the gene distal to D9S2159, but D9S2160 is homozygous 4. When these two chromosomes were typed for D9S2161, neither had the associated allele, supporting D9S2161 as the proximal border. Three other chromosomes (categories 14–16) reveal historic recombinations with more proximal markers and retained the associated allele at D9S2161. These events are consistent with positioning the DYT1 gene distal to the D9S2161locus. However, another chromosome (Table 3, category 17) had the associated alleles 12-4-4-4 between D9S159 andD9S2160 but did not have the associated allele atD9S2161. This would place the DYT1 gene proximal toD9S2161, a position that would appear to be in conflict with the other two crosses. However, this 12-4-4-4 haplotype is seen on 2% of control AJ chromosomes, whereas the associated haplotype 16-4-12 between markers D9S63 and ASS, seen in chromosomes 12 and 13 (Table 3), was never seen on any control AJ chromosomes (Table4). Therefore, it seems more likely that the gene is distal toD9S2161. The affected individual carrying only the 12-4-4-4 portion of the founder haplotype may do so by chance and instead have a different mutation at the DYT1 gene or at another locus. In summary, the haplotype analysis defines the most likely location of theDYT1 gene to be proximal to D9S63 and distal to theD9S2161, a distance of ∼150 kb as estimated from the YAC contig.
DISCUSSION
Employing both YACs and cosmids, we have generated a genomic contig spanning a 600-kb region of chromosome 9q34, including theDYT1 gene. Furthermore, we have extended our original linkage disequilibrium studies in the AJ population using 10 SSR loci within this contig (Ozelius et al. 1992a; Risch et al. 1995). The present study confirms that most cases of early-onset ITD in the AJ population result from a single founder mutation and reduces the genomic region bearing this gene to an area compatible with positional cloning. This detailed haplotype information will also serve to increase the accuracy of molecular diagnosis in the AJ population.
Haplotype analysis has been instrumental in pinpointing the genomic location of several disease genes, including those for Huntington’s disease (MacDonald et al. 1992) and diastrophic dysplasia (Hastbacka et al. 1994). Using this approach, we have identified several apparent historic recombination events that place the DYT1 gene most likely between the loci D9S2161 and D9S63 in a region of ∼150 kb. This localization should be regarded with some caution, however, as changes at the edges of the haplotype could also result from mutations (slippage) at highly polymorphic repeat markers. If the mutation rate of a marker exceeds its recombination rate with the disease-causing mutation, then the majority of apparent historical crossovers involving this marker will actually be the result of de novo mutations. Slippage events should be evident among densely spaced markers because there will usually be no change of associated haplotype alleles at flanking loci. On the proximal side, there are two AJ disease-bearing chromosomes that place the disease gene distal to theD9S2161 locus and one other that places the gene distal toD9S2158.
We have seen no evidence for slippage at the D9S2158 locus in our Jewish and non-Jewish families, but we have observed one slippage event (>400 meioses) at the D9S2161 locus. The chances that all three of these disease-bearing chromosomes represent mutational events is highly unlikely, as they all have nonassociated alleles at the proximal markers. On the distal side there is one historic recombination event defining D9S63 as the flanking marker. We have observed a slippage event at D9S63 between generations in one family, and one new mutation at this locus was reported previously in the VRP (>800 total meioses) (Kwiatkowski et al. 1992), but again, the flanking markers distal to D9S63 also show loss of associated haplotype alleles in this apparently recombinant chromosome. Thus, all of the apparent changes observed are consistent with recombination rather than slippage events.
To confirm the placement of loci and historical recombinations as well as to facilitate the positional cloning of the DYT1 gene, a YAC and cosmid contig were constructed across a 600-kb interval containing this disease gene. The genetic distance between markersD9S62a and D9S63 is small, as no recombinations have been observed between these loci in the VRP (Ozelius et al. 1992b) or in any of our dystonia families (>800 meiotic events in total). The physical map for this region, D9S62a to D9S63, spans ∼300 kb. The genetic distance between D9S62a/D9S63and the ASS locus is estimated to be 1.8 cM (Risch et al. 1995). Walking from the end of the 600-kb contig toward ASS,we were unable to find a YAC that was also positive for YACs known to contain the ASS marker, suggesting that the minimal physical distance between these loci is likely to be ⩾900 kb. The minimal region most likely to bear the DYT1 gene, as determined by linkage disequilibrium analysis in the AJ population, is ∼150 kb. Assuming 100,000 genes in the haploid genome, this region would contain on average 6–7 genes.
These studies highlight the value of linkage disequilibrium studies to direct a gene search. Using this technique, we were able to reduce the interval containing the DYT1 gene from 1.8 cM (∼1.8 Mb) to ∼150 kb, thus setting the stage for the eventual isolation of the disease gene.
Identification of the DYT1 gene should provide insights into the development and the temporal and spatial organization of the basal ganglia where the defect is believed to originate (Marsden et al. 1985). Several interesting features of this disease bear on developmental plasticity of movement control in the central nervous system, including the tendency for the disorder to progress from lower to upper body regions and the positive correlation between younger age at onset and greater severity of disease (Bressman et al. 1994b). The low penetrance in this autosomal dominant syndrome suggests that other genetic and/or environmental factors can modulate the outcome of mutations at the DYT1 locus. Because there is no apparent neuronal degeneration associated with early-onset dystonia, eventual identification of the disease gene may lead to rational therapy.
METHODS
Examination and Family Material
Subjects for this study were ascertained from several sources, including a computerized database of patients seen by the Movement Disorder Group at Columbia University (New York, NY), patients seen at the Neurologic Institute in Moscow, contacts through the Dystonia Medical Research Foundation, and referrals from other neurologists. The criteria for diagnosis of ITD and the methods of evaluation were the same as described elsewhere (Kramer et al. 1990). To reduce heterogeneity, only affected individuals who met criteria for the early-onset form of ITD were included (Bressman et al. 1994b). These criteria are onset of symptoms prior to age 28 involving a body part at or below the level of the neck in the affected proband or in relatives of the affected proband. All patients were of AJ ancestry, as defined previously (Bressman et al. 1989). A total of 80 families was ascertained. Among these, 40 were singleton affecteds, whereas the other 40 had two or more affected relatives. Clinical and pedigree information for these families has been reported in detail elsewhere (Bressman et al. 1989, 1994b; Kramer et al. 1990; M.F. Brin, S.A. Limborska, E.D. Markova, I.A. Ivanova-Smolenskaya, L.J. Ozelius, D. deLeon, and X.O. Breakefield, in prep.). Control chromosomes were obtained from two sources; the normal (nondisease bearing) chromosome from affecteds, in cases where chromosomes could be phased, and chromosomes from unaffected individuals marrying into the families. A total of 280 control chromosomes were constructed and phased, all of which were of 100% AJ origin.
DNA Methods and Polymorphic Analysis
Venous blood samples were obtained from all participating family members after obtaining informed consent. DNA was extracted from whole blood (Gusella et al. 1979) or lymphoblast lines (Anderson and Gusella 1984). PCR analysis of SSR polymorphisms was carried out on genomic DNA using oligonucleotide primer pairs reported previously (Kwiatkowski et al. 1992; Weissenbach et al. 1992; Henske et al. 1993) or provided in Table 1. PCR conditions for amplification of SSR polymorphisms and analysis of the amplified products are described in Ozelius et al. (1992a).
Libraries
YACs were identified from a total human genomic YAC library constructed by the Centre d’Etudes du Polymorphisme Humain (CEPH) (Albertsen et al. 1990) and from a chromosome 9-specific YAC library constructed from flow-sorted chromosomal DNA (McCormick et al. 1993). Cosmid clones were identified from a chromosome 9-specific library constructed at the Lawrence Livermore National Laboratory (Van Dilla and Deaven 1990). YAC and cosmid colony filters were stamped and prepared for hybridization as described (McCormick et al. 1993; Murrell et al. 1995). Library pools for PCR screening were generated from both YAC libraries and used to amplify Alu–PCR products that were spotted on filters for hybridization screening with Alu–PCR products from individual clones.
Alu–PCR
Yeast genomic DNA containing YACs, in pools or individual clones, was amplified with Alu primers S (5′-GAGGTTGCAGTGAGCCGAGAT-3′) and J (5′-GAGGCTGCAGTGAGCCGTGAT-3′), followed by purification (Murrell et al. 1995).
YAC End Isolation
YAC ends were isolated from CEPH clones by the insertion of rescue plasmids into the YAC vector (pYAC4) by homologous recombination followed by restriction enzyme digestion, circularization, and transfection into Escherichia coli by electroporation (Hermanson et al. 1991). End rescue from the chromosome 9-specific YAC clones involved restriction enzyme digestion, ligation, and transformation (McCormick et al. 1990; Shero et al. 1991). YAC end clones were purified on low melt agarose gels and used as hybridization probes.
Hybridization
Gel-purified YAC DNA, Alu–PCR products, and YAC end clones, as well as Cot1 DNA (GIBCO/BRL), and poly (dGT) (Pharmacia), were labeled by random priming (Feinberg and Vogelstein 1984). Where necessary, competition was used to saturate repeat sequences by mixing probes with 5 μg of Cot1 DNA, 2 mg of human placental DNA and 2 μg of vector DNA (pYAC4, pJS97, pJS98 or sCos) and boiling for 10–15 min followed by incubation at 65°C for 3–5 hr before addition to the hybridization buffer. Tri- and tetranucleotide 24-mers were synthesized (GIBCO/BRL) for the most common repeats and then mixed together into a cocktail. Oligonucleotides were end-labeled with T4 polynucleotide kinase (NEB). All hybridizations were performed in Church and Gilbert buffer (0.1 mm EDTA at pH 8.0, 0.5 m sodium phosphate at pH 7.2, 7% SDS, 1% BSA) at 55°C overnight. Filters were washed and exposed to autoradiographic film, as described previously (Breakefield et al. 1986), except in the case of the end-labeled primers. These filters were washed in 1× SSC for 15 min at 55°C, repeating as necessary.
Physical Mapping Strategy and Generation of SSR Polymorphisms
Primers corresponding to marker loci D9S62a, D9S62b, D9S63, and ASS were used to screen the YAC libraries by PCR. The YACs containing these loci were used to identify overlapping clones by hybridization of Alu–PCR products from the individual YAC clones to Alu–PCR products from pools of YAC clones representing the libraries. When overlapping clones were not identified, end clones were rescued from the YACs, hybridized back to the existing contig to distinguish the internal and extending YAC ends, and then used to screen the cosmid libraries. Positive cosmids were arranged into 96-well microtiter dishes, and colony filters were stamped and prepared as described (Murrell et al. 1995). The cosmid filters were probed with poly(dGT) (Pharmacia) or the tri/tetraoligonucleotide cocktail (GIBCO/BRL) to identify SSR-containing clones. These clones were digested with Sau3a (NEB), subcloned into Bluescript (Stratagene), and reprobed with the SSR primers. Positive clones were sequenced using Sequenase (U.S. Biochemical), and oligonucleotide primers were selected that flanked the repeat sequence (Table 1). These SSR polymorphisms were then used to identify new overlapping YAC or cosmid clones by PCR. This combination of procedures was repeated until a complete overlapping clone map was obtained covering the region bearing the DYT1gene, as identified by haplotype analysis.
Statistical Analysis
Marker allele frequencies were estimated by simple gene counting in the control sample. The degree of linkage disequilibrium was assessed as described in Risch et al. (1995) employing the parameter δ (Bengtsson and Thomson 1981).
Acknowledgments
We thank Drs. David Riley and John Menkes for contributing patient samples; Mr. Eric Smith, Dr. Tom Gasser, and Ms. Deborah Schuback for technical help; and Ms. Suzanne McDavitt for skilled preparation of the manuscript. This work was supported by the Dystonia Medical Research Foundation; the Jack Fasciana Fund for the Support of Dystonia Research; National Institutes of Health grants NS28384, NS26656, NS24279, NS26656, and HG00169; and Fogarty grant TW00071-91. We are particularly grateful to the families who contributed to this study and who continue to offer their support.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵8 Corresponding author.
-
E-MAIL breakefield{at}helix.mgh.harvard.edu; FAX (617) 726-5736.
-
- Received December 24, 1996.
- Accepted March 12, 1997.
- Cold Spring Harbor Laboratory Press








