
Completion of the Saccharomyces cerivisiae genome sequence and that of several bacterial genomes has led to a great rallying around the cause for sequencing of other small genomes. This has been most apparent in bacteria where sequencing projects are either complete or under way for no fewer than 38 different species. In contrast, the sequencing of small eukaryotic genomes lags far behind. The only large-scale public sequencing efforts currently under way in microbial eukaryotes are for the fission yeast Schizosaccharomyces pombe and for the protozoan parasite Plasmodium falciparum. Completion of these genomes is sure to yield greater insights into the general functions required for eukaryotic life and will permit researchers to address a number of fundamental questions. However, even coupled with the genome sequences from humans and other animals, the genome sequences from this small number of eukaryotes will not be sufficient to describe life with a nucleus.
The primary purpose of this article is to point out the merits of sequencing the genomes of additional microbial eukaryotes, in particular the genetically amenable filamentous fungi. There are an estimated >1 million filamentous fungal species and some members of this diverse group of organisms can be found in virtually every ecosystem. These organisms conduct a variety of processes not seen in yeast, many of which have significant impacts on man (Table1). For instance, some filamentous fungi are important pathogens of plants and animals. Others are major producers of materials used in the food and pharmaceutical industries. In addition to the well-known and appreciated role of yeasts in bread, beer, and wine production, filamentous fungi are used in producing citric acid and other food additives as well as serving man in the mixed fermentation processes leading to soy sauce and other Asian foods. Finally, macro-fungi, or mushrooms, are eaten throughout the world. Despite these and other important roles that fungi play in our lives, we have studied only a small number of species in detail including S. cerevisiae, S. pombe, Neurospora crassa,and Aspergillus nidulans. The primary focus of this article isA. nidulans; however, many of the arguments applied toward sequencing the genome of this fungus are also relevant to others, including N. crassa.
Some Filamentous Fungi of Special Importance to Man
| Medicine | Agriculture | Biotechnology | Basic research |
| Candida albicans | Ustilago spp. | Aspergillus niger | Aspergillus nidulans |
| Aspergillus fumigatus | Magneporthe grisea | Aspergillus oryzae | Ustilago maydis |
| Coccidiodes immitis | Fusarium spp. | Aspergillus awamori | Neurospora crassa |
| Histoplasma capsulatum | Erysipespp. | Penicillium chrysogenum | Schizophyllum commune |
| Cryptococcus neoformans | Aspergillus flavus | Trichoderma reesei | Coprinus cinereus |
| Pneumocystis carnii | Phytophthora infestans | ||
| Agaricus bisporus |
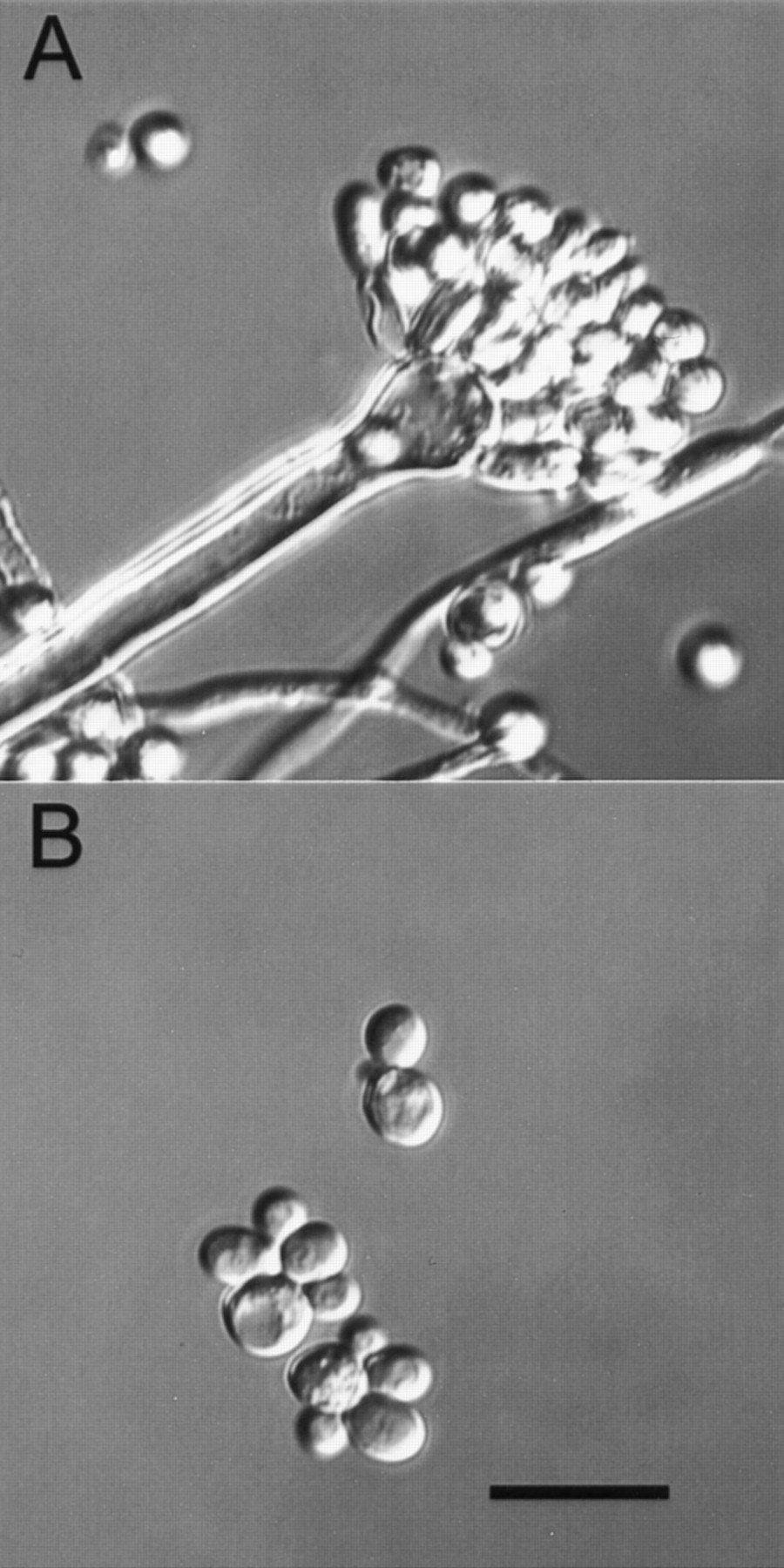
All of the fungi just mentioned are Ascomycetes, but they differ considerably from one another in many ways. Most importantly, whereasS. cerevisiae and S. pombe grow as unicellular yeasts, N. crassa and A. nidulans grow as truly multicellular filamentous fungi. The cellular architecture of these organisms is very different. Both of the filamentous fungi have a complex multicellular organization involving production of highly specialized cell types as part of their normal asexual and sexual lifecycles. An example of the complexity that filamentous fungi can exhibit is illustrated by the elaborate cellular architecture they can display (Fig. 1). Evolutionary studies of these organisms suggest that the filamentous ascomycetes diverged from the yeasts ∼1 billion years ago (Doolittle et al. 1996), not long after the fungal branch split from plants and animals. This ancient divergence raises important questions about how fungal genome organization has evolved and the number of genes that each organism requires. Moreover, fungi as experimental systems are good models for animal cell functions because of their evolutionary relatedness (Wainwright et al. 1993). This will be particularly interesting for the filamentous fungi because of their more complex life-styles. We will be able to determine whether the life-styles of the yeast are really different from those of the filamentous fungi.
Differential contrast micrographs of A. nidulans showing a condiophore, hyphae, and conidia (A) and S. cerevisiae cells (B). These micrographs were taken at the same magnification and illustrate how filamentous fungi produce a variety of complex cell types. Bar, (bottom panel), 10 μm.
A Reason to Sequence Fungal Genomes
One of the latest buzzwords in genome research is “functional genomics,” which is essentially the study of novel gene function through direct manipulation of the genome. The primary reason to do this is that knowing the sequence of yeast and other organisms has made it clear that we do not know the function of many of the predicted open reading frames. The ability to manipulate the genome of simple eukaryotes like S. cerevisiae and A. nidulans makes them attractive to begin studies that will elucidate the function of these unknown genes. This will be much more difficult to accomplish with novel genes in the human genome, except in those rare cases where nature has provided us with mutations. Similarly, functional genomics using current gene targeting methods in the mouse is difficult and costly, although new technologies are being developed that may eventually make this approach tenable. Because filamentous fungi represent a vast group of organisms in which many of the same tools available to study budding yeast genes can be applied, it will be possible to assess novel gene functions in these organisms. Their varied life-styles and the complexity of their cellular functions should provide an excellent complement to the work going on in budding and fission yeasts.
What makes A. nidulans an appropriate choice for a genome sequencing project? A. nidulans has been used extensively to address fundamental questions of biology since the 1950s. The relatively simple genetics of this filamentous fungus has been used to investigate a variety of genetic phenomena including the mechanisms regulating carbon and nitrogen metabolism, cell cycle, cytoskeletal functions, and development (Morris and Enos 1992; Marzluf 1993;Navarro-Bordonaba and Adams 1994; Osmani and Ye 1996). As such, A. nidulans has played an important role as a model eukaryotic organism. Probably more important is the role that this organism has played as a model system for the investigation of questions concerning the biology of other members of the genus that positively and negatively impact man. Members of the genus Aspergillus are used as large-scale producers of citric acid, industrial enzymes; amylases, proteases, and lipases, to name a few, and others of the genus are pathogens of man or are responsible for food spoilage and the production of toxic secondary metabolites like aflatoxin. The total economic impact of the genus Aspergillus on just the U.S. economy is estimated at ∼$45 billion. Because A. nidulanshas played an important role in our understanding of this large and diverse genus and because of the clear economic importance of these organisms, it is imperative that we obtain a more detailed picture of its genome.
Earlier aspects of the Aspergillus nidulans genome project relying on classical genetics established that its genome consists of eight linkage groups or chromosomes. These linkage groups were defined using parasexual genetic methods and mitotic recombination to demonstrate linkage between widely spaced genes on chromosome arms (Clutterbuck 1974). This establishment of eight linkage groups has been confirmed using pulsed-field gel electrophoresis, which resolves the eight linkage groups into six bands, two of which are doublets and can be separated using translocation strains. These studies estimated the genome to be 31 megabases (Brody and Carbon 1989), in good agreement with estimates of the size of the genome using reassociation kinetics and colorimetric determination (Bainbridge 1971; Timberlake 1978). Thus, the genome of A. nidulans is 2.5 × 107 to 3.0 × 107 base pairs in size and contains few repetitive DNA sequences. The relatively small size of the A. nidulansgenome, the limited amount of repetitive DNA, and its importance as a model organism make it suitable for sequencing.
In one sense, the entire genome of A. nidulans is already sequenced. Two cosmid libraries containing 5134 individual clones were constructed several years ago and stored as individual clones in microtiter plates. These libraries were later sorted by hybridization into chromosome-specific subsets that are estimated to cover ∼95% of each chromosome (Brody et al. 1991). The mapping of these libraries has been carried even further. Using a fast random cost algorithm method the two cosmid libraries were used to construct a physical map of chromosome IV of A. nidulans (Wang et al. 1994). This work has been extended to develop physical maps for all eight of the chromosomes of the A. nidulans genome. These maps and the methods used to derive them can be found at the sitehttp://fungus.genetics.uga.edu:5080/. Using these methods to build physical maps for each of the chromosomes provides the foundation on which large-scale DNA sequencing of the A. nidulans genome could begin. However, it is also important to point out that these maps have also resulted in some concern from members of theA. nidulans community because there are several instances where they are not colinear with the genetic map. The lack of colinearity may reflect the poor quality of the genetic map, which is based on assigning the order of genes from three point crosses. Alternatively, these orders could be incorrect because the genetic markers used were too far apart, preventing proper ordering. Clearly this is an issue that will be resolved in time, once the complete sequence of the genome is known.
So why are we not sequencing the genome of A. nidulans or other filamentous fungi? Current sequencing efforts are being directed at microbial organisms that are of medical importance, such as disease-causing bacteria. It is believed that the elucidation of their genome sequence has the potential to yield immediate insights into new therapeutic strategies. This is somewhat unfortunate in that we are ignoring entire groups of organisms while we are trying to determine whether whole genome sequencing will be useful. As these projects proceed, we need to consider other rationales for genome sequencing including the importance of being able to analyze gene function following identification of open reading frames by sequence analysis. This is precisely where the yeast genome effort now stands. Researchers are faced with determining novel gene functions, and the same will be true for any organism for which we determine the complete sequence. Some of these genes will be specific to yeast, others will have roles throughout the fungi, and still others will function in all eukaryotes. To learn which class each gene falls into it will be important to obtain sequences from numerous organisms. It will be very unfortunate if life scientists do not take an active role in choosing those organisms whose genomes should be sequenced. The careful selection of organisms is likely to give us greater insights into the processes of speciation and genome evolution. In the case of fungi, which are presumed to have diverged about a billion years ago, these questions are particularly relevant.
We as scientists need to be concerned about another issue that genome studies will raise. This issue is that it is not possible to derive gene function from a comparison of gene sequences. Consider the general problem of large enzyme families. The chemical reaction catalyzed by enzymes of the family may be the same but their substrates may be very different. Because of this, direct sequence comparison can provide a guide only to a gene product’s function but not the substrates it acts on. Thus, in this example we could say a gene product shares a catalytic site with that of others in the family but we cannot say what biochemical pathway it functions in. As we begin to investigate the function of novel gene products we must keep in mind that although two genes code for similar proteins, they may function differently, or as in the case of enzymes with similar activities, they may function in different pathways. This provides another reason for studying gene function in more than a small number of systems. Finally, a gene may reveal its cellular function more readily in one system than in another because the biology of the system has a special requirement for the function.
Where to Go Next
Given the great diversity of fungi and their importance to man it is important for us to consider the value of determining the sequence of several fungal genomes. For the same reasons we are sequencing several bacterial genomes there are clear reasons to sequence the genomes of the human pathogens Candida albicansand Aspergillus fumigatus. Similarly, we could argue that the genome of any number of phytopathogenic fungi should be sequenced because of their economic impact in agriculture, and the United States Department of Agriculture (USDA) has expressed an interest in fungal genomes for precisely this reason. Because of its role as a model organism for the animal and plant pathogenic fungi and industrial species, A. nidulans is an excellent choice to begin with when considering the sequencing of filamentous fungal genomes. During the later part of 1996 two meetings were held to discuss the possibility of sequencing fungal genomes. The first, hosted by Oklahoma State University and organized by Rolf Prade, led to the formation of a steering committee whose goal was to raise funds to begin a demonstration project that would begin sequencing chromosome IV, the smallest chromosome at ∼3 million base pairs. This goal is now in sight and sequencing is scheduled to begin by the fall of 1997. The second meeting, held at Tulane University in New Orleans, was to consider a broader initiative of examining additional fungal genomes, including N. crassa. At this meeting it was determined that the USDA may have a long-term interest in fungal genomics. Based on these initiatives it is clear that there is broad interest for genome studies in filamentous fungi. The only questions that remain are where and when will resources become available for sequencing fungal genomes? No governmental agency, except the USDA, has acknowledged the relative importance of filamentous fungi to the health of the general population and their importance to our national economy, let alone to the global economy. Fungi are a diverse and important group of organisms that are important to man in many ways. This group of organisms is likely to tell us much about ourselves and how cells have solved many fundamental problems in biology. It is time that filamentous fungi are put forward and considered alongside other important organisms for what they can contribute to genome level research.
Notes
[1] Corresponding author.
Notes
[2] E-MAIL [email protected]; FAX (713) 798-7799.
REFERENCES
- ↵B.W. Bainbridge(1971) Macromolecular composition and nuclear division during spore germination in Aspergillus nidulans. J. Gen. Micro. 66:319–325.
- ↵H. BrodyJ. Carbon(1989) Electrophoretic karyotpe of Aspergillus nidulans. Proc. Natl. Acad. Sci. 86:6260–6263.
- ↵H. BrodyJ. GriffithA.J. CuticchiaJ. ArnoldW.E. Timberlake(1991) Chromosome-specific recombinant DNA libraries from the fungus Aspergillus nidulans. Nucleic Acids Res. 19:3105–3109.
- ↵J. Clutterbuck(1974) Aspergillus nidulans. in Handbook of genetics, ed R.C. King(Plenum Publishing, New York, N.Y), pp 447–510.
- ↵R.F. DoolittleD.-F. FengS. TsangG. ChoE. Little(1996) Determining divergence times of the major kingdoms of living organisms with a protein clock. Science 271:470–477.
- ↵G.A. Marzluf(1993) Regulation of sulfur and nitrogen metabolism in filamentous fungi. Annu. Rev. Microbiol. 47:31–55.
- ↵N.R. MorrisA.P. Enos(1992) Mitotic gold in a mold: Aspergillus genetics and the biology of mitosis. Trends Genet. 8:32–37.
- ↵J. Navarro-BordonabaT.H. Adams(1994) The emergence of conidia and fruiting bodies in Ascomycetes. in The mycota, ed J. Wessels(Springer-Verlag, New York, NY), pp 333–349.
- ↵S.A. OsmaniX.S. Ye(1996) Cell cycle regulation in Aspergillus by two protein kinases. Biochem. J. 317:633–641.
- ↵W.E. Timberlake(1978) Low repetitive DNA content in Aspergillus nidulans. Science 202:973–975.
- ↵P.O. WainwrightG. HinkleM.L. SoginS.K. Stickel(1993) Monophyletic origins of metazoa: An evolutionary link with fungi. Science 260:340–342.
- ↵Y. WangR.A. PradeJ. GriffithW.E. TimberlakeJ. Arnold(1994) A fast random cost algorithm for physical mapping. Proc. Natl. Acad. Sci. 91:11094–11098.