
Highly accurate assembly polishing with DeepPolisher
- Mira Mastoras1,
- Mobin Asri1,
- Lucas Brambrink2,
- Prajna Hebbar1,
- Alexey Kolesnikov2,
- Daniel E. Cook2,
- Maria Nattestad2,
- Julian Lucas1,
- Taylor S. Won1,
- Pi-Chuan Chang2,
- Andrew Carroll2,
- Benedict Paten1,
- Kishwar Shafin2,
- and the Human Pangenome Reference Consortium3
- 1UC Santa Cruz Genomics Institute, University of California, Santa Cruz, California 95060, USA;
- 2Google Incorporated, Mountain View, California 94043, USA;
- 4Center for Genomic Discovery, Mohammed Bin Rashid University, Dubai Health, P.O. Box 505055, UAE;
- 5Dubai Health Genomic Medicine Center, Dubai Health, P.O. Box 505055, UAE;
- 6McDonnell Genome Institute, Washington University School of Medicine, St. Louis, MO 63108, USA;
- 7European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, UK;
- 8Center for Applied and Translational Genomics (CATG), Mohammed Bin Rashid University of Medicine and Health Sciences, P.O. Box 505055, Dubai, United Arab Emirates;
- 9Department of Genetics, Washington University School of Medicine, St. Louis, MO 63110, USA;
- 10Genome Informatics Section, Center for Genomics and Data Science Research, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA;
- 11UC Santa Cruz Genomics Institute, University of California, Santa Cruz, Santa Cruz, CA 95060, USA;
- 12Department of Genome Sciences, University of Washington School of Medicine, Seattle, WA 98195, USA;
- 13The Vertebrate Genome Laboratory, The Rockefeller University, New York, NY 10065, USA;
- 14Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN 38163, USA;
- 15Human Technopole, 20157 Milan, Italy;
- 16Institute for Genomic Health, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA;
- 17Department of Computer and Information Science and Engineering, University of Florida, Gainesville, FL 32611, USA;
- 18Canadian Center for Computational Genomics, McGill University, Montréal, QC H3A 0G1, Canada;
- 19Department of Human Genetics, McGill University, Montréal, QC H3A 0G1, Canada;
- 20Victor Phillip Dahdaleh Institute of Genomic Medicine, Montréal, QC H3A 0G1, Canada;
- 21Google LLC, Mountain View, CA 94043, USA;
- 22Department of Biomedical Informatics and Data Science, Yale School of Medicine, New Haven, CT 06510, USA;
- 23University of Florence, Department of Biology, Sesto Fiorentino (FI) 50019, Italy;
- 24Department of Ecology and Evolutionary Biology, University of California, Santa Cruz, Santa Cruz, CA 95064, USA;
- 25Arizona State University, Consortium for Science, Policy & Outcomes, Washington, DC 20006, USA;
- 26Center for Digital Medicine, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany;
- 27Department for Endocrinology and Diabetology, Medical Faculty and University Hospital Düsseldorf, Heinrich Heine University Düsseldorf, 40225 Düsseldorf, Germany;
- 28German Diabetes Center (DDZ), Leibniz Institute for Diabetes Research, 40225 Düsseldorf, Germany;
- 29University of Cambridge, Cambridge Biomedical Campus, Cambridge CB2 0QQ, UK;
- 30Wellcome Sanger Institute, Genome Campus, Hinxton, CB10 1HH, UK;
- 31Institute for Medical Biometry and Bioinformatics, Medical Faculty and University Hospital Düsseldorf, Heinrich Heine University, 40225 Düsseldorf, Germany;
- 32Howard Hughes Medical Institute, Chevy Chase, MD 20815, USA;
- 33ISEM, Univ Montpellier, CNRS, IRD, 34095 Montpellier, France;
- 34Institut Universitaire de France, 75005 Paris, France;
- 35Department of Computer Science and Engineering, University of California San Diego, La Jolla, CA 92093, USA;
- 36Department of Bioethics & Humanities, University of Washington School of Medicine, Seattle, WA 98195, USA;
- 37Department of Data Science, Dana-Farber Cancer Institute, Boston, MA 02215, USA;
- 38Department of Anthropology, University of Kansas, Lawrence, KS 66045, USA;
- 39University of Manchester, Manchester M13 9PL, UK;
- 40Traditional, ancestral and unceded territory of the Gabrielino/Tongva peoples, Institute for Society & Genetics, University of California, Los Angeles, Los Angeles, CA 90095, USA;
- 41Traditional, ancestral and unceded territory of the Gabrielino/Tongva peoples, Institute for Precision Health, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA;
- 42Traditional, ancestral and unceded territory of the Gabrielino/Tongva peoples, Division of General Internal Medicine & Health Services Research, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA;
- 43Medical and Population Genomics Lab, Sidra Medicine, P.O. Box 26999, Doha, Qatar;
- 44Montreal Heart Institute, Montreal, Quebec H1T 1C8, Canada;
- 45Center for Genomic Health, Yale University School of Medicine, New Haven, CT 06510, USA;
- 46Department of Genetics, Yale University School of Medicine, New Haven, CT 06510, USA;
- 47Department of Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA;
- 48Department of Genetics, Epigenetics Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA;
- 49Department of Computer Science, Johns Hopkins University, Baltimore, MD 21218, USA;
- 50Sun Yat-sen University, Guangzhou 510275, China;
- 51Edison Family Center for Genome Sciences & Systems Biology, Washington University School of Medicine, St. Louis, MO 63110, USA;
- 52Center for Medical Genomics, Penn State University, University Park, PA 16802, USA;
- 53Division of Medical Genetics, Department of Medicine, University of Washington School of Medicine, Seattle, WA 98195, USA;
- 54Coriell Institute for Medical Research, Camden, NJ 08103, USA;
- 55Department of Biology, Penn State University, University Park, PA 16802, USA;
- 56Department of Biomedical Science, College of Health Sciences, Qatar University, P.O. Box 2713, Doha, Qatar;
- 57Department of Genetic Medicine, Weill Cornell Medicine-Qatar, P.O. Box 24144, Doha, Qatar;
- 58IRSD - Digestive Health Research Institute, University of Toulouse, INSERM, INRAE, ENVT, UPS, 31024 Toulouse, France;
- 59MATCH biosystems, S.L., 03202 Elche, Spain;
- 60Universidad Miguel Hernández de Elche, 03202 Elche, Spain;
- 61Department of Computational Biology and Medical Sciences, The University of Tokyo, Kashiwa, Chiba 277-8561, Japan;
- 62University of Pisa, 56126 Pisa, Italy;
- 63Institute of Genetics and Biomedical Research, UoS of Milan, National Research Council, 20133 Milan, Italy;
- 64Institute for Molecular Medicine Finland, Helsinki Institute of Life Science, University of Helsinki, 00290 Helsinki, Finland;
- 65Department of Biology, Johns Hopkins University, Baltimore, MD 21218, USA;
- 66University of Amsterdam, 1012 WX Amsterdam, Netherlands;
- 67GenomeArc Inc., Mississauga, ON L4W 5M1, Canada;
- 68Department of Biology and Biotechnologies “Charles Darwin”, University of Rome “La Sapienza”, 00185 Rome, Italy;
- 69Center for Genomics, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA;
- 70PacBio, Menlo Park, CA 94025, USA;
- 71The first affiliated hospital of Xi'an Jiaotong University, Xi'an Jiaotong University, Xi'an, Shaanxi, 710049, China;
Abstract
Accurate genome assemblies are essential for biological research, but even the highest-quality assemblies retain errors caused by the technologies used to construct them. Base-level errors are typically fixed with an additional polishing step that uses reads aligned to the draft assembly to identify necessary edits. However, current methods struggle to find a balance between over- and underpolishing. Here, we present an encoder-only transformer model for assembly polishing called DeepPolisher, which predicts corrections to the underlying sequence using Pacific Biosciences (PacBio) HiFi read alignments to a diploid assembly. Our pipeline introduces a method, PHAsing Reads in Areas Of Homozygosity (PHARAOH), which uses ultralong Oxford Nanopore Technologies (ONT) data to ensure alignments are accurately phased and to correctly introduce heterozygous edits in falsely homozygous regions. We demonstrate that the DeepPolisher pipeline can reduce assembly errors by approximately half, mostly driven by reductions in indel errors. We have applied our DeepPolisher-based pipeline to 180 assemblies from the next Human Pangenome Reference Consortium (HPRC) data release, producing an average predicted quality value (QV) improvement of 3.4 (54% error reduction) for the majority of the genome.
Faithful reconstruction of the genome is increasingly essential for scientists to understand the underlying biology of any organism. Genome assembly, the process of digitally reconstructing a genome, is accomplished by piecing together segments of DNA (called sequencing “reads”) and obtaining a consensus. The past several years have seen a huge improvement in the quality of genome assembly, driven by long-read sequencing technology, which produces reads that are orders of magnitude longer than the short-read technologies that preceded them (Li and Durbin 2024; Taylor et al. 2024). Assembly algorithms have continued to advance alongside the improvements in sequencing, and it is now becoming routine to produce automated, fully phased, highly contiguous, and near-complete genome assemblies (Liao et al. 2023; Rautiainen et al. 2023; Cheng et al. 2024; Li and Durbin 2024; Porubsky et al. 2024; Taylor et al. 2024).
The quality of a genome assembly can be measured by an array of different metrics that assess different aspects of the reconstruction. Contiguity metrics describe the cumulative length and number of contigs of the assembly, whereas completeness metrics evaluate the extent of the expected sequence present in the assembly (Li and Durbin 2024). For organisms with multiple copies of their chromosomes (ploidy), fully resolving the haplotypes is a key component to accurate genome assembly, and chromosome-phasing metrics assess this (Rhie et al. 2020; Cheng et al. 2021). Another critical set of metrics evaluates the base-level accuracy of the assembled sequence (Rhie et al. 2020).
All sequencing technologies contain errors that are not completely random but are influenced by biases toward different genomic features. Even using a consensus approach, recurrent errors in the reads can propagate into the assembly. Low complexity regions like homopolymers and tandem repeats are particularly challenging for Oxford Nanopore Technologies (ONT) (Kolmogorov et al. 2023) and Pacific Biosciences (PacBio) long reads (Wenger et al. 2019), and certain motifs such as those high in GC content can cause dropout, a phenomenon particularly prevalent in short reads like Illumina (Benjamini and Speed 2012; Chen et al. 2013). Small errors can also be introduced by biases in the assembler, and different assembly algorithms differ in their limitations and error characteristics (Jarvis et al. 2022; Rautiainen et al. 2023; Cheng et al. 2024).
Base-level errors interfere with scientists’ ability to make accurate conclusions from their data. For example, they can disrupt functional elements, as in protein-coding genes, leading to frameshifts, missense, and nonsense mutations (Aganezov et al. 2022; Miller et al. 2022; Wagner et al. 2022b). For projects like the Human Pangenome Reference Consortium (HPRC) (Liao et al. 2023) and Vertebrate Genome Project (VGP) (Rhie et al. 2021), whose goal is to produce haplotype-resolved population-scale reference genomes for entire groups of species, fixing errors is especially critical to avoid propagating mistakes to all the downstream analyses that make use of them, for example, by proposing sequence variations that are, in fact, errors (Church et al. 2015; Schneider et al. 2017; Aganezov et al. 2022).
The process of removing these small-scale errors in assemblies is known as “polishing.” Most methods for assembly polishing involve aligning reads back to the draft assembly to identify potential sequence changes suggested by the reads. Because of the complex nature of sequencing and assembly error profiles, most polishing tools employ heuristic algorithms and models that need to be specific to a given sequencing platform in order to achieve high accuracy. Popular polishers like Racon (Vaser et al. 2017), Medaka (https://github.com/nanoporetech/medaka), PolishCLR (Rhie et al. 2021), and CONSENT (Morisse et al. 2021) were developed to correct assemblies using noisy long reads, whereas tools like Pilon (Walker et al. 2014), POLYPOLISH (Wick and Holt 2022), and POLCA (Zimin and Salzberg 2020) operate solely on short reads. Recently, new approaches have begun to emerge for correcting the state-of-the-art telomere-to-telomere (T2T) assemblies generated with a combination of PacBio high fidelity (HiFi) and ONT long reads. To polish the first fully complete human genome (CHM13), the T2T consortium leveraged DeepVariant (Poplin et al. 2018) calls from multiple sequencing technologies and a careful filtering strategy to avoid false-positive edits (Formenti et al. 2022; Mc Cartney et al. 2022). Algorithms like NextPolish2 (Hu et al. 2024) have also been developed to specifically target T2T genome polishing using HiFi reads. However, an ongoing major challenge for all polishing approaches is striking a balance between overcorrecting and undercorrecting assemblies.
Seq2seq transformer family models have been used to great success in many applications including language processing, machine translation tasks, and conversational AI (Brandes et al. 2022; Vaswani et al. 2023; Zhang et al. 2023). Previously, this model architecture was used in DeepConsensus to improve the quality of PacBio HiFi sequencing reads (Baid et al. 2022). In this work, we demonstrate a new adaptation of the transformer model for human and primate genome polishing called DeepPolisher, which takes HiFi sequencing reads aligned to a draft assembly as input. Along with DeepPolisher, we have developed a pipeline called PHARAOH for improving the phasing accuracy of HiFi read alignments to diploid assemblies in falsely homozygous regions using ONT ultralong (UL) reads. In this work, we show that DeepPolisher with PHARAOH outperforms current polishing approaches, reducing assembly errors by half, which is driven mostly by reductions in assembly insertion–deletion (indel) errors. We applied our pipeline to 180 haplotype-resolved assemblies from the next HPRC data release and show substantial improvements in accuracy.
Results
Overview of the DeepPolisher pipeline
An overview of the DeepPolisher pipeline is shown in Figure 1. First, PacBio HiFi reads are aligned to the diploid assembly with minimap2 (Li 2018) or winnowmap (Jain et al. 2020). For most of the genome, this alignment step is sufficient to assign the HiFi reads to the correct haplotype. However, a common error mode of modern assemblers is to mistakenly represent heterozygous sequence as homozygous. When these regions of false homozygosity are longer than the HiFi read (up to ∼25 kb), the aligner cannot phase it and will randomly assign it to a haplotype, preventing any necessary polishing edits from being made there. To address this, we developed a pipeline called PHAsing Reads in Areas Of Homozygosity (PHARAOH) that uses phasing information from ONT reads >100 kb to infer the correct haplotype for reads in long homozygous regions (Methods). For the rest of the genome, the tool Secphase (Liao et al. 2023) is used within the PHARAOH pipeline to ensure correct read phasing by revisiting secondary alignments and calculating a marker consistency score to determine whether the read should be relocated to the other haplotype.
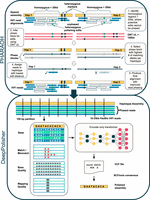
DeepPolisher pipeline overview. The PHARAOH pipeline leverages phase block information from ONT UL reads to correct the haplotype assignment of PacBio HiFi reads. The corrected alignment is passed to DeepPolisher, which is an encoder-only transformer model that predicts the underlying assembly sequence and proposes corrections in VCF format.
PHARAOH and Secphase allow us to provide improved read alignments as input to the DeepPolisher model. DeepPolisher first partitions these alignments into 100 bp segments and then represents them as a tensor object with auxiliary input features consisting of the base, whether the base is a match or mismatch to the assembly, the base quality, and the mapping quality. The tensor is provided to the encoder-only transformer neural network, which produces proposed assembly corrections in VCF format. These can be applied to the unpolished assembly with BCFtools (Danecek et al. 2021) consensus (Fig. 1; Supplemental Fig. 3). To train DeepPolisher we used a high-accuracy assembly of HG002 produced by the Q100 consortium (https://github.com/marbl/HG002) (v0.9), training across Chromosomes 1–19 (Methods). DeepPolisher can be run without PHARAOH if ONT UL reads are not available; however, assembly corrections in long stretches of false homozygosity will not be made.
Alignment-based comparison of DeepPolisher and alternate polishing approaches
We compared the performance of DeepPolisher with existing polishing approaches for HiFi reads: the T2T consortium pipeline (Mc Cartney et al. 2022), DeepVariant using HiFi alignments, and NextPolish2. To test them, we ran the UL version of hifiasm (which uses both HiFi and ONT UL reads) (Cheng et al. 2024) to generate an assembly for a sample that was not included in our training set (HG005) and polished it with each method. A comparison of the computational resources used by each polishing approach on the HG005 hifiasm assembly can be found in Supplemental Table 1. After polishing with each tool, we called variants relative to GRCh38 using dipcall (Li et al. 2018), which enabled us to assess concordance with the Genome in a Bottle (GIAB) v4.2.1 benchmark call set (Methods) (Wagner et al. 2022a). This gives an alignment-based measure of polishing accuracy that spans ∼80% of the HG005 genome, as defined by the GIAB high-confidence regions.
With DeepPolisher, we reduce the number of variant-calling errors in the unpolished assembly from 20,274 to 11,750, indicating a reduction in assembly error rate by 42% (8.14 errors per megabase to 4.72 errors per megabase) (Fig. 2A). This performance is reproduced on the held-out chromosome (20) from HG002 (Supplemental Table 2A,B). We also trained a DeepPolisher model on a Verkko (Rautiainen et al. 2023) assembly using the same data. According to the GIAB variant-calling results, the Verkko assembly for HG005 starts out at a higher quality than hifiasm (6.86 errors/megabase), and after DeepPolisher, it approaches the same quality as the polished hifiasm assembly at 4.87 errors/megabase (Supplemental Table 2E).
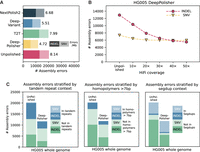
Comparison of DeepPolisher and alternate polishing methods against GIAB v4.2.1 benchmark for HG005. (A) For each polishing method, GIAB v4.2.1 variant-calling (assembly) errors are separated by indels (darker shade) and single-nucleotide variants (SNVs; lighter shade), with the number of errors per megabase to the right of each bar. (B) Total GIAB variant-calling (assembly) errors for different HiFi read coverages, with indel errors represented in pink circles and SNV errors in yellow triangles. (C) Total GIAB variant-calling (assembly) errors stratified by presence in tandem repeats (left), homopolymers >7 bp (middle), and segmental duplications (segdups; right), with SNV errors in lighter shades and indel errors in darker shades.
In comparison to DeepPolisher, the T2T polishing pipeline produces more conservative improvements, with only 3591 polishing edits passing their filters, leading to a removal of 372 errors (7.99 errors/megabase). Using DeepVariant to polish with HiFi alignments reduces assembly errors to 13,724 (5.51 errors/megabase). We note that much of the difference in GIAB variant-calling performance between DeepVariant and DeepPolisher on HG005 is owing to PHARAOH: Polishing the HG005 assembly with DeepVariant run on PHARAOH alignments produces a reduction to 11,508 assembly errors (4.62 errors/megabase). However, an important caveat is that DeepVariant was trained on several GIAB samples including HG005, with only Chr 20–22 held out, so this performance cannot be separated from training circularity. Finally, NextPolish2 removed 3260 errors from the unpolished assembly (6.68 errors/megabase) (Fig. 2A; Supplemental Table 2C).
We tested DeepPolisher across a variety of coverages and found that although 40× coverage produces optimal reduction in variant-calling errors, some assembly improvement can still be obtained with as low as 10× coverage (Fig. 2B). The DeepPolisher pipeline performance is also reproducible across HiFi read versions other than what it was trained on (DeepConsensus v1.2). This includes Sequel II, DeepConsensus v0.2, and Revio with DeepConsensus v1.1 (Supplemental Figs. 1, 2; Supplemental Tables 3, 8).
We stratified the GIAB variant-calling errors by genomic region and found that the majority of indel errors removed by DeepPolisher were in tandem repeats and homopolymers. The majority of SNV errors that were not fixed by DeepPolisher were located within segmental duplications. Some of these may be true errors, but others may be mapping artifacts or mistakes in the truth set (Fig. 2C; Supplemental Table 4). To understand DeepPolisher's performance in repetitive sequences that are excluded from the GIAB high-confidence regions, we assessed the concordance of the HG002 hifiasm assembly with the Q100 HG002 benchmark genome within centromere annotations (Methods). Although it is expected that the centromere is enriched for errors within both assemblies, DeepPolisher improves the overall concordance with the Q100 genome within centromeres, removing 85 SNVs (out of 21,073 total) and 443 INDELs (out of 8516 total). (Supplemental Table 5).
To test how DeepPolisher impacts the coding regions, we extracted genes containing frameshift and premature stop mutations in the HG005 and HG002 assemblies before and after polishing. (Supplemental Table 6A). In the unpolished assemblies, we identified a mean of 106 genes with frameshifts and 46 with stop codons per haplotype. A mean of 77.87% of genes with frameshifts and 79.31% of genes with stop codons was supported by the GIAB v4.2.1 variant calls, indicating that the substantial majority are likely true mutations. These estimates are in line with those previously reported by the HPRC (Liao et al. 2023). Across the two samples, DeepPolisher reduced false frameshifts by a net seven and stop codons by three (Supplemental Table 6A). Out of 129 total frameshift and stop codons across both samples that are unvalidated by GIAB and that DeepPolisher is not fixing, 100 are outside of GIAB high-confidence regions, and 15 are coming from known polymorphic gene families (such as OR, KRT, FAM). We manually investigated 14 of these putative errors and noted that in all cases the HiFi reads appear to support the assembly.
To directly assess the impact of polishing on every gene, we extracted all transcripts that were altered after polishing, and calculated their DNA identity and protein identity to the transcript in GRCh38. Of these, we excluded transcripts with only synonymous or nonsynonymous mutations and examined those for which the transcripts were impacted significantly by frameshifts or in-frame insertions and deletions (Supplemental Table 6B). Polishing improved the protein and DNA identity to the original transcript in GRCh38 for seven out of eight of these transcripts. In the one transcript in which polishing decreased the protein identity, the read alignments looked reliable and supported the polishing edit.
To assess the performance benefits of the PHARAOH pipeline, we ran DeepPolisher directly on minimap2 alignments. Without PHARAOH, DeepPolisher removes 8005 variant-calling errors from the unpolished HG002 assembly (32% error reduction) and 6917 errors from the unpolished HG005 assembly (39.5% error reduction). Using PHARAOH alignments as input to DeepPolisher removes an additional 1915 errors for HG002 (9% error reduction) and 519 for HG005 (2.5% error reduction). (Supplemental Table 2B,G). The impact of PHARAOH is expected to differ between assemblies based on the amount of false homozygosity that is present.
k-mer-based comparison of DeepPolisher and alternate polishing methods
k-mer-based methods assess the accuracy of an assembly using an alignment-free approach (Rhie et al. 2020). To compare the k-mer and alignment-based assessments, we first restricted the k-mer assessment to just the assembled sequence contained within the GIAB high-confidence regions, which span ∼80% of the HG005 assembly (Methods). k-mer methods generally report a quality value (QV), a log base 10–scaled measure of assembly error, with higher values indicating more accurate assemblies. We find that DeepPolisher improves the k-mer-based assembly QV by 1.8, equivalent to a 34% reduction in errors, which is approximately consistent with the quantity of improvement suggested by the GIAB variant-calling performance (Fig. 3A; Supplemental Table 7). Out of all the polishing methods compared, DeepPolisher induces the least amount of error k-mers to the assembly (Fig. 3A). NextPolish2, which uses Illumina k-mers in its algorithm for assembly polishing, produces the highest QV improvement, yet with the trade-off of inducing the highest number of new error k-mers to the assembly. The introduction of new error k-mers likely lies behind the increase in switch and hamming error rate produced by NextPolish2 (0.03% and 0.06%, respectively). In contrast, the other polishing methods manage to reduce switch and hamming error rates (Fig. 3B).
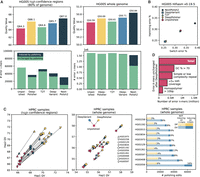
k-mer-based comparison of DeepPolisher and alternate polishing approaches for HG005. (A) Top panels display QV scores for each polishing method. Bottom panels depict total error k-mers, divided by error k-mers induced by polishing (dark blue) and error k-mers unchanged after polishing (green). Left panels show results for the GIAB high-confidence regions; right panels, whole genome. (B) Switch (x-axis) and hamming (y-axis) error rates for each polishing method. (C) Comparison of DeepVariant and DeepPolisher for eight HPRC samples. Left and middle panels show Hap1 (x-axis) and Hap2 (y-axis) QV for eight HPRC samples, with an arrow connecting the unpolished QV (pink) to the QV after polishing with DeepVariant (blue) and DeepPolisher (yellow). The left panel is within the GIAB high-confidence regions; middle panel, whole genome. The right panel shows the number of polishing edits from DeepPolisher (yellow) and DeepVariant (blue). Lighter shades indicate edits not inducing error (FP) k-mers; darker shades show edits that induce error k-mers. (D) Number of error k-mers unchanged by polishing with DeepPolisher falling into sequence annotation categories.
Because the DeepVariant model was trained on the HG005 GIAB variants, it is critical to also compare its performance against DeepPolisher on samples that were completely held out from training in both methods. We applied both tools to the same PHARAOH alignments for eight of the HPRC samples and assessed their QV. For all samples, DeepPolisher produces a higher QV improvement (average 3.95 QV, 60% error reduction) within the high-confidence regions compared with DeepVariant (average 2.26 QV, 41% error reduction) (Fig. 3C). In addition, the percentage of edits inducing error k-mers is more than twice as high for DeepVariant (average 9%) than DeepPolisher (average 4%) (Fig. 3C; Supplemental Table 9).
k-mer-based assessments underestimate assembly error rates
Relative to alignment-based metrics like those used in the GIAB assessment above, k-mer-based QVs are unbiased by alignment and easy to assess. However, k-mer-based methods lack an underlying ground truth, instead relying on consistency between assembly and sequencing technology. This results in a potential limitation in which results could be consistent but not correct owing to technology-specific sequencing bias. Because k-mer-based quality metrics are agnostic to the sequence context outside of the k-mer window, they also are limited in their ability to capture errors in repetitive sequence or in regions with a loss of heterozygosity.
For the unpolished HG005 assembly within the high-confidence regions, we see a QV estimate of 66.97 using Illumina k-mers (k = 31), suggesting 0.811 errors/megabase, whereas the GIAB variant analysis suggests the same assembly has about 10× more errors per megabase (8.14; equivalent to a QV of 50.9) (Supplemental Table 10). Using shorter k-mer sizes or a hybrid k-mer set built with both Illumina and HiFi reads as proposed by Mc Cartney et al. (2022) led to substantially more inflated estimates of accuracy (0.258 and 0.201 errors/megabase, respectively; Methods) (Supplemental Table 10). To explore the source of this difference in error rate, we performed a manual analysis investigating the concordance between the errors reported by the QV and the GIAB variants for HG005 after polishing with DeepPolisher (Methods) (Supplemental Table 11). We found that error k-mers reported by QV were almost always validated by errors reported using GIAB, indicating that most of the QV errors within the high-confidence regions are likely true. However, for 35% of the cases examined, the QV did not flag the pre- nor postpolishing sequence as an error. Because it is impossible for both versions of the assembly sequence to be correct, we attribute this to the fact that k-mer-based quality estimates often miss errors in repetitive sequences, owing to their presence in the “truth” k-mer set coming from other regions of the genome. Therefore, we conclude that the nonunique k-mer sequence is likely the cause of the underestimated error rates in QV metrics.
k-mer-based methods identify residual errors in unpolished sequence
Looking at the k-mer QV whole genome, we found little difference between the unpolished assembly and the four different polishing methods, with all QV scores in the range of 50.56–50.84. This is because the vast majority of the error k-mers leading to this QV score is unchanged after polishing (97.3% for DeepPolisher) (Fig. 3A). Investigating these unchanged error k-mers, we found that 41% have >70% GC content, and when we plotted the distribution of their GC content, we saw a clear bias relative to randomly permuted k-mers (Methods) (Supplemental Fig. 4). This indicates that the observed GC bias (Benjamini and Speed 2012; Chen et al. 2013) in short reads is likely inducing a substantial number of falsely reported errors. However, this still leaves a majority of these error k-mers that may be real, unaddressed errors. Indeed, we find that 22.8% of predicted error k-mers are located in regions with <5× coverage of HiFi data. These regions were likely bridged by ONT sequence in the assembly stage and therefore have a higher error rate but are inaccessible to DeepPolisher owing to the lack of HiFi read coverage. Additionally, we found that 36% of the unchanged error k-mers overlapped simple and low-complexity repeat annotations, with 10% overlapping homopolymers >10 bp. These regions tend to have higher error rates in HiFi reads (Wenger et al. 2019; Stoler and Nekrutenko 2021), which makes it likely these are true errors that DeepPolisher was unable to fix. Together, these observations indicate that although the residual predicted error k-mers contain many false positives, the k-mer-based methods are identifying significant sequence outside of GIAB high-confidence regions that is erroneous and unchanged by our current polishing methods (Fig. 3D; Supplemental Table 12).
Polishing the HPRC release 2 assemblies
We applied our polishing pipeline to 180 HPRC assemblies made available as part of the second HPRC data release. Within the high-confidence regions (81.4% of the genome on average), and consistent with our analysis of HG005, we noted an average QV improvement of 3.4 (54% reduction in errors), with the average unpolished QV of 66.66 moving to an average of 70.05 after polishing (Fig. 4A). Whole genome, we improved the QV by 0.306 points on average (7% reduction in errors) (Fig. 4B), owing to the presence of the lower-quality ONT-only regions that cannot be polished with the current model and regions subject to sequencing bias as previously discussed. Polishing improved phasing accuracy for all 111 samples that contained trio data: On average, it reduced switch error by 0.039 and hamming errors by 0.037 (Fig. 4C; Supplemental Table 13).

Polishing results for 180 HPRC assemblies. (A) Hap1 QV (x-axis) and Hap2 QV (y-axis) in the high-confidence regions for 180 HPRC samples from the second release. For each sample, unpolished QV is in blue with an arrow pointing to the polished QV. (B) The same as A but for whole-genome QV. (C) Switch (x -axis) and hamming (y-axis) error rate for the 107 samples with trio data. Unpolished in pink with an arrow pointing to polished in yellow.
Application to non-human primate assemblies
We generated hifiasm assemblies for two marmoset individuals, Baguette and Maisie, using HiFi and ONT long-read and Illumina short-read data derived from fibroblast cell lines, in order to test the DeepPolisher pipeline's performance on non-human species. Whole genome, DeepPolisher improved the QV for Baguette from 57.1495 to 57.5501 (9% error reduction) and for Maisie from 57.0333 to 57.5419 (11% error reduction), which is comparable to the whole-genome error reduction observed for the HPRC assemblies (Supplemental Table 14). To assess the performance in regions accessible to the current DeepPolisher model, we excluded from the QV calculation the regions in which HiFi coverage of mapping quality greater than one was lower than 5×. For Baguette, this excluded 5.8% of the genome and resulted in a QV improvement from 59.0761 to 59.7212 (14% error reduction). For Maisie, this excluded 3.3% of the genome and resulted in a QV improvement from 59.3859 to 60.2831 (18.7% error reduction).
Removing additional errors with element data
Element Biosciences AVITI (Element) short-read data have gained popularity recently for its high accuracy in tandem repeats and homopolymers. We tested whether further polishing with Element data after DeepPolisher could fix residual errors. Because of the challenges associated with phasing short-read data, we restricted our initial experiment to polishing just homozygous locations. To obtain potential homozygous edits, we aligned 50× Element Cloudbreak reads (Carroll et al. 2023) to each haplotype of the HG002 and HG005 polished assemblies. We ran DeepVariant on each alignment and selected just the homozygous-alternate calls with a GQ greater than seven. This approach removed 9% (1182) of the remaining variant-calling errors after polishing with DeepPolisher for HG002 and 5% (600) for HG005, a promising number of errors removed given the relatively low number of polishing edits applied (5586 for HG002 and 8795 for HG005) (Supplemental Table 15).
Discussion
Rapid advancements in long-read sequencing and assembly algorithms in recent years are ushering in a new era of population-scale, T2T genome assembly projects (Ebert et al. 2021; Rhie et al. 2021; Liao et al. 2023). Many of these assemblies are being used as reference genomes, making it important that the sequences be highly accurate. To remove the remaining errors in these sequences, deep learning approaches are a promising way to model sequencing bias in regions like homopolymers and low-complexity repeats. In this work, we developed a new polishing tool called DeepPolisher, an encoder-only transformer model, that takes in HiFi reads aligned to a diploid assembly and predicts the underlying sequence. Accompanying this, we introduced a pipeline called PHAsing Reads in Areas Of Homozygosity (PHARAOH) that ensures read alignment inputs to DeepPolisher are assigned to the correct haplotype and phases potential polishing edits in long homozygous assembly regions.
Using an alignment-based assessment of assembly accuracy measured against the GIAB v4.2.1 benchmarking variant calls, we demonstrate that DeepPolisher reduces assembly errors by approximately half, largely driven by reductions in indel errors, across the large majority of sequence that is mappable by HiFi sequencing. Although alignment-based assessments of high-quality benchmark sets are close to a gold standard, current benchmarks do not cover the full genome and are dependent on accurate alignment. To get around these issues, we also assessed assembly quality with k-mer methods. Comparing k-mer methods to the alignment-based assessment within the same genomic regions predicted similar percentage reductions in errors, which is consistent and reassuring, but also revealed that the k-mer methods miss around ≥90% of residual errors. Although some of these may be false positives in the alignment benchmark, our manual analysis suggests that most are simply missed by the k-mer methods owing to their limitations regarding modeling repeat k-mers that are likely often enriched in assembly errors. We would therefore urge users to recognize this and note that such QV estimates are likely overly optimistic because they have a high false negative rate.
However, k-mer methods also reveal large numbers of predicted errors in regions that remain untouched with the current iteration of DeepPolisher and are located outside of the regions assessed by the alignment-based benchmarks. Although nearly half of these predicted errors may be false positives caused by GC content bias, it is probable that many are real. We attribute a quarter of these errors to regions of HiFi coverage dropout in which ONT was primarily used during the assembly stage. We plan to expand the DeepPolisher model to work on ONT reads in the future to address this fraction of errors. Around one-third of the residual errors lie in homopolymers and tandem repeats, sequences that are challenging even for highly accurate HiFi reads. We experimented with using Element Biosciences data to fix these errors and demonstrate that we can remove a minority of errors using the approach. However, Element reads are currently relatively short, preventing accurate mapping across repetitive sequence and limiting phasing. We predict the remaining class of errors will be challenging to fix.
To resolve the limitations of current alignment and k-mer-based approaches to assessing base-level assembly accuracy, the Q100 project (https://github.com/marbl/HG002) aims to create a “perfect” genome assembly to be used as a benchmark for assembly methods. Although achieving a completely perfect assembly will be challenging, the direct comparison of a draft assembly to an assembly benchmark will help expand the alignment-based assessment of assembly quality to more difficult regions of the genome than are covered by the GIAB high-confidence regions. In the future, we hope that genome benchmarks for more samples will be produced to expand the training and testing sets to multiple genomes for models like DeepPolisher.
Despite the difficulties inherent in assessing the performance benefits of the DeepPolisher model, we demonstrate that it provides the best balance between over- and underpolishing for high-quality assembly projects like the HPRC. We have applied the DeepPolisher and PHARAOH pipeline to the next release of HPRC assemblies and demonstrate consistent improvements to QV across all samples, which will be critical for downstream applications of this new reference data set. We have demonstrated that the current iteration of DeepPolisher, although trained on human data, is applicable to non-human primate species, which may be useful for projects like the VGP (Rhie et al. 2021) and the T2T consortium's T2T-Primates project (Yoo et al. 2025). We anticipate that for expansion into more distantly related clades, new models will need to be trained.
Methods
Assembly
To create HG002 and HG005 hifiasm assemblies for training and testing DeepPolisher, we ran hifiasm (UL) v0.19.5 in trio mode with the default parameters. To create HG002 and HG005 Verkko assemblies for training and testing DeepPolisher, we ran Verkko v2.0 in trio mode with the ‐‐correct-overlap-batches 64 and ‐‐screen human and otherwise default parameters. As input we used 40× HiFi DeepConsensus v1.2 and 40× ONT UL >100 kb reads from the HPRC. The ONT reads for the HG005 and HG002 samples were sequenced using R9.4.1 flow cells and basecalled using Guppy software (HG005 with version 5.0.7 and HG002 with version 6.0.6). For phasing assembly haplotypes in the trio mode, both assemblers needed k-mer databases that were created from parental short reads, and Verkko required k-mer databases from the child's short reads. For hifiasm we used yak and for Verkko we used meryl to create the related k-mer databases. For HG002, we used the 300× parental and child Illumina data from the HPRC to create input yak and meryl databases (using default settings for both) and used the 100× Illumina data for HG005 parental and child databases. All data used to generate and polish the assemblies were downloaded from the HPRC (https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=working/HPRC_PLUS/). The HPRC release 2 assemblies and their associated data were downloaded (https://github.com/human-pangenomics/hprc_intermediate_assembly/tree/main/data_tables).
DeepPolisher development and training
DeepPolisher uses an encoder-only transformer model to predict potential errors present in a haplotype-resolved genome assembly. The framework of DeepPolisher is adopted from our previous work DeepConsensus (Baid et al. 2022). The input to DeepPolisher is an alignment file in which the reads are aligned to the correct haplotype assembly. Then, it performs the following steps to identify potential errors:
-
Make_images. In the make_images step
-
DeepPolisher takes a 25 kb window of the assembly and tries to find potential positions at which there could be an error. This is done by comparing the reads to the assembly. If at any position, there are more than two reads containing a sequence that does not match the assembly (mismatched base, insertion or deletion), we pick that position to be a potential error site.
-
The positions that are within 50 bp or closer are grouped together to create a window that does not exceed 100 bp sequence.
-
For each window, we take alignment features (base, base quality, mapping quality, match, or mismatch) and create a tensor-like representation of the window that potentially contains assembly errors.
-
This tensor-like representation of the alignment is then fed to the transformer-model for prediction.
-
-
Inference. In the inference step, we take the tensor-like representation of the 100 bp window with potential sequencing errors and use that as an input to the encoder-only transformer model. The model predicts a sequence based on the read alignments. We then compare the predicted sequence to the assembly, and any difference between the predicted sequence and the assembly is reported in a VCF format file, which would indicate potential errors in the assembly. These VCF edits can be applied to the assembly after DeepPolisher in a separate step using BCFtools (Danecek et al. 2021) consensus -H2.
We trained DeepPolisher on the HG002 hifiasm assembly taking the HG002-T2T-v0.9 (2024) as the truth. We took HiFi reads aligned to the hifiasm assembly using the PHARAOH pipeline. Then, we aligned the hifiasm diploid assembly to the HG002-T2T-v0.9 assembly to create an assembly-to-truth alignment, which we use as the truth sequence for training the model. During training, the predicted sequence is compared against the truth sequence, and the loss is calculated via the alignment loss function we introduced in our previous work DeepConsensus (Baid et al. 2022). We use a similar approach to train a model for Verkko assembly, in which we use the Verkko assembly for training.
PHARAOH pipeline
The first step in the PHARAOH workflow involves aligning all HiFi reads to the diploid assembly using minimap2 with parameters “-L ‐‐eqx ‐‐cs -c -k19 -x map-hifi.” Next, we identify stretches of homozygosity in the assembly by aligning the two haplotype FASTA files to each other with minimap2 using the parameters “-L ‐‐eqx ‐‐cs -c -x asm5.” The resulting PAF file is passed into a Python script (https://github.com/mobinasri/secphase/blob/main/programs/src/find_homozygous_regions.py) that parses the cigar string to return stretches of 100% identical sequence >20,000 bp (the average length of HiFi reads). Next, the reads within the homozygous regions are extracted and aligned separately to the maternal and paternal haplotypes using minimap2 with parameters “-L ‐‐eqx ‐‐cs -c -k19 -map-hifi.” All reads with gap-compressed mismatch ratio exceeding 0.02 are removed to avoid calling spurious variants in reads too diverged from the assembly. DeepVariant is then used to detect heterozygous variants in these alignments, and all variants with a genotype quality less than 10 are filtered out. We align ONT UL reads >100,000 bp separately to each haplotype assembly using minimap2 with parameters “‐‐cs ‐‐eqx -L -Y -map-ont.” These UL alignments are passed to WhatsHap (Martin et al. 2016), which is used to phase the heterozygous variants called by DeepVariant in homozygous regions.
To reassign reads to the correct haplotype assembly using the phased variants from WhatsHap, we added a new mode to the tool Secphase. From WhatsHap, we have phased variants grouped into phase blocks. Each phase block contains two haplotypes, and Secphase picks the haplotype with a higher number of reference alleles (i.e., the one that is more similar to the assembly sequence) covered by that phase block. Secphase then retains the variants with alternative alleles in the selected haplotype of the phase block. Next, a variant block is created around each selected variant. The minimum block size can be set as a parameter (default = 100), and the actual size is adjusted to be twice the variant size if the minimum block size is smaller than twice the variant size. Overlapping variant blocks are merged, so a single block may encompass multiple variants. Each variant block, defined in assembly coordinates, is then projected onto the read coordinates. A single read may have multiple alignments, typically one per assembly haplotype, with each alignment having its own projected blocks. To create nonoverlapping blocks crucial for distinguishing assembly haplotypes, all projected variant blocks are merged on the read coordinates. These merged blocks serve as proxies to compare haplotypes. The variants are then applied to the assembly haplotypes, effectively “pseudopolishing” the assembly only in the created blocks. The pseudopolished sequence of each haplotype is then compared against the read sequence within each variant block, with edit distances calculated using the Edlib (Šošić and Šikić 2017) library. The edit distances across all merged variant blocks are summed for each alignment. The haplotype with the lower total edit distance is selected as the correct haplotype for the related read.
In the PHARAOH pipeline, this new variant mode of Secphase is used for the homozygous regions >20 kb containing a potential heterozygous polishing edit, and the original marker mode is used for the rest of the genome. After reassigning reads to the new location determined by Secphase, we remove alignments with a gap-compressed mismatch ratio exceeding 0.002, producing the final alignment for input to DeepPolisher. PHARAOH is implemented as a wdl workflow and is publicly available at GitHub (https://github.com/miramastoras/PHARAOH). The tool Secphase may be found at GitHub (https://github.com/mobinasri/Secphase). The entire pipeline for running PHARAOH and DeepPolisher may be found at GitHub (https://github.com/human-pangenomics/hpp_production_workflows/tree/master/polishing).
DeepPolisher GQ filter optimization
We selected four HPRC samples (HG01975, HG04115, HG02129, HG01993) with varying QV improvements after polishing (Supplemental Table 16) to optimize a set of GQ filters for DeepPolisher. First, we annotated the error k-mers in the assemblies by whether they were induced, fixed or did not change with polishing. To accomplish this we first aligned each haplotype of the unpolished assembly to the corresponding haplotype of the polished assembly with minimap2 and parameters -x asm5 -L ‐‐eqx ‐‐cs -c. We took the polished assembly *_only.bed file produced by Merqury (which contains the assembly coordinates for the error k-mers), merged the k-mers with BEDTools (Quinlan and Hall 2010) merge -c 1, and projected them to the unpolished assembly using the script https://github.com/mobinasri/flagger/blob/main/programs/src/project_blocks_multi_thread.py. This allowed us to have the polished assembly error k-mers in unpolished assembly coordinates. We then subtracted the polished projected error k-mer BED file from the raw error k-mer BED file with BEDTools subtract -A to extract the error k-mers fixed by polishing. We performed the reverse to obtain the error k-mers induced by polishing. Finally, we used BEDTools intersect to produce the error k-mers unchanged by polishing, which were common to both BED files. To annotate the polishing edits by whether they induced, fixed, or did not change error k-mers, we intersected the polishing VCF with the error k-mer BED files labeled in the previous step.
We found that the majority of DeepPolisher edits inducing error k-mers were 1 bp insertions or deletions (Supplemental Fig. 4). Based on this observation, we tested three filtering scenarios: (1) a single GQ filter for all variants, (2) a GQ filter for 1 bp insertions and a separate one for the remaining edits, and (3) a separate GQ filter for 1 bp insertions, 1 bp deletions, and all other edits. We combined the annotated polishing edits from all four samples, and for each of these scenarios, we swept the GQ filter cutoff from zero to 25 and calculated the estimated QV improvement using the formula −10 × log10(error_k-mer_after/error_k-mer_before). We found the optimal GQ cutoff to be GQ 20 for 1 bp insertions, GQ 12 for 1 bp deletions, and GQ 5 for all other variants (Supplemental Table 16). All DeepPolisher results shown in the paper have this filter applied unless otherwise stated.
Implementing alternate polishing methods
We adapted the T2T polishing pipeline described by Mc Cartney et al. (2022) for diploid assemblies to compare against DeepPolisher. We aligned 40× HiFi DeepConsensus v1.2 reads from the HPRC for both cell lines to the diploid hifiasm assemblies with winnowmap v2.03 and parameters ‐‐cs ‐‐eqx -L -Y -I8g -map-pb. We aligned ONT R9 40× reads from the HPRC to the diploid hifiasm assembly using winnowmap v2.03 and parameters ‐‐cs ‐‐eqx -I8g -Y -L -p0.5 -map-hifi. We also aligned 30× Illumina data to the diploid hifiasm assembly with BWA-MEM v.0.7.17 and default parameters. HiFi and Illumina BAM files were merged with SAMtools, and DeepVariant v1.6.1 with parameter –model-type=HYBRID_PACBIO_ILLUMINA was used to call variants. PEPPER-Margin-DeepVariant was used to call variants for the ONT alignments. All GQ and VAF filters described by Mc Cartney et al. (2022) were applied. Merfin mode -polish was also used to further filter polishing edits. Edits were applied to the assembly with BCFtools consensus -H1.
We used the same 40× HiFi DeepConsensus v1.2 winnowmap read alignments described above as input to NextPolish2 and DeepVariant. For NextPolish2, 30× Illumina data from the HPRC were first preprocessed with the tool fastp (Chen et al. 2018) with parameters -f 5 ‐‐cut_front ‐‐cut_tail as suggested by the authors (https://github.com/Nextomics/NextPolish2). The preprocessed data were then used to create a yak k=21 and k=31 database as input to NextPolish2, using default settings. NextPolish2 was run with the default settings. DeepVariant v1.6.1 was run with the default settings and flag ‐‐model_type=PACBIO. For DeepVariant, BCFtools consensus -H1 was used to apply edits to the original assembly.
Measuring GIAB variant-calling performance
To assess the accuracy of the assemblies against the GIAB v4.2.1 benchmark set, we first ran dipcall (Li et al. 2018) with the default settings against GRCh38 obtained from https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/references/GRCh38/. We next ran the tool hap.py (https://github.com/Illumina/hap.py) to calculate performance metrics of the dipcall VCF file against the GIAB v4.2.1 benchmark VCF.
For HG002, we restricted the hap.py analysis to regions within the GIAB high-confidence regions subset that were also validated by the T2T Q100 v1.0.1 (https://github.com/marbl/HG002) assembly. To create this subset, we ran dipcall (Li et al. 2018) on the T2T v1.0.1 assembly against GRCh38 using minimap parameters -z200000,10000 (to align across larger SVs and more divergent regions like the MHC). We next intersected the HG002 high-confidence regions BED file provided by GIAB with the BED file produced by dipcall in the previous step to produce a BED file of high-confidence regions that are alignable between GRCh38 and T2T v1.0.1. We ran hap.py on the output VCF of dipcall VCF containing variants between T2T v1.0.1 and GRCh38 against the GIAB v4.2.1 VCF, restricting the analysis to the high-confidence regions that are alignable between GRCh38 and T2T v1.0.1 by passing that BED file in with parameter -f. Using BEDTools subtract, we removed 50 bp surrounding any FP or FN designated variants produced in that hap.py run from the GIAB high-confidence regions BED file in order to remove regions that were not concordant with the T2T v1.0.1. Eight hundred thirty total variants were subtracted, and 63.7% of them were located within the GRCh38 segmental duplication track.
To create the final BED file to use with hap.py -f in benchmarking the HG002 assemblies, we intersected the BED file containing regions concordant between the Q100 T2T v1.0.1 assembly and GIAB v4.2.1 (described in the previous paragraph) with the BED file produced from the dipcall output of the unpolished assembly to GRCh38. For HG005, we repeat this step but with the original HG005 high-confidence regions BED file provided by GIAB. For all hap.py runs, we used the options ‐‐pass-only ‐‐no-roc ‐‐no-json ‐‐engine=vcfeval and the GIAB v3.3 stratifications to obtain performance statistics per genomic region.
To investigate the performance of DeepPolisher in the centromere, we ran dipcall of our HG002 assembly (pre- and postpolishing) against Q100 HG002 v1.0.1 with the default parameters. Using samtools -q 60, we subset the output BAM file from dipcall to regions where mapping quality between the assemblies was 60. We used BEDTools to convert this BAM file to BED and then intersected it with the provided cenSat annotations BED file (https://github.com/marbl/HG002) to define centromere regions alignable between the Q100 assembly and our HG002 hifiasm assembly. Using BEDTools again to intersect the dipcall VCF file with these regions, we counted the number of SNVs and INDELs relative to the Q100 genome before and after polishing with DeepPolisher.
Stratifying QV by whole genome and within GIAB high-confidence regions
For QV calculations, we ran Merqury and Yak with the default settings, using ∼30× coverage Illumina data for all samples. For trio samples, we used Yak trioeval along with parental Illumina data to calculate switch and hamming rates. To assess QV just within the high-confidence regions, we first took an intersection of the GIAB high-confidence regions for HG002 and HG005 using BEDTools to define a subset of high-confidence regions that is not genome specific. We aligned each haplotype assembly to GRCh38 with minimap2 and parameters -x asm5 -L ‐‐eqx ‐‐cs -c. We then used the script https://github.com/mobinasri/flagger/blob/main/programs/src/project_blocks_multi_thread.py to project the GIAB high-confidence regions bedfile from GRCh38 to the assembly. Next, we used BEDTools getfasta with the default settings to extract only the sequence found within this BED file and passed it to Merqury and Yak for QV calculation. To count the number of error k-mers induced, fixed, or unchanged after polishing and to obtain the polishing edits that were the cause of inducing, fixing, or failing to change the error k-mers, we performed the same annotation procedure described under the section “DeepPolisher GQ filter optimization.” To transform QV into an estimate of error per megabase, we used the formula (10−QV/10) × 106.
Comparing different methods for estimating QV
We compared several different methods for calculating QV on our HG005 hifiasm assembly and found them to be highly variable (Supplemental Table 9). Merqury (Rhie et al. 2020) and Yak (Cheng et al. 2021) typically report values two to three QV points apart (approximately a 2× difference in error rate). We found that using k-mers of size 31 led to more conservative scores than k of 21 (between two and four points lower, equivalent to an ∼50% difference in error rate). These longer k-mers can capture differences in longer repeats, which likely accounts for the higher predicted error rates. We also tested Merqury with a hybrid of HiFi and Illumina k-mers following the approach suggested by Mc Cartney et al. (2022). We found that the hybrid k-mer database led to QV scores around 10 points higher (a predicted order of magnitude lower error rate) than when we used just Illumina. Although using hybrid k-mers may remove some Illumina-specific bias, specifically known issues with GC bias (Benjamini and Speed 2012; Chen et al. 2013), we observed that it introduces too many HiFi-specific sequencing errors to the “truth” k-mer space and so reduces the apparent error rate by masking true errors. We also experimented with using Element cloudbreak (Carroll et al. 2023) short-read k-mers to calculate QV and found it predicted double the error rate relative to Illumina (Supplemental Table 9). However, we found the same impact of GC bias on the error k-mers calculated by Merqury QV as was described for Illumina (Supplemental Fig. 4). Given that Element data were not available for the HPRC samples, and to be conservative, we report Merqury QV with Illumina k-mers of size 31 for this paper unless otherwise stated.
Manual investigation of concordance between GIAB variant calls and Illumina QV
We performed a manual investigation of the concordance between the GIAB variant call errors and the Illumina QV reported errors for HG005 (polished by DeepPolisher) within the high-confidence regions using IGV (Robinson et al. 2011). To obtain BED tracks for viewing the GIAB variant call errors in respect to the raw assembly coordinates, we used BCFtools (Danecek et al. 2021) isec to obtain the intersections, unions, and complements between the raw and polished VCFs output by hap.py (these VCFs contain variants relative to GRCh38 labeled by true positive, false positive, or false negative). We used the tool vcf2bed from BedOps (Neph et al. 2012) to convert the VCF files to BED format, expanded the intervals by 10 bp on each side to enable projecting of the indels over to the raw assembly coordinates with the script https://github.com/mobinasri/flagger/blob/main/programs/src/project_blocks_multi_thread.py.
For viewing the QV errors in IGV, we took the *_error.bed file (containing the locations in the assembly of the error k-mers) produced by Merqury using Illumina k-mers of size 31 and used the procedure described in the section “DeepPolisher GQ filter optimization” to annotate the polishing VCF by whether the edits induced, fixed, or failed to change the error k-mers, as well as to label the error k-mers by whether they were fixed, induced, or unchanged by polishing.
We used the files described above to randomly select 10 GIAB errors induced by DeepPolisher, 10 GIAB errors fixed by DeepPolisher, 10 polishing edits inducing error k-mers, and 10 polishing edits removing error k-mers. We carefully evaluated all 40 examples in IGV alongside the PHARAOH read alignments. For the GIAB errors, we looked at both haplotypes to understand if one or both alleles were considered an error, and in two cases, there was a polishing edit made to both haplotypes for a given GIAB error, so edits on both haplotypes were assessed. For all 42 polishing edits, we recorded the error status in both GIAB and QV. All details for the 42 edits may be found in Supplemental Table 10.
In summary, out of 42 polishing edits manually inspected, there were three cases in which GIAB indicated the edit induced an error, whereas the QV indicated that it fixed an error. For 24/42 polishing edits, the QV and GIAB metrics both agreed on whether the edit fixed or induced an error. For 15/42 polishing edits, the QV reported no error k-mer in either the pre- or postpolishing sequence, indicating that both were correct according to the QV metric. Because it is impossible for both versions of the sequence at a given location to be correct, this 33% of inspected edits likely falls in nonunique sequence, in which either the pre- or postpolishing sequence is found elsewhere in the genome, so the QV failed to flag it as false. This proportion may also be caused by noise from sequencing errors in the Illumina reads. In addition, all but two of the 42 edits examined were found within long tandem repeats or homopolymers, which are more likely to be nonunique in the genome. We propose that this category accounts for the much lower error rate reported by the QV than by the GIAB analysis within the high-confidence regions. Because all of the QV reported errors we examined were corroborated as false by GIAB (and there were only three cases in which QV indicated an error was fixed whereas GIAB indicated it was induced), we conclude that the majority of the QV errors within the high-confidence regions are most likely true errors.
Characterizing the context of unchanged error k-mers
After obtaining the error k-mers unchanged by polishing as described in the section “DeepPolisher GQ filter optimization,” we sought to establish how many were located within coverage dropouts. We ran the tool mosdepth with parameter ‐‐quantize 0:5:10:150: on the PHARAOH alignment to obtain locations with less than 5× coverage and then BEDTools intersect to obtain the error k-mers unchanged by polishing within those regions. To obtain the numbers that were within simple or low complexity repeats, we ran dipcall with the default parameters to align the HG005 raw assembly against CHM13 and then the script https://github.com/mobinasri/flagger/blob/main/programs/src/project_blocks_multi_thread.py to project the repeat annotations produced for T2T-CHM13 at https://github.com/marbl/CHM13 to HG005 coordinates. We then used BEDTools intersect with the unchanged error k-mers and the projected repeat annotations. To calculate the GC content of all error k-mers, we used BEDTools nuc. To obtain regions in the HG005 raw assembly with homopolymers >10 bp, we used a custom Python script that may be found at GitHub (https://github.com/miramastoras/DeepPolisher_manuscript/blob/main/paper_analysis/quantify_error_kmers_HG005.md) and BEDTools intersect.
Gene impact analysis
We annotated the HG002 and HG005 assemblies before and after polishing with the Comparative Annotation Toolkit (CAT2.0) (Fiddes et al. 2018) using the transMap and liftoff modules based on the GENCODE v46 gene annotations for hg38 as the reference. For these annotation sets, we identified the locations of frameshifts by iterating over the coding sequence of every transcript and looking for gaps in the alignment. If the gap length was not a multiple of three or if the length was >30 bp, the gap was determined to be a frameshift. To identify the number of nonsense mutations that would cause early stop codons in the annotation sets, we iterated through each codon in the coding sequence and looked for an early stop codon before the canonical stop codon at the end of the transcript. We then sought to validate how many of the mutations were likely true mutations, rather than errors, by checking for their presence in an orthogonal set of variant calls (GIAB v4.2.1). The mutations were lifted over to the GRCh38 reference using halLiftover (Hickey et al. 2013) on a GRCH38-based Cactus alignment and then intersected with the GIAB set using BEDTools intersect (Quinlan and Hall 2010). To study the impact of the polishing edits on the coding regions of the genome, we intersected the total set of edits made to the assembly with the CDS regions of the assembly. The DNA and protein sequences of transcripts that were affected by these edits were extracted from the raw and polished assemblies. These were aligned against the DNA and protein sequences extracted from the GRCh38 transcripts using parasail (Daily 2016) to look at changes in DNA and protein sequence identity for these transcripts caused by polishing.
Marmoset assembly polishing
To create the hifiasm assemblies for the two marmosets (Baguette and Maisie), we ran hifiasm (UL) v0.24.0 in HiC mode with the default parameters. As input, we used 60× PacBio HiFi DeepConsensus reads and 30× ONT UL >100 kb reads. The ONT reads were sequenced using R10.4.1 flow cells and basecalled using Dorado v4.3.0. For phasing this assembly, we used HiC data at 50× coverage. The assemblies were polished using the HiFi and ONT data as input to the PHARAOH + DeepPolisher pipeline. To exclude regions of low HiFi coverage from the QV calculation, we ran mosdepth (Pedersen and Quinlan 2018) with parameters -Q1 ‐‐quantize 0:5:10:150: on the PHARAOH BAM files for Baguette and Maisie. We extracted regions from zero to 5× coverage from the resulting BED file and used bedtool subtract to obtain the inverse of these regions. We then used BEDTools getfasta with the default settings to extract only the sequence with greater than 5× coverage of mapping quality greater than one. We ran Merqury with illumina k-mers of size 31 for the QV calculation.
Element polishing
We obtained 50× element data for HG002 and HG005 from Carroll et al. (2023) and aligned all reads to each polished haplotype assembly separately. We used DeepVariant v1.6.1 with ‐‐model_type = WGS to call variants. We selected only homozygous alt PASS variants with genotype quality greater than seven and applied them to the assembly with BCFtools consensus -H 2.
Software availability
DeepPolisher code is available publicly on GitHub (https://github.com/google/deeppolisher). PHARAOH is implemented in a pipeline available on GitHub (https://github.com/miramastoras/PHARAOH). Scripts for analysis and benchmarking are available on GitHub (https://github.com/miramastoras/DeepPolisher_manuscript). Code used to generate the HPRC assembly data release 2 may be found at GitHub (https://github.com/human-pangenomics/hprc_intermediate_assembly). Source code for all of these repositories is also available as Supplemental Code.
Data access
The HG002 and HG005 assemblies and DeepPolisher VCFs used for benchmarking are available at https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=publications/DeepPolisher_2024/.
Competing interest statement
A.C., D.E.C., P.-C.C., A.K., L.B., M.N., and K.S. are employees of Google and own Alphabet stock as part of the standard compensation package.
Acknowledgments
B.P. and UCSC personnel were in part supported by National Institutes of Health (NIH) grants R01HG010485, U01HG010961, U24HG010262, OT2OD026682, and U24HG011853. M.M. was in part supported by NIH grant T32HG012344. The HPRC acknowledges the National Human Genome Research Institute (NHGRI) for funding the following grants supporting the creation of the human pangenome reference: U41HG010972, U01HG010971, U01HG013760, U01HG013755, U01HG013748, U01HG013744, R01HG011274, and the Human Pangenome Reference Consortium (BioProject ID: PRJNA730823).
Author contributions: K.S. and B.P. devised the study. M.M., B.P., and K.S. drafted the manuscript. A.C., D.E.C., P.-C.C., A.K., L.B., M.N., and K.S. developed DeepPolisher. M.A. and M.M. developed PHARAOH. M.M., M.A., and T.S.W. performed data analysis. P.H. performed gene impact analysis. M.A. and J.L. generated assemblies.
Footnotes
-
↵3 A complete list of the HPRC authors appears at the end of this paper.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280149.124.
- Received October 22, 2024.
- Accepted April 30, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








