
Regeneration alters open chromatin and cis-regulatory landscape of erythroid precursors
Abstract
Stress erythropoiesis elevates the rate of red blood cell (RBC) production as a physiological response to stressors such as anemia or hypoxia. In acute anemia, RBC progenitors and precursors temporarily rewire their transcriptome, up- and downregulating hundreds of genes to accelerate the production of mature RBCs. Effective regeneration requires communication between critical cytokine signals (e.g., BMP4) and cis-regulatory elements on chromatin which coordinate transcriptional changes. To identify cis-regulatory changes that underlie anemia-specific gene expression and cellular responses, we analyzed chromatin accessibility in populations of cells enriched for red blood cell precursors isolated from mice at a range of time points after anemia induction. Early in the anemia response, chromatin is transiently open at AP-1-containing regions, correlated with increased Jun and Fos transcript/protein levels. Jun knockdown ex vivo decreases the percentage of KIT+ erythroid precursors after anemia induction. We observe a second rewiring event at time points consistent with anemia resolution, involving repression of GATA factor-accessible regions and activation of ETS factor-accessible regions. In both mouse in vivo models and human CD34+ cells stimulated with BMP4, accessibility changes at regions with prior associations to human blood phenotypes. Dozens of BMP4- and anemia-activated loci are sensitive to natural human variation. The representation of red blood cell trait–associated loci in ATAC-seq data remains durably elevated more than 1 month after anemia resolution. Together, these findings provide a framework to understand the early establishment and late resolution of a regeneration-dependent transcriptome in RBC precursors.
Quick regeneration of damaged tissues is a vital function of multicellular organisms. In acute anemia, multipotent progenitors differentiate into erythroid-specified precursors to regenerate lost red blood cells (RBCs) via a process termed “stress erythropoiesis.” Self-renewing spleen-resident progenitors are responsible for the transient acceleration of erythropoiesis in acute anemia, with the involvement of multipotent progenitors (MPPs) and hematopoietic stem cells (HSCs) (Perry et al. 2007; Porayette and Paulson 2008; Inra et al. 2015; Tseng et al. 2024). The process of stress erythropoiesis involves transcriptional changes that regulate progenitor cell expansion and differentiation, coinciding with increased accessibility and usage of cis-regulatory elements at distal enhancers (Zhou et al. 2023). Establishing key chromatin changes occurring in erythroid precursors at cis-regulatory elements genome-wide has the potential to identify new regulators of stress erythropoiesis in the context of acute anemia.
Among molecular mechanisms needed to accelerate and decelerate erythropoiesis in anemia, erythroid stress progenitors and precursors require critical extracellular signals (Bresnick et al. 2018; Paulson et al. 2020). Bone morphogenetic protein 4 (BMP4), hypoxia inducible factors (HIFs), erythropoietin (EPO), and other inflammatory cytokines are involved in stages of activation, expansion, and differentiation of stress progenitors (Perry et al. 2007; Xu et al. 2014; Bennett et al. 2019; Hao et al. 2019). To link signaling molecules to transcriptional changes, prior work has demonstrated that cis-regulatory elements involved in anemia-induced transcriptional changes include elements that bind transcription factors GATA1 and TAL1 at critical red blood cell-promoting genes (Hewitt et al. 2017; Zhou et al. 2023). Through the anemia recovery period, a central question emerges: What are the key cis-regulatory mechanisms, intrinsic to the erythroid precursors, that promote functional responses to acute stress? Using a model of acute anemia, mice become rapidly anemic (within 12 h) and recover in 5–7 days (Lee et al. 2015; Hewitt et al. 2017). The pool of erythroid precursors required for regeneration is not fully renewed until several weeks later (Perry et al. 2007, 2009), highlighting the importance of the post-recovery period to remodel and regain the ability to respond to subsequent stressors.
Human genetic variants identified by genome-wide association studies have linked thousands of polymorphisms (SNPs) to erythropoietic traits (Astle et al. 2016; Vuckovic et al. 2020). However, mechanisms that explain the role of SNPs in regulating blood traits are not understood for most associations. Within specific contexts (e.g., globin expression in erythroid cells), base editing screens have identified new noncoding variants involved in gene regulation (Martin-Rufino et al. 2023). Because many SNPs are located within predicted enhancers and promoters, cataloging the changes in chromatin accessibility and occupancy at trait-associated SNPs during periods of specific environmental stressors (e.g., anemia stress) provides an important clue towards understanding mechanisms and possible causes for distinct inter-individual stress responses. Anemia stress-dependent transcriptional regulation can be useful for developing clinical strategies to regenerate RBCs in chronic conditions. SNP regions annotated for associations to specific hematologic traits within anemia-sensitive chromatin accessible regions may represent new genetic identifiers of susceptibility to chronic anemia at noncoding sites.
Results
Increased accessibility, expression, and activity of AP-1 during erythroid regeneration
Cells enriched for erythroid precursors (LIN−KIT+TFRC+LY76−)3 were isolated from mouse spleen 24 h after treatment with the red blood cell lysing chemical phenylhydrazine (PHZ) and processed for ATAC-sequencing (Fig. 1A). This population contains anemia-activated erythroid burst-forming units (stress BFU-E) with distinctive properties compared to bone marrow BFU-E found in steady-state conditions. A unique property of stress BFU-E is that cells respond to the BMP4 growth factor, which is upregulated in anemia (Lenox et al. 2005). Comparing samples isolated 24 h after PHZ treatment to control, untreated mice (referred to as “Day 0”), 1786 ATAC-seq peaks were gained in erythroid precursors, while 1242 peaks were lost (Fig. 1B). Examining known motifs, we saw expected increases in accessibility at STAT-binding γ-activated sites (GAS motifs) and GATA motifs, as well as increases in accessibility peaks containing AP-1 motifs (Fig. 1C). To evaluate whether chromatin accessibility changes were related to transcription factor occupancy and anemia-induced transactivation at these motifs, we conducted transcription factor occupancy prediction by investigation of ATAC-seq signal (TOBIAS) “footprinting” genome-wide (Bentsen et al. 2020). Significant increases in footprint scores were found at STAT motifs and at sites predicted to bind JUN/FOS transcription factors, indicating higher transcription factor motif use (Fig. 1D; Supplemental Fig. 1A). Among these, an anemia-activated chromatin accessibility peak is present upstream of the anemia-regulated oncostatin M (Osm) gene, which is required for erythropoiesis (Tanaka et al. 2003). The Osm locus contains an AP-1 motif and increased AP-1 footprint score at day 1 post-PHZ (Fig. 1E). Along with the oncostatin M receptor (OSMR), the OSM protein coordinates mRNA splicing during acute stressors in other contexts, for example, inflammation (Tanaka et al. 2003; Böing et al. 2006; Kubin et al. 2011, 2022). A similar anemia-induced AP-1 footprint was detected at the locus encoding kinesin family member 3B (Kif3b)—a gene predicted to control intracellular transport (Fig. 1F). Although the mouse Kif3b knockout is embryonic-lethal (Nonaka et al. 1998), little is known about the function and regulation of this protein in physiological or stress contexts. These results predict that transient, early chromatin occupancy at AP-1 motifs is promoting transcription of anemia-activated genes.
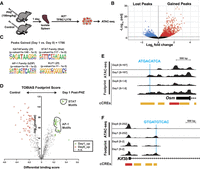
Chromatin accessibility and footprint changes at early time points in erythroid regeneration. (A) Experimental layout. (B) Volcano plot depicts the significantly gained and lost ATAC-seq peaks 24 h post-PHZ. (C) Motif analysis (HOMER) at regions where ATAC-seq peaks were gained 24 h post-PHZ. (D) Volcano plot of transcription factor motif differential binding scores between 0 and 24 h post-PHZ on the x-axis (calculated using TOBIAS) and −log10 (P value) on the y-axis. Each dot represents one transcription factor. Circled areas correspond to consensus AP-1 or STAT family motifs with similar sequences. (E) ATAC-seq and TOBIAS Footprint scores at the Osm locus. Blue line demarcates a region with changing footprint score and underlying sequence. Candidate cis-regulatory elements (cCREs) were classified by ENCODE (The ENCODE Project Consortium 2012). Red = promoter, orange = proximal enhancer-like signature. (F) ATAC-seq and TOBIAS Footprint scores at the Kif3b locus. Blue line demarcates region with changing footprint score and underlying sequence. cCREs were classified by ENCODE (The ENCODE Project Consortium 2012). Red = promoter, orange = proximal enhancer-like signature.
In KIT+ erythroid precursors isolated from day 0 versus day 1 post-PHZ, single- cell RNA-seq revealed 246 upregulated genes (fold change >1.5, adj-P < 0.05), 10 of which are TFs, and three of the 10 are AP-1 TFs (Jun, Junb, and Fos) (Fig. 2A; Supplemental Table S1). Jun is expressed in many non-erythroid cell types in the hematopoietic system. In erythroid precursor cells, Jun expression increases in acute anemia (Supplemental Fig. 1B). Next, we evaluated the expression level of all AP-1 transcription factor family members in erythroid and HSPC clusters monitoring changes over a 7 day period post-PHZ. The transcriptomes of spleen isolated erythroid-committed cells clustered into four distinct populations and were analyzed separately unless otherwise noted (see Zhou et al. 2023). Corroborating protein-level data, three AP-1 family genes (Jun, Junb, Fos) were transiently upregulated in erythroid and HSPC populations (Fig. 2B). Other upregulated AP-1 genes (e.g., Atf4 and Mafg) exhibited a distinct pattern of expression at day 3 post-PHZ. In a similar analysis of STATs, several STAT family mRNAs increased at day 3 post-PHZ in HSPCs (Supplemental Fig. 1C,D). We next evaluated the extent to which AP-1 motif utilization changes at early time points post-PHZ. At day 0 versus day 1, 180 open chromatin regions exhibited either increased or decreased AP-1 footprints (Fig. 2C; Supplemental Table S2). Likewise, we identified 433 footprints containing STAT motifs changing at day 1 post-PHZ (Supplemental Fig. 1D; Supplemental Table S3). Overall, a small proportion of AP-1 and STAT motif footprints overlap (26 out of 433) at the day 1 time point, indicating distinct mechanisms. Annotated genes that were found nearby the sites gaining AP-1 footprints included transcription factors Zfpm1 (also known as Fog1), Ctbp2, Lmo2, Zbtb7a, and Tead2 (Supplemental Table S2). Jun, Junb, and Fos may act as early trans-activating factors driving stress erythropoiesis. To infer AP-1-dependent activities in the transcriptome after PHZ treatment, we performed single-cell regulatory network inference and clustering (SCENIC) analysis. “Regulon” scores of JUN, FOS, and FOSB increased in HSPCs and erythroid clusters 3 and 4, specifically and transiently at day 1 post-PHZ and were decreased at day 3 post-PHZ (Fig. 2D; Supplemental Fig. 1E). Representative gene loci in this analysis included Kif3b and Osm. Both loci contained nearby open chromatin regions with AP-1 occupancy, increased AP-1 footprint score post-PHZ (Fig. 1E,F), and transient increases in transcription at day 3 post-PHZ (Fig. 2E). Osm, Kif3b, and Zfpm1 loci contain distinct cis-elements with AP-1 and STAT footprint scores increased at day 1 post-PHZ (Supplemental Fig. 2A). To evaluate whether observed footprint changes correlated with known AP-1 chromatin occupancy sites, we compared to JUN, JUNB, and FOS ChIP-seq data in primary human CD34+ cells and K562 erythroleukemia cells. Out of 180 sites exhibiting anemia-induced changes to AP-1 footprints, 42 overlap with known occupancy of JUN/JUNB or FOS (23%). To evaluate the significance, we compared the number of experimentally validated overlaps in ChIP-seq versus inferred ATAC footprints to randomly sampled ATAC-seq peaks. AP-1 ATAC footprints were more common at known AP-1 occupancy sites established by ChIP-seq than random sites (χ2 test: P < 0.001) (Supplemental Fig. 2B). Palindromic TGA[G/C]TCA motifs can be occupied by any of the AP-1 transcription factor family members, making them indistinguishable by TOBIAS footprint alone. However, transient increases in Jun, Junb, and Fos family members (while other AP-1 family members are not significantly higher at day 1 post-PHZ) suggest they contribute to day 1 footprint changes at erythroid-promoting anemia-upregulated genes.
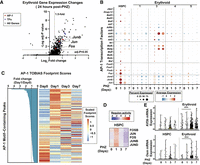
Anemia-induced changes to AP-1 expression correspond with accessibility at erythropoiesis-promoting gene loci. (A) Volcano plot in single-cell RNA-seq data from PHZ-treated mice depicts expression changes in erythroid cells after 24 h, TF = transcription factor. (B) Dot plot depicting single-cell RNA-seq analysis of HSPC and erythroid-specific expression of all detectable components of the AP-1 transcription factor family at 0, 1, 3, and 7 days post-PHZ. (C) Heat map of genomic regions with changes to TOBIAS footprint scores between 0 and 1 day post-PHZ, ranked by fold change. (D) SCENIC analysis comparing the regulon activity of AP-1 components in HPSCs at 0, 1, 3, and 7 days post-PHZ. (E) Kif3b and Osm mRNA expression plots from single-cell RNA-seq data in cells annotated as HSPCs and erythroid clusters. Units of expression are relative log normalized. Error bars represent SD. (***) P < 0.001 (Wilcoxon signed-rank test).
Erythroid recovery involves early, transient, and deferred chromatin changes
To evaluate anemia-induced JUN expression and activity in erythroid cells, we isolated and lineage-depleted cells from PHZ-treated mice at several time points and measured total JUN and phospho(Ser73)-JUN expression by flow cytometry. Within erythroid-committed KIT+TFRC+LY76+ cells, total JUN levels increased 2.0-fold (P = 0.04), consistent with mRNA results, and phospho-JUN levels increased 2.9-fold (P = 0.04) at 24 h post-PHZ (Fig. 3A). After 72 h, JUN protein levels decreased to pre-treatment control levels in the erythroid (TFRC+LY76+) fraction of the spleen (Fig. 3A). In non-erythroid cells (TFRC−LY76−), no changes were observed in either the total JUN or phospho-JUN levels, indicating that transient JUN upregulation is specific for cells expressing erythroid markers. To test AP-1 function in stress erythroid precursors, we lineage-depleted spleen after 24 h PHZ treatment and infected with shRNA targeting Jun (co-expressing GFP) or an shControl in an erythropoiesis-promoting media (Fig. 3B). Knockdown efficiency from protein isolated after 3 days culture was 88% (shJun1) and 84% (shJun2) (Fig. 3C). Jun knockdown decreased the percentage of KIT+ cells in cultures by 1.5-fold (Fig. 3D). Upon treatment with JUN inhibitor SR11302 (20 µM) for 24 h, the percentage of uninfected KIT+ cells in culture decreased significantly (P = 0.002) (Fig. 3E). Next, we quantitated the percentage of cells terminally differentiating to the erythroid lineage. The percentage of LY76+ cells increased from 30.6% to 42.4% after Jun knockdown (Fig. 3F). A similar increase in LY76+ cell percentages was seen using the JUN inhibitor (Fig. 3F). One potential regulatory mechanism for controlling AP-1 gene expression and the AP-1-dependent genetic network as an early anemia response is BMP4 signaling (Xu et al. 1996). BMP4 stimulation of LIN− erythroid cells isolated from spleen increased Jun expression (Fig. 3G). Jun expression at 24 h post-PHZ maintains the KIT+ population of erythroid precursors. High Jun may oppose terminal erythroid differentiation during early stages of erythroid recovery in acute anemia.
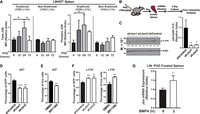
AP-1 maintains KIT+ erythroid precursors and opposes differentiation. (A) Intracellular flow cytometry for total JUN and phospho(Ser73)-JUN in cells isolated from spleens at up to 72 h post-PHZ. N = 3. (B) Experimental layout for testing JUN function in stress precursors. (C) Western blotting of JUN and actin, beta protein in G1E proerythroblasts infected with Jun-targeting shRNA (shJun1 and shJun2) or shControl. (D) Flow cytometry analysis of the percentage of shRNA-infected (GFP+) cells expressing the KIT surface receptor. (E) Flow cytometry analysis of the percentage of live (DAPI−) cells expressing the KIT cell surface receptor. SR11302 treatment 20 µM for 24 h (see Methods). (F) Flow cytometry analysis of shRNA-infected cells after 3 days culture, stained with the erythroid-specific cell surface marker LY76 (also known as TER-119). (G) Quantitative RT-PCR of Jun mRNA expression in LIN− cells isolated from spleens of PHZ-treated mice and cultured for 3 days, followed by stimulation with BMP4 (50 ng/mL). Statistical significance was determined by two-tailed unpaired Student's t-test. (*) P < 0.05, (**) P < 0.01, (***) P < 0.001.
To compare early chromatin-level changes to later steps in recovery from acute anemia, ATAC-seq was performed on erythroid precursors (LIN−KIT+TFRC+LY76−) isolated from mouse spleen over a 35 day time course following anemia induction (Fig. 4A). PHZ significantly altered global chromatin accessibility in erythroid precursors at day 1 through day 14 before returning to a state reminiscent of pre-treated samples (Fig. 4B). Days 1, 3, 7, and 14 samples each segregated separately in principal component analyses, reflecting the establishment of multiple unique chromatin states from stress erythroid precursor specification through complete resolution of anemia.
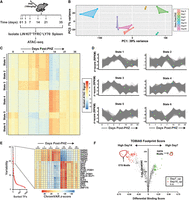
Broad chromatin accessibility changes at intermediate stages of anemia resolution. (A) Experimental layout. (B) Principal component analysis of ATAC-seq peak data comparing control spleen to post-PHZ time points at days 1, 3, 7, 14, 21, and 35 (N = 3). (C) Heat map of Z-score changes in ATAC-seq peak signal among a cohort of sites identified as differentially accessible using DESeq2 (likelihood ratio test) and k-means clustered. (D) Line graphs of ATAC-seq peak accessibility within each state (Z-score). (E) Ranked variability scores of individual transcription factor (TF) motifs in anemia-regulated ATAC-seq peaks. Inset depicts a heat map representation of ChromVAR Z-scores at the top 20 motifs identified by variability. (F) Volcano plot of TOBIAS differential binding footprint scores between days 7 and 14 post-PHZ.
Hierarchical clustering of chromatin state changes over the 35 day time course identified six distinct states. We describe these states in the following ways: Increased accessibility early during recovery (state 3); increased accessibility in mid-recovery (states 1+2); and delayed accessibility during recovery (states 4–6) (Fig. 4C,D). Most clusters showed a sharp change (increase or decrease) in accessibility between days 7 and 14, indicating a rearrangement of chromatin during the recovery and post-recovery transition (Fig. 4C,D). Chromatin changes between these time points corresponded to a decreased fraction of KIT+ erythroid (TFRC+LY76−) cells (Supplemental Fig. 3A). To confirm these results, we analyzed data in several distinct ways. First, we asked how similar the accessibility profiles were between each replicate at all time points analyzed. Days 7 and 14 were the least similar (based on scaled sample distance) among all samples tested (Supplemental Fig. 3B). At later time points, the reduced sample distance scores relative to controls indicates that chromatin profiles more closely resembled the pre-anemia state. To predict and evaluate transcription factor occupancy between these time points, we utilized ChromVAR (Schep et al. 2017). The highest variability scores at dynamically regulated accessibility regions were primarily GATA motifs and ETS motifs, with the starkest transition between days 7 and 14 (Fig. 4E; Supplemental Table S4). Confirming this result, TOBIAS footprint scores between days 7 and 14 post-PHZ showed a switch between a strong preference for GATA-factor occupancy at day 7 and ETS factors on day 14 (Fig. 4F).
To test whether chromatin changes mimicked cellular differentiation events, we compared PHZ-induced accessibility changes to those seen during transitions from common myeloid progenitors (CMP) to megakaryocyte-erythrocyte progenitors (MEP) and from MEP to EryA (Lara-Astiaso et al. 2014). PHZ-induced changes negatively correlated with CMP to MEP transitions (R = −0.52) and positively with MEP to EryA (0.43), indicating that the anemia-induced flux in chromatin state occurred at many of the same chromatin sites associated with cell type–specific differentiation (Supplemental Fig. 4A). Finally, we evaluated the extent to which chromatin fully resolved at post-recovery stages. A large majority of ATAC-seq peaks that were lost post-PHZ were regained in post-recovery stages (Supplemental Fig. 4B). While 6065 sites were significantly changed (gained or lost) at day 7 post-PHZ and then resolved, 893 sites remained differentially accessible at day 35 (Supplemental Table S5). Examples of regions with acute changes included intronic sites within the Stk3, Erg, Itpkb, and Tmem176 loci (Supplemental Fig. 5). Our findings indicate that anemia resolution is coordinated with a loss of GATA-associated ATAC-seq peaks and a gain of ETS-associated peaks.
BMP4-sensitive enhancers contain polymorphisms linked to anemia-dependent gene expression
Prior work showed that stimulation of human erythroid progenitors with the BMP4 growth factor during erythroid differentiation increased accessibility at enhancers nearby crucial stage-specific genes. BMP4-induced enhancers contained prevalent numbers of SNPs associated with clinically relevant RBC traits (Choudhuri et al. 2020). BMP4 promotes expansion and differentiation of erythroid progenitors in fetal, adult, and stress-specific contexts (Paulson et al. 2011), leading to effective erythroid regeneration in acute anemia. Although many environmental factors likely contribute to anemia-induced chromatin changes, we reasoned that BMP4-sensitive and anemia-induced chromatin changes may overlap at select sites. To evaluate BMP4-induced chromatin changes within a genetically heterogeneous human population, 18 healthy human donor CD34+ cells were differentiated to erythroid precursors, stimulated with 25 ng/mL rhBMP4 for 3 h, and processed for ATAC-seq (Fig. 5A). Footprint analysis identified a BMP4-induced shift in occupancy from ETS factors to GATA factors at accessible sites (Supplemental Fig. 6A). Thirty-nine evolutionarily conserved cis-elements were both BMP4-sensitive and PHZ-sensitive (Supplemental Fig. 6B). Peaks were then filtered based on the conditions that BMP4 stimulation induced a significant overall change in chromatin accessibility and that a BMP4-sensitive human polymorphism was identified in at least two donor genomes. We identified 110 sites where a polymorphism associated with increased or decreased BMP4-induced chromatin accessibility (Supplemental Table S6). Thirty-four of the 110 sites were located nearby gene transcription start sites that were anemia-regulated in mouse erythroid cells (Fig. 5B). Among these genes, anti-silencing function 1B histone chaperone (ASF1B) was previously shown to be downregulated in hereditary persistence of fetal hemoglobin (HPFH) (Papadopoulos et al. 2020). In addition to anemia regulation in HSPCs, Asf1b is strongly downregulated by GATA1 (Tanimura et al. 2018), and GATA1 is occupied near the SNP site (Fig. 5C). Carboxypeptidase D (CPD) is anemia-upregulated in HSPCs and erythroid cells, and promoter footprint scores were transiently elevated at days 1 and 3 post-anemia (Fig. 5D). A BMP4-sensitive SNP (rs7220175 C/T) is within the JUN-occupied CPD promoter (Fig. 5E). Therefore, chromatin accessibility changes induced by BMP4 are commonly found near anemia-induced genes within genomic regions containing common human polymorphisms.
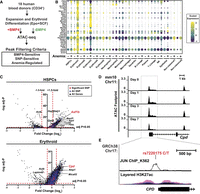
Anemia- and BMP4-sensitive chromatin accessibility changes associated with common human SNPs. (A) Experimental layout. (B) Dot plot representing scaled single-cell mRNA expression per cell (color) and fraction of cells expressing (size) within each annotated cell population isolated from mice 3 days post-PHZ versus control cells. Gene list was filtered to those located at BMP4 and anemia-sensitive genomic regions, containing SNPs which are sensitive to BMP4 stimulation. (C) Volcano plot depicts all anemia-regulated genes in HSPCs and erythroid cells, annotated by loci containing human polymorphisms that may alter expression profiles and SNPs that were significantly associated with differential sensitivity to BMP4 stimulation. (D) TOBIAS Footprint scores at the Cpd locus at days 1, 3, and 7 post-PHZ mapped to the mouse mm10 genome. (E) Inset represents the corresponding region in the human genome (GRCh38) depicting the location of the BMP4-sensitive SNP within the CPD promoter, JUN ChIP in K562 (GEO: GSM935355), and layered H3K27ac from ENCODE (The ENCODE Project Consortium 2012).
To test whether human SNPs in genome-wide association studies correlated with acute anemia-induced chromatin changes, we performed a human-mouse genome lift-over of GWAS data (Ulirsch et al. 2019). We identified 1857 chromatin accessible sites in mouse which overlap with individual variants associated with human RBC traits (Supplemental Fig. 7A). At 618 sites linked to human RBC trait variation, ATAC-seq peak signal significantly changed in response to PHZ treatment (Supplemental Fig. 7B,C; Supplemental Table S7). To investigate whether acute anemia-induced chromatin accessibility changes occurred at or near RBC trait-linked SNPs, we used g-ChromVAR (Ulirsch et al. 2019). At SNP-containing regions associated with RBC traits, we observed substantial changes in chromatin accessibility over the 35-day time course (Fig. 6A). SNP-containing regions linked to red blood cell traits (mean reticulocyte volume [P = 0.002], MCH [P = 0.003], MCV [P = 0.01], RBC count [P = 0.006], hematocrit [P = 0.007], hemoglobin [P = 0.002]) were more accessible at day 1 versus day 0, with a sharp decrease in accessibility at these sites between days 7 and 14 (Fig. 6B). Hemoglobin scores were also significantly decreased at day 35 (P = 0.03), indicating that some RBC trait–associated SNPs remained durably changed. At SNP regions associated with white blood cell parameters (WBC count, lymphocyte count, monocyte count), gChromVar Z-scores were decreased at days 1–7 post-PHZ, followed by a sharp increase between days 7 and 14 (Fig. 6C). ATAC peaks at SNPs associated with platelet volume and platelet count did vary throughout the timeline but were not significantly impacted by PHZ treatment (Fig. 6D). Overall, these analyses identified trait-associated SNPs at chromatin accessible and anemia-sensitive cis-elements, facilitating new experimental directions aimed at testing their role in hematopoietic lineage commitment and differentiation mechanisms.
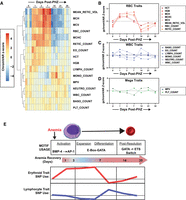
Transiently increased accessibility of anemia-sensitive cis-elements at RBC trait–associated SNPs. (A) Clustered heat map of gChromVAR Z-scores across a 35-day time course post-PHZ. (RBC) Red blood cell, (MCH) mean corpuscular height, (MCV) mean corpuscular volume, (MCHC) mean corpuscular hemoglobin concentration, (Retic) reticulocyte, (Eo) eosinophil, (HCT) hematocrit, (HGB) hemoglobin, (MPV) mean platelet volume, (WBC) white blood cell, (PLT) platelet. (B) Line graph of gChromVAR Z-scores specific to RBC traits. (C) Line graph of gChromVAR Z-scores specific to WBC traits. (D) Line graph of gChromVAR Z-scores specific to megakaryocyte traits. (E) Modeling cis-regulatory usage across the erythroid recovery timeline. At early time points in anemia recovery, erythroid precursor cells upregulate expression and activity of JUN and FOS transcription factors at AP-1 motifs on chromatin. Following activation events, erythroid expansion and differentiation includes the use of canonical E-box-GATA elements. Later in the recovery timeline—after anemia has largely resolved—GATA element use drops sharply and ETS motifs are more widely utilized. These changes are associated with transient increase in the use of cis-elements containing single nucleotide polymorphisms previously linked to RBC traits.
Discussion
Coordinated checks and balances maintain steady rates of red blood cell production and allow for the rate of production to accelerate in contexts of acute anemia. The mechanisms driving this accelerated rate and phenotypic diversity of anemia responses are poorly understood. To identify DNA elements controlling increased regenerative activities involved in recovery from acute anemia, we profiled chromatin accessibility in populations of cells enriched for RBC precursors (LIN−KIT+TFRC+LY76−) isolated from mice at various time points after anemia induction. Multiple rewiring events occurred during the process of anemia recovery, including early increases in the usage of AP-1 sites, followed by E-box-GATA sites, and finally an increase in ETS motif usage at post-recovery time points (Fig. 6E). These findings track genome accessibility during the early establishment and late resolution of a regeneration-dependent transcriptome in RBC precursors. Chromatin accessibility and transcriptional landscapes can be used to define mechanisms of hematopoietic differentiation within distinct developmental stages (Corces et al. 2016; Papadopoulos et al. 2020; Mende et al. 2022). One intriguing aspect of various published chromatin maps is the stark distinctions drawn between fetal and adult erythropoietic programs (Chaand et al. 2023). Within the frame of acute responses to anemia, our data contributes to a body of work seeking to reconstruct the cis-regulatory logic of hematopoiesis and erythropoiesis (Ludwig et al. 2019; Liao et al. 2020; Xiang et al. 2020).
AP-1 regulates transcription during stress and inflammation (Karin et al. 1997). AP-1 cis-elements have been previously linked to regulation of apoptosis during erythropoiesis (Jacobs-Helber et al. 1998) and may cooperate with GATA factor activities on chromatin (Linnemann et al. 2011). Whereas our data suggest transient up- and downregulation may control AP-1 activities in stress erythropoiesis, AP-1 activity is also controlled post-translationally by temporally staged phosphorylation events (Waudby et al. 2022). An elevated ETS footprint at post-resolution time points in acute anemia corresponds with an established role for ETS protein SPI1 (also known as PU.1) in attenuating erythropoiesis (Moreau-Gachelin et al. 1988; Subramanian et al. 2005, 2008; Rimmelé et al. 2007). Because GATA factor deregulation can cause leukemia (Katsumura et al. 2018), and AP-1 exhibits oncogenic activity (Eferl and Wagner 2003), establishing cooperative functions of AP-1 with GATA or ETS factors in acute anemia (or other biological contexts) will elaborate to what extent this mechanism is disrupted in human pathologies.
In the hematopoietic system, inter-individual phenotypic variation has been associated with thousands of independent variants in noncoding regions of the genome. What distinguishes individuals who can successfully adapt to stressors from those who cannot? Prior work has shown that genetic trait-sensitive SNPs are commonly found at regions where chromatin accessibility changes during cell differentiation, providing a useful way to track lineage fates and understand critical gene regulatory mechanisms in erythropoiesis (Ludwig et al. 2019). In response to specific environmental stimuli required for erythroid regeneration, our data indicate that BMP4-sensitive and anemia-activated cis-elements are sensitive to natural human variation, with hemoglobin trait–associated loci remaining durably elevated more than 1 month after anemia resolution. Among the important mechanisms driving durable and adaptable stress responses at the molecular level, changes in gene expression programs, cell surface receptor activation, intracellular signaling pathways, and transcription factor occupancy modulate chromatin structure at stress-responsive genes (Costa-Mattioli and Walter 2020). Genome-wide accessibility profiling combined with single-cell transcriptomics provide data mining resources to discover how organisms respond to different stresses.
Limitations to this study design include the lack of individual cell tracking along this timeline. Therefore—especially at later —we are not able to distinguish whether changes to genome organization in anemia are intrinsic to spleen resident erythroid progenitors or precursors, or whether these changes represent a repopulation of progenitor cells originating from bone marrow. The second model is supported by evidence suggesting that a repopulation event may occur at post-resolution stages of anemia (Harandi et al. 2010). Investigating this system at single-cell resolution to address possible distinctions in cellularity and using lineage tracing methods to address cell source may resolve this. It is important to note that, although our approach corrected for Tn5 transposition bias, false positive hits in ATAC footprint analysis are possible at specific sites, and our data cannot distinguish specific proteins occupied within the ATAC footprint. Cis-elements which are predicted to control erythroid regeneration should be validated using knockout or loss-of-function approaches.
Methods
ATAC-seq sample preparation
Animal experiments received ethical approval from the Association for the Assessment and Accreditation of Laboratory Animal Care at the University of Nebraska Medical Center. Hemolytic anemia was induced by phenylhydrazine (Sigma-Aldrich) administered subcutaneously with a single dose of 100 mg/kg. LIN− (CD3E−ITGAM [CD11B]−CD19−PTPRC [CD45R]−LY6G [GR1]−TER-119 [LY76]−) cells from 8- to 13-week-old C57BL/6J mice (1, 3, 7, 14, 21, 35 days post-PHZ treatment with untreated control, three replicates each condition) spleen were isolated using MojoSort streptavidin-conjugated magnetic nanobeads (BioLegend #480015). Fifty thousand cells were sorted using Fluorescence-Activated Cell Sorting (FACS) using the following antibodies purchased from BioLegend: PE-Cy7-conjugated anti-mouse CD117 (#135111), PE-conjugated anti-mouse CD71 (#113807), and APC-conjugated anti-mouse TER-119 (#116211). We performed ATAC-seq on these as described in Grandi et al. (2022). Briefly, 50,000 sorted cells in each sample were resuspended in cell lysis buffer (containing 10% NP-40, 10% Tween-20, and 1% Digitonin). After washing, samples were treated with Tn5 Transposase containing transposition reaction mix using the Nextera DNA Library Prep kit (NEB). DNA fragments were purified using the Qiagen MinElute Reaction Cleanup kit. During library prep, unique adapters were added to transposed samples using NEBNext Ultra II Q5 2× Master Mix (NEB). PCR cycles for library amplification were determined by qPCR using NEBNext Library Quantification analysis (NEB). After amplification and purification, the quantity and library quality were determined by QuantiFluor ONE dsDNA System and High Sensitivity DNA ScreenTape Analysis, respectively.
ATAC-seq data analysis
ChIPseeker was used to annotate differential peaks with TxDb.Mmusculus.UCSC.mm10.knownGene (Yu et al. 2015). Sample distances were measured using Euclidian distance measurements calculated from peak counts and scaled for use in heat map visualization. Footprint annotation was conducted using Universal Robust Peak Calling (UROPA) (Kondili et al. 2017). enrichGO in clusterProfiler org.Mm.eg.db was used for Gene Ontology (GO) analysis (Yu et al. 2012). For motif identification within specific subsets of peaks, we used HOMER findmotifsgenome.pl t using the mm10 genome build. Motifs were from a core database of all vertebrate motifs in JASPAR 2024 (Sandelin et al. 2004). Differential analysis among time points used DESeq2 (Love et al. 2014), and the P value cutoff for significantly changed peaks across all time points was P < 0.01. To identify TF binding motifs across all chromatin accessibility across all samples, we used the motifmatch function in chromVAR package (Schep et al. 2017). The motif file used in chromVAR is all vertebrate motifs from JASPAR 2020. Footprinting analysis was conducted using merged BAM and BAM files from different biological replicates in TOBIAS, corrected for Tn5 cut bias (Bentsen et al. 2020). The database of all vertebrate motifs in JASPAR 2024 was used to annotate TF binding motifs at scored footprint coordinates and compared between conditions. To correlate chromatin accessibility with genome-wide association studies (GWAS), we used g-chromVAR enrichment analysis from 115,000 individuals of European ancestry from the UK Biobank (Ulirsch et al. 2019). To annotate, the human variants described above were converted to their coordination position in mouse using the liftOver function built in UCSC Genome Browser (Nassar et al. 2023). The individual ATAC-seq peaks from our data were correlated with specific cell traits based on the presence of the converted variants. Then, the score of the possibility that those ATAC-seq peaks were associated with cell traits was calculated based on the sequencing counts of each peak and the GWAS fine-mapping score. Weighted deviations using the scores across each peak generates bias-corrected Z-scores to estimate trait relevance for each sample.
For additional methodological information on ATAC-seq analysis, see Supplemental Methods.
scRNA-seq data analysis
We used previously published scRNA-seq data collected from 4809 KIT+ control cells from mouse spleen, compared to cells collected 1, 3, and 7 days post-PHZ injection in littermate mice. Information regarding sequencing reads, mapping percentage, and gene coverage can be found in a prior publication (Zhou et al. 2023). Single-cell regulatory network inference and clustering analysis (HSPCs and five erythroid clusters) was performed in Python and R (Aibar et al. 2017).
Murine primary cell isolation and culturing
Spleens from PHZ-treated mice were lineage-depleted to generate primary cell cultures. Flow cytometric cell phenotyping, cell cycle, and proliferation analyses were conducted on 3-day-old cultures of PHZ-treated and retrovirally-infected erythroid precursors isolated from mice at indicated time points post-PHZ. Serum-starved cultures were stimulated with BMP4 (50 ng/mL) for 2 h prior to mRNA isolation. For mouse Jun knockdown, shRNA was synthesized and cloned into pMSCV PIG (Addgene #21654). Intracellular flow cytometry for Jun and phospho-Jun protein levels was conducted by 4% formaldehyde fixation of LIN-KIT+ spleens isolated at indicated time points, followed by permeabilization in 95% methanol and analysis using primary rabbit anti-c-Jun (Cell Signaling Technology #9165S) and phospho-c-Jun (Cell Signaling Technology #3270S) antibodies, and a secondary APC anti-rabbit antibody (Jackson ImmunoResearch # 111-136-144), co-stained with PE anti-mouse CD71 (#113807) and FITC anti-mouse TER-119 (#116206) (each at 1:200 dilution). JUN inhibition experiments were performed by treating cells isolated on day 1 post-PHZ with SR11302 for 2 days at 1 µM followed by treatment for 1 day at 20 µM. Cells were analyzed on the BD LSRII flow cytometer.
Human CD34+ cell culture
Human CD34+ cells were isolated from the peripheral blood of granulocyte colony-stimulating factor-mobilized healthy volunteers (Fred Hutchinson Cancer Research Center, Seattle, WA). The cells were expanded in StemSpan medium supplemented with StemSpan CC100 cytokine mix (Stem Cell Technologies) and 2% P/S for 6 days. To promote erythropoietic differentiation, cells were reseeded in differentiation medium (StemSpan SFEM Medium with 2% P/S, 20 ng/mL SCF, 1 U/mL Epo, 5 ng/mL IL-3, 2 mM dexamethasone, and 1 mM b-estradiol) at a density of 0.5–13 106 cells/mL. Cells were treated for 2 h with 25 ng/mL human recombinant BMP4 or vehicle control prior to ATAC-sequencing.
Statistical analysis
Wilcoxon signed-rank test was used to test significance of transcriptional changes in scRNA-seq data. Adjusted P value is based on Bonferroni correction. Statistical analysis for quantitative RT-PCR used GraphPad Prism v8 as indicated. The results are mean ± standard error of the mean. Multiple independent cohorts were used in each experiment. Statistical comparisons between two groups were performed using two-tailed unpaired Student's t-test, with a significance cutoff of P < 0.05.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE275430.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank all members of the Hewitt, Rowley, and Zon labs who assisted in discussions of this project. This work was supported by National Institutes of Health/National Heart, Lung, and Blood Institute (NIH/NIGMS, R01 HL155439 and R01 HL144780), funding for K.J.H. as a project leader in the Nebraska Center for Molecular Target Discovery and Development (1P20GM121316-01), the NIH/NIGMS MIRA award (R35 GM147467) for M.J.R., and a UNMC Graduate Studies Assistantship for Yc.Z. We thank the UNMC DNA Sequencing Core, which receives partial support from the National Institute for General Medical Science (NIGMS) INBRE – (P20GM103427-19) and the UNMC Flow Cytometry Research Facility, supported by the Nebraska Research Initiative (NRI) and The Fred and Pamela Buffett Cancer Centers’ National Cancer Institute Cancer Support Grant (P30 CA036727). Research reported in this publication was also supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number U24DK126127.
Author contributions: Yc.Z. and K.J.H. conceived the study. Yc.Z., A.C., L.I.Z., and K.J.H. designed the methodology. Yc.Z., A.C., S.R., S.Y., and Yi.Z. collected data and performed experiments. Yc.Z., V.R.D., A.C., S.R., H.L.H., S.Y., and M.J.R. performed statistical analyses and data curation. K.J.H., M.J.R., and L.I.Z. supervised the experiments and analysis for the study. Yc.Z. and K.J.H. wrote the original draft, and all authors reviewed and edited.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279949.124.
-
↵3 Lymphocyte antigen 76 (LY76) is also known as TER-119, which is commonly used for the detection antibody.
- Received August 21, 2024.
- Accepted April 30, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








