
Homozygous editing of multiple genes for accelerated generation of xenotransplantation pigs
- Xiaoyue Duan1,2,6,
- Chaolei Chen2,6,
- Chang Du2,6,
- Liang Guo1,2,6,
- Jun Liu2,
- Naipeng Hou1,
- Pan Li1,2,
- Xiaolan Qi3,
- Fei Gao1,2,
- Xuguang Du1,2,
- Jiangping Song1,4 and
- Sen Wu1,2,5
- 1Sanya Institute of China Agricultural University, Sanya, 572024, China;
- 2State Key Laboratory of Animal Biotech Breeding, College of Biological Sciences, China Agricultural University, Beijing 100193, China;
- 3State Key Laboratory for Animal Disease Control and Prevention, College of Veterinary Medicine, Lanzhou University, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Lanzhou 730000, China;
- 4Beijing Key Laboratory of Preclinical Research and Evaluation for Cardiovascular Implant Materials, Animal Experimental Center, Fuwai Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China;
- 5State Key Laboratory of Animal Biotech Breeding, College of Biological Sciences, National Engineering Laboratory for Animal Breeding, Frontiers Science Center for Molecular Design Breeding, China Agricultural University, Beijing, 100193, China
-
↵6 These authors contributed equally to this work.
Abstract
Although CRISPR-Cas-based genome editing has made significant strides over the past decade, achieving simultaneous homozygous gene editing of multiple targets in primary cells remains a significant challenge. In this study, we optimized a coselection strategy to enhance homozygous gene editing rates in the genomes of primary porcine fetal fibroblasts (PFFs). The strategy utilizes the expression of a surrogate reporter (eGFP) to select for cells with the highest reporter expression, thereby improving editing efficiency. For simultaneous multigene editing, we targeted the most challenging site for selection, whereas other target sites did not require selection. Using this approach, we successfully obtained single-cell PFF clones (three of 10) with seven or more homozygously edited genes, including GGTA1, CMAH, B4GALNT2, CD46, CD47, THBD, and GHR. Importantly, cells edited using this strategy can be efficiently used for somatic cell nuclear transfer (SCNT) to generate healthy xenotransplantation pigs in <5 months, a process that previously required years of breeding or multiple rounds of SCNT.
Porcine pancreatic islets, corneas, skin, and solid organs such as hearts and kidneys have served as valuable substitutes for human tissues and organs in transplantation. Recently, significant progress has been made in the transplantation of genetically modified pig organs, including hearts, kidneys, and livers, into humans, offering hope for improved survival times and better functional adaptation of pig organs for xenotransplantation. This progress may ultimately help alleviate the global shortage of human organs for transplantation (Griffith et al. 2022; Mohiuddin et al. 2023; Mallapaty 2024; Mallapaty and Kozlov 2024). The generation of genetically modified donor pigs is a crucial step in xenotransplantation, typically achieved through somatic cell nuclear transfer (SCNT) using gene-edited primary porcine fetal fibroblasts (PFFs) (Lai et al. 2002; Phelps et al. 2003). However, simultaneous homozygous editing of multiple genes in primary cells remains a significant challenge owing to limitations in editing efficiency and the capacity to propagate cells.
Multiple gene editing is required to address critical issues such as immune rejection, coagulation dysregulation, complement activation, and functional adaptation of pig organs (Zhao et al. 2019; Fu et al. 2020; Boulet et al. 2022; Sykes and Sachs 2022; Wu et al. 2023). Although successful editing of up to four genes in pigs has been reported through SCNT, using phenotypic enrichment strategies such as isolectin B4 or toxin A selection (Fischer et al. 2020; Tanihara et al. 2021), the generation of pigs with more extensive genetic modifications, such as 10 genes edited (10GE) or 13 genes edited (13GE), has required years of iterative rounds of SCNT and selective breeding (Reardon 2015; Cooper et al. 2019). To achieve successful xenotransplantation of genetically modified organs, tissues, and biomaterials, it is estimated that more than 100 xenotransplantation-related genes will need to be targeted (Cooper 2022; Singh et al. 2022; Wolf and Reichart 2024). Achieving the homozygous modification of these genes in a time-efficient manner remains a major bottleneck in xenotransplantation (Pan et al. 2019; Yang et al. 2021; Yue et al. 2021; Anand et al. 2023).
Current multigene editing methods primarily rely on the simultaneous delivery of multiple sgRNAs targeting different genes. Various strategies have been explored, including using all-in-one plasmids containing multi-sgRNA/crRNA arrays (Zetsche et al. 2017; Breinig et al. 2019; Campa et al. 2019; Zhang et al. 2019b; Yuan and Gao 2022), synthesizing multi-crRNA:tracrRNA complexes (Khan et al. 2019), employing multitargeting RNPs (Xu et al. 2018), or transfecting a mixture of gRNAs expressing plasmids (Cong et al. 2013; Fujii et al. 2023). However, these attempts to edit multiple genes simultaneously have been inefficient and often dependent on the specific gene or cell line being targeted.
To improve the efficiency of multiplex genome editing in primary cells, it is essential to develop enrichment or selection strategies that can identify and isolate successfully edited cells. Single-gene editing has benefited greatly from such selection strategies. For example, positive and negative selection methods have been used to enrich for targeted cells in knockout mouse models (Capecchi 2005). Editing genes such as HPRT1, HBEGF, or ATP1A1 confers resistance to 6-thioguanine, diphtheria toxin, or ouabain, respectively, and these properties have been utilized for enriching targeted cells (Liao et al. 2015; Agudelo et al. 2017; Li et al. 2021; Levesque et al. 2022). However, most genes do not possess such selectable traits. To overcome this limitation, researchers have developed surrogate reporter systems, such as mRFP-eGFP reporters for Cas9-based editing (Kim et al. 2011; Ramakrishna et al. 2014), “TREE” reporters for base editors (Standage-Beier et al. 2019), “fluoPEER” reporters for prime editors (Schene et al. 2022), and CRISPRa selection for gain of function (Heidersbach et al. 2023). For these methods, the gene of interest is typically an endogenous gene located on the chromosome, whereas the surrogate selection gene is located episomally on a plasmid (Mikkelsen and Bak 2023). However, these methods have shown suboptimal efficiency, with enrichment efficiency varying significantly across different target genes (10% to 55%) (Ramakrishna et al. 2014; Schene et al. 2022), making them insufficient for multiplex genome editing in primary cells.
In this study, we developed an optimized coselection strategy for multiplex genome editing in primary PFFs. By incorporating a surrogate reporter (eGFP), we were able to select for cells with the highest editing efficiency. This approach led to the successful generation of xenotransplantation pigs with multiple homozygously edited genes in a much shorter time frame than previously achievable.
Results
Surrogate reporters for genomic targeting
Efficient selection methods are essential for enhancing multiplex genome editing. To address this need, we optimized a surrogate reporter system to detect genome editing events (Fig. 1A). In this system, sequences containing an alternative target site (3n ± 1 nt) are inserted after the ATG start codon within the open reading frame of the reporter gene on the surrogate plasmid, causing a frameshift. It is important to note that the inserted sequence must not contain a start or stop codon in frame, either before or after the editing site. If this condition cannot be avoided, the targeted sequence can be inserted in reverse orientation or adjusted by a few base pairs to shift the open reading frame. When Cas9 targets the endogenous gene, it also targets the surrogate site, leading to in-frame reporter expression in the presence of insertions or deletions (indels), as previously reported (Kim et al. 2011; Ramakrishna et al. 2014; Mikkelsen and Bak 2023). However, further evidence of the correlation between genomic targeting and surrogate plasmid targeting at the single-cell level was lacking.
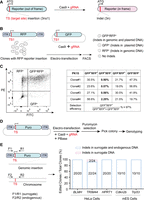
Detection of targeting events using surrogate reporters. (A) The principle of the surrogate reporter system. The reporter gene is initially frameshifted owing to the insertion of a CRISPR target site adjacent to the “ATG” start codon. CRISPR-Cas9 mediated editing results in insertions and deletions (indels) that restore the correct reading frame of the reporter gene. (B) Workflow for assessing the correlation between genomic and plasmid DNA targeting at the single-cell level. Colonies containing an integrated surrogate reporter (eRFP) were electro-transfected with Cas9, sgRNA, and a surrogate reporter (eGFP) plasmid. Both genomic (eRFP) and plasmid (eGFP) surrogate reporters incorporate the same target site (TS1). (ITR) Inverted terminal repeat sequence. (C) Flow cytometry analysis distinguishing colonies edited in genomic DNA (PE positive) and plasmid DNA (FITC positive). The selection efficiency is calculated by the ratio of GFP and RFP double-positive cells to GFP-positive cells. (D) Workflow for assessing the correlation between endogenous and surrogate DNA targeting at the single-cell clone level. Cells were electro-transfected with Cas9, sgRNAs, a surrogate reporter (Puro), and PBase. Following puromycin selection, cell colonies were picked for genotyping. (E) Primers (F1/R1) amplify the surrogate target site (Puro) integrated into the genome, and primers (F2/R2) amplify the endogenous locus. The right panel illustrates the ratio of edited clones.
To evaluate the accuracy of the surrogate plasmid in reflecting targeting events within endogenous genes, we conducted a single-cell analysis. Initially, we integrated a surrogate reporter gene (eRFP), which includes a CMAH target site, into the genome of PK15 cells using the piggyBac transposon system. Subsequently, we isolated and identified single-cell clones with successful integration. These clones were then transfected with Cas9, an sgRNA targeting the CMAH site, and a surrogate reporter (eGFP) plasmid containing the same CMAH target site. We analyzed the cells using flow cytometry to assess the correlation between genomic targeting events and those in the surrogate plasmid (Fig. 1B). The presence of RFP fluorescence in the cells signified successful genomic targeting events, whereas GFP fluorescence indicated targeting events within the surrogate plasmid. Our findings revealed that 97% of the cells selected for GFP fluorescence were also RFP-positive, suggesting that plasmid targeting (GFP+) can effectively identify genomic targeting (RFP+) (Fig. 1C). Moreover, the purity of the selected cells could be increased to 100% by selecting those with higher FITC intensity (>104) (Supplemental Fig. S1A).
To further analyze the correlation and targeting sequence outcomes between the endogenous target site and the surrogate reporter in other cell types, we transfected Cas9, sgRNAs, a surrogate reporter (Puro), and PBase into HeLa cells and mouse embryonic stem cells (mESCs). This allowed us to sequence the outcomes of both the endogenous target site and the related surrogate target site in single-cell clones (Fig. 1D). After puromycin selection, we found that all mESC clones (10/10) were targeted at Cdkn2b and Trp53, and in HeLa cells, all clones (20/20) were targeted at BLMH and HPRT1, with 22/24 clones targeting TRIM44 (Fig. 1E). Notably, the outcomes for plasmid DNA and genomic DNA were not always consistent in single cells, as confirmed by TA cloning and Sanger sequencing (Supplemental Fig. S1B). These results, obtained at the single-cell level, demonstrate that the surrogate reporter can reliably reflect genome editing events at the endogenous target site.
Cells selected by coselection strategy are mostly homozygously targeted
To assess the ability of the surrogate reporter system in selecting targeted cells, we cotransfected HeLa cells with Cas9, sgRNAs, a surrogate reporter (Puro), GFP, and PBase plasmids to target the endogenous genes BLMH, TRIM44, and HPRT1, along with their corresponding target sites in surrogate plasmids. In this experiment, the surrogate reporter plasmid was integrated into the genome, conferring successive drug selection. After sorting the GFP+ transfected cells, cells were divided into two groups: One group was subjected to puromycin selection, whereas the other remained untreated. We then determined the targeting efficiency by Sanger sequencing and the ICE Analysis Tool. The results demonstrated significantly higher targeting efficiencies in puromycin-selected cells across all three genes, with cell clones from the puromycin group achieving a 100% genomic editing rate (Fig. 2A).
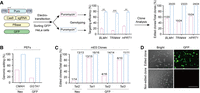
Efficient enrichment of genome-targeted cells using a surrogate reporter. (A) Enrichment of genome-targeted cells using a surrogate reporter (Puro). HeLa cells were cotransfected with Cas9, sgRNAs, a surrogate reporter (Puro), GFP, and PBase plasmids. Transfection-positive cells (GFP+) were divided into two groups: One group was treated with a high dose (2 µg/mL) of puromycin for 5 days, and the other group remained untreated. Targeting efficiency of these two group cells was determined by ICE analysis from Sanger sequencing. Cell colonies were picked for further genotyping. (**) P ≤ 0.01, (***) P ≤ 0.001 (Student's two-tailed t-test). (B) Enrichment of CMAH- and GGTA1-targeted PFFs by surrogate reporter (Neo) and surrogate reporter (GFP), respectively. (C) Enrichment of Tet2-targeted mESCs by surrogate reporter (Neo) and of et1-, Tet2-, and Tet3-targeted mESCs by surrogate reporter (eGFP), respectively. (D) The GFP expression in genomic targeted and nontargeted mESCs clones selected by the surrogate reporter (eGFP) plasmid. Scale bars, 1000 μm.
To explore the applicability of the surrogate reporter system across different cell types, we performed a similar test in PFFs and mESCs, both of which typically exhibit limited targeting efficiency (Fig. 2B–D). In PFFs, cells targeted for CMAH and GGTA1 were selected using Neo and eGFP surrogate reporters, respectively. The genomic editing efficiencies for CMAH and GGTA1 increased to 85%–98%, compared with 16%–44% in unselected groups. In mESCs, cells targeted for Tet2 were selected using a Neo surrogate reporter, whereas those targeting Tet1, Tet2, and Tet3 were selected with an eGFP surrogate reporter. All selected clones were successfully targeted, even though the initial targeting efficiency for Tet2 was only 7% (1/14). GFP-positive clones corresponded to targeted cells, whereas GFP-negative clones remained unmodified.
The surrogate reporter system demonstrated a robust ability to enrich for genome-edited cells, regardless of cell type or target site, outperforming previous technologies. We speculate that its success may be owing to the integration of the reporter, which allows extended drug selection to enhance selection pressure. However, for studies requiring a clean genomic profile, the presence of an integrated reporter gene may be undesirable. Therefore, we removed the ITR sequences from the surrogate reporter plasmid, resulting in a nonintegrative system, version 2.1. When GFP was used as the reporter gene, it was termed version 2.1-eGFP (V2.1G) (Fig. 3A).
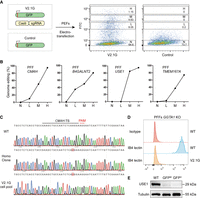
Selection of homozygously edited cells based on highest surrogate reporter expression. (A) Schematic of selection strategy using version 2.1-eGFP (V2.1G), a nonintegrated surrogate reporter system. PFFs were cotransfected with V2.1G, Cas9, and sgRNA plasmid, whereas the control group was only transfected with the V2.1G plasmid. FACS analysis revealed varying intensities of FITC expression among the cells. (H) The highest FITC intensity population, (M) the middle FITC intensity population, (L) the lower FITC intensity population, and (N) the negative FITC intensity population. (B) Correlation between genome editing efficiency and FITC intensity of endogenous targeting in PFFs. (C) Representative Sanger sequences of CMAH target site in wild-type cells, homozygous cell clones, and cell pools selected by V2.1G. (D) FACS analysis of GGTA1 targeting in PFF pools detection by IB4 lectin staining. (E) Western blot analysis of USE1-targeted Vero E6 cell pools with middle and high FITC intensity, respectively.
Using V2.1G to target four endogenous genes (CMAH, B4GALNT2, USE1, TMEM167A) in PFFs, we observed a positive correlation between gene-targeting efficiency and FITC intensity. Specifically, cells with high FITC intensity (H group) achieved efficiencies of 74%–100%, surpassing those with medium (M group), low (L group), or no FITC intensity (N group) (Fig. 3B; Supplemental Fig. S2A). Genotyping of H-group cells targeting CMAH, B4GALNT2, or GGTA1 revealed similarities to clones with homozygous modifications (Fig. 3C; Supplemental Fig. S2B,C). FACS analysis after IB4 lectin staining confirmed a reduction in α-Gal epitopes in GGTA1-targeted cells (H group), with complete elimination observed after 12 days (Fig. 3D; Supplemental Fig. S3). For conducting western blot analysis, we performed the knockout of USE1 in Vero E6 cells and sorted the M and H groups of cells using V2.1G. The results confirmed a complete knockout of USE1 in both the M and H groups (Fig. 3E). These results highlight V2.1G's strong capability to enrich homozygously edited cells efficiently. A streamlined workflow using V2.1G is provided in Supplemental Figure S4.
Editing multiple genes simultaneously in PFFs using V2.1G
To test the efficiency of V2.1G for multigene editing, we conducted experiment MV014, targeting seven genes simultaneously in PFFs (Fig. 4A; Supplemental Tables S1, S2). These genes (GGTA1, CMAH, B4GALNT2, THBD, CD46, CD47, and GHR) require either knockout or targeted insertion/replacement, playing critical roles in xenotransplantation. Given that the B4GALNT2 gene comprises four alleles, in the seven-gene-targeting experiment MV014, a total of 16 alleles needed to be targeted. Initially, two sgRNAs were tested for each gene, and the B4GALNT2-1 sgRNA exhibited the lowest targeting efficiency. Thus, the B4GALNT2-1 site was incorporated into the V2.1G surrogate reporter plasmid, under the hypothesis that successfully targeting the least efficient site would ensure other sites are also targeted. For challenging genes (CMAH, THBD, GGTA1, CD46, and B4GALNT2), paired sgRNAs spaced 50 bp apart were used, as prior studies indicate this configuration enhances multigene-targeting efficiency (Ran et al. 2013; Gopalappa et al. 2018).
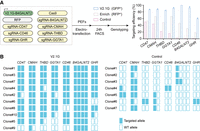
Simultaneous targeting of seven genes using V2.1G. (A) Schematic for targeting seven endogenous genes in PFFs using V2.1G. A surrogate reporter (eGFP) plasmid is used for selecting B4GALNT2-targeting cells. After 24 h of transfection, cells were sorted into three groups: V2.1G (FITC-highest), Enrich (PE-highest), and Control (PE-positive), for genotyping and colony formation. (B) Genotyping of the clones for seven genes at 16 alleles. Each box represents one allele, filled boxes represent a targeted allele, and empty boxes represent a nontargeted allele.
To compare V2.1G with traditional enrichment methods, RFP plasmids were cotransfected as controls to select cells with high PE intensity. Cells with the highest FITC intensity were sorted 24 h after transfection for genotyping and colony formation. V2.1G yielded remarkable results, with targeting efficiencies for all seven genes significantly improved. Among 10 colonies analyzed, eight successfully targeted five to seven genes, and three exhibited homozygous modifications across all seven genes (Fig. 4B). This contrasts with a homozygous colony efficiency of two of 91 for simultaneously targeting three genes (GGTA1, CMAH, and B4GALNT2) using conventional methods (Supplemental Table S3).
To assess potential off-target effects from multiple sgRNAs, we analyzed three predicted off-target sites for each sgRNA using PCR and high-throughput sequencing. No significant off-target effects were detected by this method (Supplemental Fig. S5). RNA-seq analysis of seven-gene-edited cells revealed increased expression of genes involved in DNA replication, repair pathways, p53 signaling, and cancer-related pathways (Supplemental Fig. S6), consistent with observations from other Cas9-mediated editing studies (Haapaniemi et al. 2018; Enache et al. 2020). Despite these changes, cell morphology and colony formation efficiency were unaffected, underscoring V2.1G's capability to generate robust multigene-targeting clones. A streamlined workflow of enrichment for knockout of multiple genes using V2.1G is provided in Supplemental Figure S7.
Drug-resistant surrogate reporters for multigene editing
Next, we assessed drug-resistant surrogate reporters in a seven-gene editing experiment MV020. The GFP reporter was fused with Cas9 via F2A. Three drug-resistant surrogate plasmids were used: Neo for B4GALNT2, Puro for CD46, and Hygro for GHR (Supplemental Fig. S8A; Supplemental Tables S1, S2). Drug-resistant plasmids were integrated using piggyBac transposition, enabling long-term selection. After treatment with the corresponding drugs, all three targeted genes were edited successfully, and the remaining genes showed improved efficiencies of 79.8%–100% (Supplemental Fig. S8B).
To apply this to practical applications without integration of selection elements into the genome, we conducted episomal selection experiments MV021–MV025 targeting the same seven genes as in MV020 but without transfecting PBase expression plasmid (Supplemental Fig. S9). Notably, when cells were subjected to a 3-day drug selection period, only a few cells survived, exhibiting poor colony formation. Subsequently, we adopted a shorter selection period with various drug selection conditions and found that a high drug dose combined with a short selection period yielded optimal results. In the context of our experiment targeting seven genes in PFFs, the use of 2 μg/mL puromycin, 1200 μg/mL G418, and 800 μg/mL hygromycin for a 4 h selection period led to improved targeting efficiencies of 57.7%–100% (MV024 experiments). In short, the drug-resistant surrogate reporter demonstrated its ability to select cells with multiple gene modifications through a stringent, transient drug treatment approach.
Enhancing the efficiency of knockout and knock-in events with a surrogate reporter
To determine whether our surrogate reporter coselection strategy could be employed for both gene knockout and knock-in applications, we conducted an experiment that knocked out the GGTA1 and CMAH genes while simultaneously knocking in the hTHBD and hCD55 genes into PFFs. For gene targeting, we used the Puro surrogate plasmid to select GGTA1 gene targeting and used the Neo surrogate plasmid to select CMAH gene targeting. The knock-in strategy employed was Cas9-mediated nonhomologous end joining (NHEJ), allowing the expression cassette (EF1a-hTHBD-P2A-hCD55-pA) to be inserted into either the GGTA1- or CMAH-target sites. After 2 days of treatment with puromycin (1 μg/mL) and G418 (1200 μg/mL), clones were picked after 10 days and subjected to PCR and sequencing for identification. The efficiency of obtaining homozygous targeting for two genes and knock-in for two genes was determined to be 10.9%, as analyzed from 211 colonies (Fig. 5A).
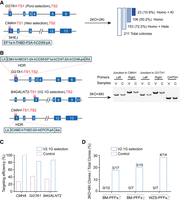
Enhanced knockout and knock-in efficiency through coselection strategy. (A) Generation of PFF clones with GGTA1 and CMAH knockouts, and hTHBD and hCD55 knock-ins via Cas9-mediated NHEJ. The target sites, located in the first exon of GGTA1 and CMAH, are indicated by arrows. Puro and Neo serve as surrogate reporters for GGTA1 and CMAH targeting, respectively. The EF1a-hTHBD-P2A-hCD55-pA sequence is used for insertion. Clones are categorized as follows: (Homo + Hete) one gene homozygously edited, one heterozygously edited in GGTA1 and CMAH, (Homo) both genes homozygously edited, and (Homo + KI) both genes homozygously edited with hTHBD and hCD55 knock-ins. (B) Generation of PFF clones with triple knockout of GGTA1, B4GALNT2, and CMAH and with knock-in of six genes—hHOMX1, hCD55, hCD47, hCD46, hTHBD, and hEPCR—via Cas9-mediated HDR. V2.1G is used for B4GALNT2 targeting. The CMV-hHOMX1-T2A-hCD55-EF1a-hCD47-P2A-hCD46-pA cassette is inserted into the GGTA1 locus, and the ICAM2-hTHBD-P2A-hEPCR-pA cassette is inserted into the CMAH locus, using ∼1 kb of homologous arms. (Right) Junction PCR for V2.1G-selected (V; V2.1G) and control (C; control) cell pools. Control pools consist of transfected positive cells. Primers detect left and right junctions in CMAH and GGTA1 loci, with “GAPDH” as a normalization control. (C) Analysis of triple gene knockout in “3KO + 6KI” cell pools. (D) Ratio of triple knockout and six genes knock-in PFF clones in V2.1G-selected and control groups. BM-PFFs♂ and BM-PFFs♀ represent PFFs from male and female Bama miniature pigs, respectively. WZS-PFFs♂ indicates PFFs from male Wuzhishan pigs.
Next, we expanded this strategy by knocking out the GGTA1, CMAH, and B4GALNT2 genes while simultaneously knocking in six human genes (hHOMX1, hCD55, hCD47, hCD46, hTHBD, and hEPCR) into PFFs. We used the V2.1G surrogate plasmid to select B4GALNT2 gene targeting. The knock-in strategy employed was Cas9-mediated homology-directed repair (HDR), with 1 kb of homologous arms facilitating the insertion of the expression cassette CMV-hHOMX1-T2A-hCD55-pA-EF1a-hCD47-P2A-hCD46-pA inserted into the GGTA1-targeting locus, and the ICAM2-hTHBD-P2A-hEPCR cassette inserted into the CMAH-targeting locus. Cells with the highest FITC fluorescence were sorted 24 h after transfection. After 10 days, clones were picked and subjected to PCR and sequencing for verification. The results showed increased knockout efficiencies for the three pig genes (GGTA1, B4GALNT2, and CMAH) and increased knock-in efficiencies for the six human genes (HOMX1, CD55, CD46, CD47, THBD, and EPCR) (Fig. 5B,C). Clones with modifications in all nine genes were readily generated with efficiencies ranging from 17.6% (three of 17) to 28.6% (four of 14) across three PFF cell lines (Fig. 5D).
One-step generation of multiple gene–edited pigs for xenotransplantation
To verify whether our surrogate reporter coselection strategy can be used for the efficient one-step generation of pigs with multiple gene modifications (Fig. 6A), PFFs were transfected with plasmid expression Cas9; sgRNAs targeting GGTA1, B4GALNT2, and CMAH; a donor plasmid containing an EF1a-hTHBD-P2A-hCD55-pA expression cassette; and a V2.1G surrogate plasmid encoding GFP to select B4GALNT2-targeted cells. Cells with the highest FITC fluorescence were sorted 24 h after transfection, and cell clones were sequenced to verify knockout and knock-in events. These cells were then transferred into enucleated pig oocytes as nuclear donors, and the reconstructed embryos were transferred to a sow for gestation. We obtained five live piglets with a genotype consistent with that of the donor cells and photographed at 25 days of age (Fig. 6B). Genotyping revealed that GGTA1 had one allele with a 1 bp deletion and another with a 1 bp insertion; CMAH had one allele with a 1 bp deletion and another with the EF1a-hTHBD-P2A-hCD55-pA cassette insertion; and B4GALNT2 had two alleles with 1 bp insertions, one with a 2 bp insertion, and one with a 10 bp deletion.
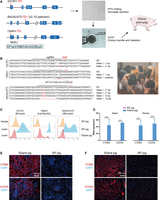
One-step generation of multiplex genome editing pigs for xenotransplantation using coselection strategy. (A) Schematic for generating five genes editing xenotransplantation pigs. (Left) The gene-targeting strategy for triple gene knockout (GGTA1, B4GALNT2, and CMAH) and dual gene knock-in (hTHBD and hCD55) in PFFs. The target sites, located in the first exon of GGTA1 and CMAH and the third exon of B4GALNT2, are indicated by arrows. The V2.1G surrogate plasmid is used to select for B4GALNT2 targeting, with selected cells serving as donors for somatic cell nuclear transfer (SCNT) to produce 5Gene xeno-pigs. (B, left) Overview of the DNA sequences at the modification loci in the piglets shown in the right panel. (C) FACS analysis validating the three-gene knockout (3KO) in peripheral blood mononuclear cells (PBMCs). (D) RT-qPCR validation of the two genes knock-in (2KI) in heart and kidney tissues. (E,F) Immunofluorescence (IF) validation of human thrombomodulin (hTBM) and human decay-accelerating factor (hCD55) expression in the kidney (E) and heart (F) of 5Gene pigs.
We further confirmed the protein expression in the edited pigs. Peripheral blood mononuclear cells (PBMCs) were isolated from these pigs and stained with IB4 lectin, anti-Neu5Gc, and DBA lectin to detect α-Gal, Neu5Gc, and SDa glycan epitopes encoded by GGTA1, CMAH, and B4GALNT2 genes, respectively. FACS analysis revealed a reduction in glycan markers, indicating the functional elimination of the three genes responsible for synthesizing these glycan epitopes in the pigs (Fig. 6C). RT-qPCR and IF analysis validated the expression of the knock-in genes (hTHBD and hCD55) in the heart and kidney (Fig. 6D–F). In short, the coselection strategy combined with SCNT offers a faster and more efficient alternative compared with the previously extended pig model breeding method.
Discussion
In this study, we developed a surrogate coselection strategy that allows for high pressure selection to efficiently enrich multiplex genome editing cells. This strategy is based on our validation that the surrogate plasmid can report the genome-targeting events of CRISPR, resulting in an enrichment efficiency of up to 100%.
The selection pressure might be the key to the remarkable enrichment efficiency. Theoretically, when plenty of surrogate plasmids are electro-transfected into cells, the more plasmids are targeted, the higher intensity of fluorescence exhibited, providing a greater opportunity for genome targeting. We found a positive correlation between selection pressure and the detected genome-targeting efficiency, allowing us to leverage this to select gene-targeted cells. Across five cell lines and 16 gene loci, mixed-cell pools selected after using surrogate reporters included many cells homozygous at all loci, whereas no previous tools were able to achieve this. This achievement might be because of the surrogate selection strategy effectively enriching for cells with the highest concentrations of Cas9 and gRNA plasmids, or it could be selecting a subset of cells that are inherently more susceptible to genetic manipulation. To further elucidate the mechanism of selection, we propose conducting experiments with RNP transfections to express Cas9 and gRNA at different concentrations. Furthermore, examining the expression levels of DNA repair pathway proteins, as well as assessing the epigenetic modifications of DNA, could provide insights into the cellular propensity for genetic editing.
This transient method is safer than integrated reporters or cotargeting endogenous marker genes. The efficiency of this strategy offers a simple and effective way to obtain a targeted cell pool that can proceed directly to the next stage of experimentation, bypassing the need for labor-intensive colony picking. This method is particularly valuable in situations in which cells are difficult to edit or form colonies, such as in chicken primordial germ cells (PGCs), chimeric antigen receptor T cells (CAR-T), and organoids (Deng et al. 2022; Yang et al. 2022; Geurts et al. 2023; Allen et al. 2024).
Although a few enrichment tools have been developed for single-gene targeting in 293T cells, their efficiency has been limited, especially for multiplex genome editing (Ramakrishna et al. 2014; Niccheri et al. 2017). In contrast, V2.1G demonstrated excellent selection efficiency and cell viability in multiplex genome editing, requiring only a single reporter gene to select cells with edits in seven genes simultaneously. We also explored drug-resistant surrogate selection and found that successful selection requires rigorous drug treatments with optimized doses and treatment periods. Multiplex genome editing in primary cell lines holds great potential to accelerate agricultural trait research and disease model generation (Zhang et al. 2019a; Xu et al. 2020; Ren et al. 2022). The preparation of coselection strategy in PFFs for multiplex genome editing is straightforward, requiring only one additional surrogate plasmid beyond the standard Cas9 and sgRNA plasmids typically used in gene editing experiments. Only the single most challenging target sequence is constructed into the surrogate plasmid. The construction process for both sgRNAs and surrogate plasmids primarily involves the ligation of linkers to vector backbones, simplifying the procedure and making it more accessible for multiplex genome editing. Once various single plasmids are prepared, any combination of multiplex targeting can be straightforwardly conducted, without the need to build a large and complex all-in-one plasmid. Electroporation of dozens of small plasmids showed better transfection efficiency and expression than a large plasmid (Søndergaard et al. 2020).
Based on our current experiments, we have successfully edited up to eight biallelic genes simultaneously. By optimizing the selection strategy to target the site with the lowest editing efficiency, we estimate that up to 15 biallelic genes (30 loci) can be modified in one step. However, the upper limit of this method depends on the cell's tolerance to double-strand breaks (DSBs), which requires further investigation.
It is well established that CRISPR-Cas systems can induce a broad range of off-target modifications, sometimes at unexpectedly high frequencies (Tao et al. 2023). For example, studies have shown that CRISPR-Cas9 can induce large structural variants at both on-target and off-target sites, with a significant degree of individual-level variation in genome editing outcomes (Höijer et al. 2022). To evaluate potential off-target effects, we examined three near-cognate gRNA sites for each gRNA used, but we did not detect any deletions at these sequences. This suggests that off-target issues are not significant with our methods. However, we acknowledge that more comprehensive analyses of possible off-target rearrangements should be performed in animals engineered using this method in order to better assess potential off-target consequences.
To alleviate the damage caused by off-target and chromosome translocations, other gene editing tools such as base editors and prime editors can be considered as alternatives to avoid excessive DSBs. However, it is difficult for base editors to use the endogenous target sites as reporters, and the editing efficiency of prime editors may not be sufficient for multigene editing. High-fidelity and efficient Cas9 variants, such as HiFi-Cas9 or Sniper2L, may offer superior performance for the simultaneous editing of multiple genes (Vakulskas et al. 2018; Kim et al. 2023). Although we cannot entirely prevent damage from DSBs, we can select the cells repairing the DSBs through HDR by combining surrogate reporter with HDRobust, a safe selection method developed by Riesenberg and colleagues that kills cells with unrepaired DNA damage while leaving the cells with precision mutations (Riesenberg et al. 2023). Furthermore, this might facilitate the selection of long-range sequence knock-in as well as multiple genes knockout. We expect that the coselection strategy will be valuable in disease model generation and gene function studies.
Methods
Ethics statements
All animal procedures were approved by the Animal Welfare Committee of China Agricultural University (approval no. AW03111202-3-1).
Editing plasmids construction
Cas9-expressing plasmids (pM3-Cas9) and sgRNA plasmids (pCRISPR-sg6) were obtained from previously published vectors in our laboratory (Xu et al. 2017; Lu et al. 2021). Gene-specific sgRNA vectors were constructed by designing sgRNAs for each gene using CHOPCHOP v3 (Labun et al. 2019). Oligonucleotides were synthesized, annealed, and inserted into the BbsI site of pCRISPR-sg6. Donor vectors for insertion were generated by synthesizing and cloning the coding sequences (CDSs) of human genes into backbones containing homologous arms and a target site for linearization. Homologous arms, ranging from 800 to 1000 bp, were PCR-amplified from the DNA of the intended cell line for gene insertion. Primers are listed in the Supplemental Table S4.
Surrogate reporter plasmids construction
Integration surrogate reporter plasmids were constructed using a backbone containing the U6-ccdB-scaffold cassette, as well as EF1a-driven reporter elements such as Puro, Neo, Bsd, Hygro, and eGFP via restriction enzyme cloning. Primers containing a specific target site and its gRNA were used for PCR, and the products were assembled into the surrogate reporter plasmid backbone using Gibson assembly (NEB). As an example of construction of the surrogate reporter (Puro) for targeting the TRIM44 gene, pVerispr-Puro-TRIM44, the pVerispr-Puro was digested by BbsI and NheI, and PCR is amplified using pVerispr-Puro as a template, with the forward primer “tatatcttGTGGAAAGGACGAAACACCgAGCTTGATATAATCCAGTATgttttagagctaGAAAtagcaa” and the reverse primer “CACCGTGGGCTTGTACTCGGTAGCTTGATATAATCCAGTATTGGGGATCCCATGGTGGCgatcGCTAGCT,” containing the gRNA and target site highlighted in bold and the underlining 6 bp inserted between the target site and the start codon ATG, protecting the ATG signal from indels. The pVerispr-Puro-TRIM44 was generated by Gibson assembly of the plasmid backbone and the PCR products. Primers are listed in the Supplemental Table S4.
The nonintegrating version 2.1 GFP (V2.1G) was constructed from pMax-GFP. A ccdB flanked with two BbsI sites was inserted into the start codon ATG of GFP. The BbsI digestion will produce two 5′ sticky ends, right on the ATG and the first amino acid of GFP. The oligos containing a targeting site with PAM sequence were annealed, ligated to the backbone, and resulted in gene-specific V2.1 plasmids. For instance, pV2.1G-B4GALNT2 was constructed by BbsI digestion of pV2.1G; annealing of a linker using specific primers, the forward primer 5′-CATGTCCTCAGGTTCACTGCGGGGAGG-3′ and the reverse primer 5′-TCACCCTCCCCGCAGTGAACCTGAGGA-3′; and subsequent ligation. For additional details, refer to Supplemental Figure S4 and Supplemental Sequences.
Cell culture and electroporation
mESCs established by our laboratory were cultured in 2i/LIF on gelatin-coated plates. HEK293T cells, HeLa cells, and Vero E6 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Gibco). PFFs were isolated from day 28–30 porcine embryos of laboratory minipigs and cultured in minimum essential medium α (MEM α) containing 15% FBS, 1% nonessential amino acids (Invitrogen), 1% penicillin–streptomycin (Gibco), and 10 ng/mL bFGF (Proteintech). PFFs were used within five passages as donor cells for SCNT. Electroporation was performed using the Nucleofector 2b device (Lonza) following the manufacturer's protocols. Endotoxin-free plasmids were prepared and mixed with prewarmed Nucleofector solution, followed by electroporation according to the cell-specific electroporation program (U-023 for PFFs, B-016 for mESCs, Q-001 for HEK293T cells, T-030 for PK15 cells and Vero E6 cells, and I-013 for HeLa cells). For the seven-gene editing experiment MV014, 106 PFF cells were electroporated with the plasmids listed in Supplemental Table S2 using the U-023 program. The medium was changed 12 h after transfection.
Coselection strategy
When using the nonintegrating surrogate reporter plasmids, selection needs to be quick and powerful. As an example of the V2.1G surrogate reporter plasmids selection, GFP-positive cells were sorted using BD FACS Aria III 24–36 h after transfection. Typically, compared with the control group cells, the proportion of the highest intensity FITC cells ranges from 0.1 to 5%, varying according to the efficiency of the sgRNAs used. These cells can be harvested for efficiency analysis and proceed directly to the subsequent experimental steps.
When using the integration surrogate reporter plasmids that integrate into the genome facilitated by PBase, selection can be extended over a longer period. The concentration of drugs and treatment duration are cell line dependent. For instance, in PFFs, PK15, and Vero E6 cells, the concentrations used were 2 μg/mL puromycin, 1200 μg/mL G418, and 800 μg/mL hygromycin, respectively. In mESC cells, the concentrations were 1 μg/mL puromycin and 150 μg/mL hygromycin.
Flow cytometry
Cells were enzymatically digested and prepared as single-cell suspensions in Dulbecco's Phosphate Buffered Saline (DPBS) before flow cytometry cell sorting or analysis. In the coselection strategy using a V2.1G or V4.1R reporter, cells were expressing GFP or RFP fluorescence. For cell gating, a combination of forward scatter (FSC) and side scatter (SSC) was used to exclude debris, with SSC employed to gate on single cells. Subsequently, FITC or PE was used to gate on fluorescence-positive cells, utilizing control cells for setting gates. Before conducting two-color analysis or sorting, compensation was adjusted using cells with a single color. To enrich for homozygously edited cells, fluorescence-highest cells were gated.
Cell staining and flow cytometry
For cell staining prior to flow cytometry, cells were first centrifuged at 200g for 5 min, washed with DPBS containing 2% FBS, and centrifuged again at 200g for 5 min. They were subsequently resuspended in DPBS containing appropriate stains, incubated for 30 min at 4°C, and then washed and resuspended in DPBS for filtration into single-cell suspensions for flow cytometry. The following lectins and antibodies were used for staining: IB4 lectin (Vector FL-1201, 1:50 dilution), DBA lectin (Vector FL-1031, 1:50 dilution), AntiNeuGC (BioLegend 146901, 1:50 dilution), and thrombomodulin antibody (Abmart T55279, 1:100 dilution). Samples were sorted on a BD FACS Aria III system and analyzed using FlowJo V10 software.
Targeting efficiency detection
Following coselection, approximately 1000 cells were lysed in 40 μL of mouse tissue lysis buffer (Vazyme, PD101) and 0.8 mg/mL Proteinase K for 45 min at 55°C, followed by 5 min at 99°C. The lysate was used as templates for PCR, with ∼4 μL in a 20 μL reaction. If the cell number was less than 1000, nested PCR was used for successful detection with at least 50 cells. Sanger sequencing data and ICE Analysis were employed to calculate outcomes and targeting efficiency. For colony validation, PCR products were ligated into the pMD19-T vector (TakaRa 6013) via TA cloning and sequenced. The Sanger traces were analyzed using the SnapGene software (version 2.3.2).
For measuring on-target editing in seven-gene targeting, two rounds of PCR were performed for high-throughput sequencing. Round 1 PCR was performed in a 10 μL reaction volume containing 2 μL DNA lysis, following the instructions of Ex Premier DNA polymerase (Takara RR371A). Round 2 PCR was performed for 12 cycles to add Illumina adapters (P5 and P7), in a 20 μL reaction volume containing 1 μL of PCR round 1 product. PCR products containing different index were gel-purified, mixed, and sequenced using the DNBSEQ-T7 platform. Primers are listed in the Supplemental Table S4. CRISPResso2 (Clement et al. 2019) was used to analyze the sequencing read percentage of the wild type and indels.
Knock-in detection
Genomic DNA was extracted as previously described (Wu et al. 2008). Junction PCR across homologous arms was used to detect successful knock-in events. Primers are listed in the Supplemental Table S4. PCR was performed in a 20 μL reaction volume containing 1 μL of gDNA using Ex Premier DNA polymerase. PCR products were analyzed and quantified by agarose gel electrophoresis.
RT-qPCR and western blot analysis
Total RNA was extracted using the RaPure total RNA kit (MAGEN) and was reverse-transcribed into cDNA using TransScript one-step gDNA removal and cDNA Synthesis supermix (TransGen Biotech). RT-qPCR was conducted with TB Green Premix Ex Taq II (Takara) on a QuantStudio 3. Primers used for RT-qPCR are listed in Supplemental Table S4. Cultured cells were harvested and resuspended in RIPA lysis buffer (Beyotime P0013B). Western blotting followed using standard protocols. The following antibodies were used: anti-USE1 primary antibody (Proteintech 25218-1-AP, 1:1000 dilution), anti-tubulin primary antibody (Beyotime AT819, 1:1000).
Off-target analysis
For each sgRNA, three potential off-target sites were predicted and subjected to PCR and high-throughput sequencing analysis. Cells were lysed, and two rounds of PCR were performed for high-throughput sequencing, as described above. Primers are listed in the Supplemental Table S4.
Immunofluorescence staining
Formalin-fixed, paraffin-embedded kidney and heart tissue sections of 5Gene xeno-pigs and wild-type pigs were prepared. Antigen retrieval was conducted by heating slides in 0.01 M citrate buffer (pH 6.0) for 10 min in a microwave oven. Slides were then blocked in Tris-buffered saline (TBS) containing 3% bovine serum albumin (BSA) for 30 min at room temperature, followed by incubation with primary antibodies overnight at 4°C. The primary antibodies utilized in this study were thrombomodulin antibody (Abmart T55279, 1:200 dilution), PE antihuman CD141 (thrombomodulin) antibody (BioLegend 344104, 1:100 dilution), anti-CD55 antibody (Abcam ab133684, 1:4000 dilution), and APC antihuman CD55 antibody (BioLegend 311312, 1:100 dilution). After incubation with primary antibodies (unlabeled thrombomodulin antibody Abmart and anti-CD55 antibody Abcam), slides were incubation with secondary antibodies (Cy3-labeled goat antirabbit IgG [H + L]) and counterstained with DAPI in mounting media.
Production of cloned embryos and pigs via SCNT
SCNT was performed as previously described. In brief, pig ovaries were collected from a local abattoir and transported to the laboratory. After maturation of oocytes, a single donor cell was injected into the perivitelline space of the enucleated oocyte. The oocyte-cytoplasm-cell complexes were fused using a BTX electro-cell manipulator 200. The reconstructed embryos were incubated overnight in PZM3 at 38.5°C with 5% CO2 and then transferred into surrogate mother pigs. The pregnancy status of the surrogates was monitored by ultrasonography 1 month later, and piglets were delivered via natural birth.
Statistics and reproducibility
The coselection experiments in cells by drugs or flow cytometry were performed at least three independent experiments. Statistical significance was determined by two-tailed Student's t-test, and P < 0.05 was considered significant: (*) P < 0.05, (**) P < 0.01, and (***) P < 0.001.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the Wu laboratory members for continuous support. This work was supported by the National Key Research and Development Program of China (2023YFC3404301, 2023YFF0724700), the Innovative Project of State Key Laboratory of Animal Biotech Breeding (grant no. 2024SKLAB 1-8), the Chinese Universities Scientific Fund (grant no. 2024TC167), the Natural Science Foundation for Youths of Hainan Province, China (grant no. 324QN290), and Pinduoduo-China Agricultural University Research Fund (grant no. PC2023A01004).
Author contributions: S.W. and X.Duan conceived and designed the study. X.Duan, X.Q., and J.L. performed the surrogate reporter coselection strategy development and optimization experiments. X.Duan, C.D., J.L., and L.G. performed the single-gene enrichment and multiple-gene enrichment experiments. X.Duan, C.C., N.H., and F.G. performed the multiplex modified xenotransplantation pig generation experiments. X.Duan, X.Du, and S.W. wrote the paper with comments from all authors. S.W., J.S., and X.Du supervised the work.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279709.124.
-
Freely available online through the Genome Research Open Access option.
- Received June 19, 2024.
- Accepted February 18, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








