
Multisample motif discovery and visualization for tandem repeats
- Yaran Zhang1,
- Marc Hulsman1,2,
- Alex Salazar1,
- Niccolò Tesi1,2,
- Lydian Knoop1,
- Sven van der Lee1,2,3,
- Sanduni Wijesekera1,
- Jana Krizova1,
- Erik-Jan Kamsteeg4 and
- Henne Holstege1,2,3,5
- 1Section Genomics of Neurodegenerative Diseases and Aging, Department of Clinical Genetics, Vrije Universiteit Amsterdam, Amsterdam UMC, 1081HV Amsterdam, The Netherlands;
- 2Delft Bioinformatics Lab, Delft University of Technology, 2628CD Delft, The Netherlands;
- 3Amsterdam Neuroscience, Neurodegeneration, 1081HV Amsterdam, The Netherlands;
- 4Department of Human Genetics, Radboud University Medical Center, 6525GA Nijmegen, The Netherlands;
- 5Alzheimer Center Amsterdam, Neurology, Vrije Universiteit Amsterdam, Amsterdam UMC location VUmc, 1081HV Amsterdam, The Netherlands
Abstract
Tandem repeats (TRs) occupy a significant portion of the human genome and are a source of polymorphisms due to variations in sizes and motif compositions. Some of these variations have been associated with various neuropathological disorders, highlighting the clinical importance of assessing the motif structure of TRs. Moreover, assessing the TR motif variation can offer valuable insights into evolutionary dynamics and population structure. Previously, characterizations of TRs were limited by short-read sequencing technology, which lacks the ability to accurately capture the full TR sequences. As long-read sequencing becomes more accessible and can capture the full complexity of TRs, there is now also a need for tools to characterize and analyze TRs using long-read data across multiple samples. In this study, we present MotifScope, a novel algorithm for the characterization and visualization of TRs based on a de novo k-mer approach for motif discovery. Comparative analysis against established tools reveals that MotifScope can identify a greater number of motifs and more accurately represent the underlying repeat sequences. Moreover, MotifScope has been specifically designed to enable motif composition comparisons across assemblies of different individuals, as well as across long-read sequencing reads within an individual, through combined motif discovery and sequence alignment. We showcase potential applications of MotifScope in diverse fields, including population genetics, clinical settings, and forensic analyses.
A large part of the human genome consists of repetitive elements. One such class of repeats is tandem repeats (TRs), which are DNA sequences characterized by the contiguous repetition of at least one nucleotide, accounting for ∼6%–8% of the human genome (Cui et al. 2024; English et al. 2024; Rajan-Babu et al. 2024). TRs are broadly classified based on the size of their repetitive motif: TRs with motif size ≤ 6 bp are referred to as short tandem repeats (STRs), while those with larger motifs and variability in copy numbers are categorized as variable number tandem repeats (VNTRs) (Tautz 1993; Eslami Rasekh et al. 2021).
TRs are highly polymorphic, making them a major source of diversity in human genomes (Jeffreys et al. 1985). In fact, 13–17 STRs are currently used in North America and in the United Kingdom to uniquely identify a person (Hammond et al. 1994; Opel et al. 2007; Glynn 2022; Mallinder et al. 2022). Due to the high genetic variability and lack of linkage disequilibrium with each other, the probability of two unrelated individuals sharing a perfect match of this set of STRs is <1 in 1 billion (Reilly 2001). This variability has led to the adoption of these TRs as standard tools in forensics analyses, where they play a crucial role in DNA profiling and identifying individuals with a high degree of accuracy (Moretti et al. 2001; Jobling and Gill 2004).
TRs have been associated with a range of neurological disorders (Tang et al. 2017; Hannan 2018; Chintalaphani et al. 2021). For instance, Friedreich ataxia (FRDA) can be caused by homozygous expansion of a GAA repeat in the first intron of frataxin (FXN) gene, while the expansion of a GGGGCC repeat intronic of C9orf72 gene has been linked to increased risk of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (Pandolfo 2009; DeJesus-Hernandez et al. 2011). VNTR expansions have also been implicated in several diseases (De Roeck et al. 2018; Hannan 2018; Song et al. 2018). For example, the expansion of a 25 bp repeat in the intronic region of the ATP-binding cassette subfamily A member 7 (ABCA7) gene has been linked to an increased risk of Alzheimer's disease (AD) (De Roeck et al. 2018). However, it is not just the repeat length that is associated with disease. In most pathogenic TRs, the motif composition of the repeat is also important (Seixas et al. 2017; Ishiura et al. 2018; Cortese et al. 2019; Chen et al. 2020; Wright et al. 2020). For instance, in patients with cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS), expansions of the repeat in replication factor C subunit 1 (RFC1) gene are composed primarily of AAGGG or GACAG, variations from the more common AAAAG motif found in nonexpanded alleles (Cortese et al. 2019; Scriba et al. 2020).
Furthermore, TRs can provide insight into (the evolution of) population structure due to their high mutability (Rosenberg et al. 2002; Ellegren 2004; Course et al. 2021; Lu et al. 2023). Course et al. demonstrated significant differences in repeat length among VNTRs in genes, including ADP-ribosyltransferase 1 (ART1), PROP paired-like homeobox 1 (PROP1), and dynein 2 intermediate chain 1 (DYNC2I1), as well as substantial differences in motif organization in poly(rC) binding protein 3 (PCBP3) between superpopulations (Course et al. 2021). Using a pangenome-based approach, Lu et al. (2021) discovered that more than 8000 VNTRs show differential motif usage across populations. These findings emphasize the importance of considering both repeat length and motif organization in TR genotyping.
With new sequencing technologies emerging that combine longer read length and higher accuracy, it is now possible to deeply characterize TRs. As such, multiple methods have been developed to genotype and profile TRs. One class of methods has been proposed that relies on databases of TRs, describing for each TR the motifs that are to be considered. This has an advantage that discovered motifs are more homogenous across individuals. For instance, Ren et al. (2023) developed a toolkit, vamos, that generated a representative set of motifs for over 460,000 TRs in the human genome. This was achieved by selecting motifs for each TR in a reference set of genomes, while allowing for some sequence divergence. Then, vamos uses this motif database to annotate TRs in the query genome. Similarly, Dolzhenko et al. (2024) developed the Tandem Repeat Genotyping Tool (TRGT), specifically for Pacific Biosciences (PacBio) HiFi sequencing data. It uses prespecified motifs to genotype simple TR regions, while more complex repeats are defined using hidden Markov models (HMMs). Additionally, TRGT comes with a visualization module, the TRVZ tool, which displays read-level evidence supporting the genotype calls made by TRGT as well as the TR motifs. While the use of a motif database can work well for relatively stable TRs, it may result in the loss of important motifs for more variable TRs, for instance, rare motifs that are relevant to diseases. In addition, relying on a database hinders the application to novel repeats, or application to species not covered by the database (currently databases are only available for the human genome). Therefore, there remains a need for more versatile methods that can accurately capture the complexity of TR sequences across diverse genomic contexts.
Another class of methods detects motifs in a given sequence using a de novo approach. This allows them to handle extensive genomic diversity, including complex TRs in which motifs can be highly variable. A drawback of these methods is that in a setting in which multiple individuals are analyzed together, it can be hard to canonicalize the discovered motifs across individuals. For instance, the widely used tandem repeats finder (TRF) program generates a separate annotation for each motif it identifies, leaving it to the user to select the optimal motif representation, and to canonicalize these representations across different individuals (Benson 1999). Recently, Masutani et al. (2023) proposed an algorithm, uTR, to decompose TRs after selecting a better set of motifs according to maximum parsimony that minimizes replication slippage events. Still, this method will analyze each allele sequence individually. Furthermore, these methods are not sensible to small mutations (e.g., single-nucleotide polymorphisms [SNPs]). These small variations can, however, be biologically important, for instance, in clinical settings in which some pure repeats are considered more pathogenic than interrupted repeats (Rafehi et al. 2023).
Here, we present MotifScope, a flexible toolkit for motif annotation and visualization of TRs from sequencing data, that uses a de novo k-mer-based approach for motif discovery. To evaluate MotifScope performances, we compared it to the three existing tools for motif discovery: uTR, TRF, and vamos. Our findings indicate that MotifScope identified a greater number of motifs and reflected the actual repeat sequence more accurately than other tools. Additionally, we show potential applications of MotifScope in population genetics to explore population stratification due to TRs, in clinical studies to study pathogenic TRs, and in a forensic setting.
Results
MotifScope is a tool for characterizing and visualizing the motif composition of TRs. The input for MotifScope is a FASTA-formatted file containing the sequence(s) to annotate (Fig. 1). This can be a single repetitive sequence, individual reads from one individual, or a collection of assembled alleles from multiple individuals. There are three major algorithmic steps in MotifScope: (i) iteratively identifying and annotating motifs using a k-mer-based approach; (ii) color mapping of motifs using a dimensional reduction technique, and (iii) clustering and aligning sequences based on their motif composition. MotifScope iteratively discovers and annotates motifs that make up long-continuous sequences within a single sequence without error correction, but also across sequences to enable joint discovery and annotation across multiple genomes. The output consists of a FASTA-formatted file reporting each input sequence with motif information, representing motifs as sequences followed by their respective counts. Additionally, MotifScope provides a tab-separated file summarizing the amount of sequence covered by each motif and their corresponding count in each input sequence. Finally, a graphic representation of the motif composition for each TR in each sample can be generated.
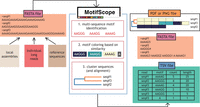
Overview of MotifScope. Input to MotifScope consists of a set of sequences. MotifScope first identifies motifs by evaluating the length of consecutive sequence stretches formed by each k-mer present in the sequences. It then iteratively annotates consecutive occurrences of k-mers as motifs and masks them in the sequences. Subsequently, single occurrences of identified motifs are annotated and masked. To visualize the motif composition, motifs are colored based on their sequence similarity to each other. The sequences are then clustered based on their motif composition. It also offers the option to perform multiple sequence alignment (MSA) based on motif compositions.
Accuracy and efficiency
To evaluate the performance of MotifScope in characterizing TRs, we conducted a comparative analysis alongside recently developed TR-analysis methods: TRF, uTR, and vamos (with both the original motif set, “vamos original,” and an efficient motif set, “vamos efficient”) on the HG002 genome sequenced with PacBio HiFi technology (see Methods). We used a set of TRs from the PacBio repeat catalog, which contains 171,146 TRs and benchmarked the tools using long-read whole-genome sequencing of the HG002 genome with PacBio HiFi technology (see Methods).
It is important to note that vamos can only be applied to locations in its own repeat catalog, consisting of 467,104 VNTRs. The size distribution of TRs within the PacBio catalog differed from that in the vamos VNTR catalog. Specifically, repeats were smaller in the PacBio catalog, with 99.97% of repeats being ≤100 bp in the GRCh38 human reference genome, while in the vamos VNTR catalog, 80.42% of repeats are ≤100 bp (Supplemental Fig. 1A). The motif sizes are also larger for the TRs in the vamos catalog compared to the PacBio catalog (Supplemental Fig. 1B). The PacBio catalog also showed reduced sequence complexity, as illustrated by the distinct k-mer (k = 10) counts observed in each repeat in the GRCh38 human reference genome (Supplemental Fig. 1C).
Hence, we evaluated the performances of the tools based on a subset of 5486 TRs that exactly overlapped between these two catalogs (i.e., same start and end coordinates for each TR). This set of TRs is more similar to the TRs in PacBio catalog based on length, motif size, and sequence complexity. Additionally, to provide a comprehensive evaluation, we also randomly sampled 5000 repeats from the vamos VNTR catalog and compared the performances of the tools on this set of repeats. MotifScope identified a greater number of motifs compared to other tools (Fig. 2A). To evaluate the quality of motif identification, we assessed the normalized edit distance between the concatenation of the motif representations generated by these tools and the true repeat sequences (Fig. 2B). By design, MotifScope consistently achieves an edit distance of 0, indicating an exact match between its motif description and the underlying repeat sequence. In contrast, other methods exhibited increasing edit distances, for example, by sorting the edit distance in ascending order, at 90th percentile, the edit distance was 0 for MotifScope, 0.022 for TRF, 0.032 for vamos original, 0.029 for vamos efficient, and 0.098 for uTR. This shows that MotifScope's description of TRs reflects the actual repeat sequences more accurately compared to the other three tools.
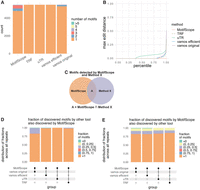
Comparative analysis of TR characterization. Four tools are tested in the analysis, MotifScope, TRF, uTR, and vamos. For vamos, here we use both the original motif set (vamos original) and the efficient motif set (vamos efficient). (A) The number of motifs discovered by each of the four tools. (B) The edit distance between the actual sequence and the results obtained from the four tools, normalized with respect to repeat length. (D,E) The intersection of motifs between MotifScope and other tools (dots connected by lines below the X-axis): in the bar plot, each column shows the results obtained from both MotifScope and the respective other tools (Method X) for all 5486 loci; for D, the stacked bar plot shows the fraction of intersected motifs over the total number of motifs found by the other tool for each loci, as shown in C, A / Method X, and the segments in the bar denote the distribution of the ratio among all these loci; for E the stacked bar plot shows the fraction of intersected motifs over the total number of motifs found by MotifScope, as shown in C, A / MotifScope, and the segments in the bar denote the distribution of the ratio among all these loci.
We further evaluated the extent to which different tools captured the same motifs within the studied TRs: we found that MotifScope identified all motifs found by TRF in 97.89% of loci, by uTR in 99.75%, by vamos original in 79.64%, and by vamos efficient in 98.34% of the loci (Fig. 2D). The relatively higher fraction of motifs identified by vamos original but not by MotifScope is attributed to the presence of nonrepeated motifs that occur only once in vamos original motif sets, which are characterized differently by MotifScope as single nucleotides. Moreover, MotifScope was also able to pick up motifs that were not detected by the other tools. For example, in 15.30% of TRs, MotifScope found motifs not identified by TRF, in 16.49% not identified by uTR, in 17.70% and 17.94% not identified by vamos original and vamos efficient, respectively (Fig. 2E). These additional motifs found by MotifScope are largely composed of single-nucleotide motifs. In the 3544 comparisons between MotifScope and other methods, where new motifs were detected by MotifScope, 87.4% contained at least one single-nucleotide motif. However, these single-nucleotide motifs make up a small percentage of the repeat sequences (1.74%). Altogether, 86.4% of sequences had no single-nucleotide motifs, and the percentage increased with sequence complexity, as demonstrated by distinct k-mer count (Supplemental Fig. 2). These additional motifs allow MotifScope to more accurately represent the underlying repeat sequence. When using 5000 VNTR randomly sampled from the vamos catalog, MotifScope identified more motifs and reflected the sequence more accurately compared to other tools. Additionally, the overlap of motifs identified by MotifScope and other tools decreased in this subset, reflecting the higher complexity of this set of TR (Supplemental Fig. 3).
We evaluated computational performances in terms of running time and memory usage using 48 known pathogenic TR from PacBio catalog on 1, 5, 10, 20, and all 47 genomes from the Human Pangenome Reference Consortium (HPRC), respectively (see Methods). We found that running time of MotifScope was mainly driven by figure generation and startup time. The motif discovery step took on average 5 sec, and only marginally increased when multiple genomes were considered (Supplemental Fig. 4). When coupled with the figure generation, this took 60.94 sec for 48 TRs for a single genome, increasing to 538.13 sec for 47 genomes. The maximum memory usage was 0.3 GB when coupled with the figure generation on a single genome, increasing to 4.70 GB for 47 genomes.
Merit of de novo motif discovery
MotifScope uses a de novo motif discovery approach to enable the discovery of rare, possibly pathogenic motifs. In Figure 3A, we present the motif characterization results of a TR within intron 2 of RFC1 (Chr4:39,348,424–39,348,483), where biallelic AAGGG or GACAG repeat expansions have been associated with an autosomal recessive neurological disorder, CANVAS. We employed MotifScope to characterize this repeat on HG002 and a Dutch CANVAS patient (Wang et al. 2022; van de Pol et al. 2023). The patient is a compound heterozygous carrier of two different AAAAG expansions of 6.28 and 7.69 kb. In HG002, all four tools annotated the repeat using the motif AAAAG or its cyclic shifts. In the CANVAS patient, MotifScope identified three major motifs: GACAG, GACAA, and AAAAG. Using these motifs, the TR can be characterized as (GACAG)1136(GACAA)118GAAG(AAAAG)14 with four indels for haplotype 1 and (GACAG)1420(GACAA)114GAAG(AAAAG)15 with 20 indels for haplotype 2. Note that the results of MotifScope also represent the indels through additional motifs (total of six motifs), resulting in a fully accurate representation of the actual repeat sequences (edit distance = 0.0). In contrast, uTR annotated the sequences using GACAG and GACAA, (GACAG)1137(GACAA)133 for haplotype 1, and (GACAG)1422(GACAA)130 for haplotype 2, with an average normalized edit distance of 0.005, mislabeling the AAAAG motifs at the end of the alleles. TRF explained the sequences with a single GACAG motif, (GACAG)1255 for haplotype 1 and (GACAG)1535 for haplotype 2, resulting in an average normalized edit distance of 0.019. Finally, vamos original primarily characterized the sequence with motifs GGGAC and AAAAG, (GGGAC)1177(AAAAG)130 for haplotype 1 and (GGGAC)1418(AAAAG)130 for haplotype 2 generating an average normalized edit distance of 0.212, while vamos efficient predominantly used motifs GGAAA, GGCAA, and AAAAG for annotation, resulting in (GGAAA)1181(GGCAA)115(AAAAG)14 for haplotype 1 and (GGAAA)1422(GGCAA)115(AAAAG)14 for haplotype 2, with an average normalized edit distance of 0.214.
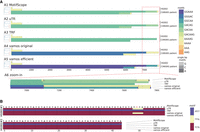
Motif characterization with different methods. (A) The motif characterization of the RFC1 repeats in HG002 and a CANVAS patient. The results from (A1) MotifScope, (A2) uTR, (A3) TRF, (A4) vamos original, (A5) vamos efficient of the RFC1 repeat in the HG002 assembly and the assembly of a Dutch CANVAS patient, and (A6) zoomed-in view of the circled region of one patient allele are visualized. In each subfigure, the left panel shows the clustering of the sequences, the right panel shows the composition of the repeat, with distinct motifs represented in different colors, as indicated by the color bar on the right side of the figure. (B) The motif characterization of forensic loci D2S441 in HG002. This shows the visualization of the decomposing result from MotifScope, TRF, uTR, vamos efficient, and vamos original of the assembly of the forensic D2S441 locus in HG002 genome assembly, with distinct motifs represented in different colors, as indicated by the color bar on the right side of the figure.
To further illustrate the motif discovery results, we also present the characterization of a forensic locus, D2S441 (Chr2:68,011,946–68,011,994), in the HG002 genome assembly (Fig. 3B). MotifScope and vamos original successfully characterized the two alleles as (TCTA)11 and (TCTA)12TTTA(TCTA)2 while uTR, TRF, and vamos efficient failed to identify the TTTA motif in the second allele.
Joint motif discovery and annotation across multiple genomes
MotifScope offers the capability to analyze multiple samples simultaneously, which can improve the characterization and comparison of TR haplotypes. For example, when MotifScope was applied to the RFC1 repeat in a single genome HG01175 from the HPRC, it initially failed to identify the starting motif AAAAG and motif GACGG immediately after the AAAGG repeat in haplotype 1. Instead, these motifs were represented as single nucleotides (e.g., A, A, A, A, G for AAAAG) (highlighted in the red box in Fig. 4A). As the GACGG and AAAAG motifs are in the original motif set, vamos original was able to identify these two single motifs on HG01175 (Supplemental Fig. 5). However, when jointly analyzing HG01175 with HG01109 and HG00733, MotifScope managed to also identify the single copies of these motifs in HG01175, as these were recurring motifs across samples (Fig. 4B–D). In addition, the joint analysis also revealed that HG01175 haplotype 1 and HG01109 haplotype 1 share the same motif structure: AAAAG(AAGGG)7(GACGG)1/2(AAAGGG)n(AAAGGGAAGG)2AAAG(GAAA)2AAG (Fig. 4D).

Motif characterization of the RFC1 repeats in three HPRC genomes. The results from MotifScope on the assemblies of three genomes, (A) HG01175, (B) HG01109, and (C) HG00733. These three genomes are Admixed Americans. (D) The joint analysis result of these three genomes. The sequence “GACGG” in these sequences is highlighted in red boxes. The left panel shows the clustering of sequences along with genome identifiers, represented by the corresponding color bar on the second-to-right side. The right panel shows the motif composition of the repeat, with distinct motifs represented in different colors, as indicated by the color bar on the right side of the figure.
We assessed the stability of the set of discovered motifs for known pathogenic TRs by sampling subsets of 1–47 genomes from the HPRC samples, and comparing the similarity of discovered motifs between subsets. The motifs identified were generally stable, with increased stability observed as more genomes were included (Supplemental Fig. 6A). In fact, this can be seen as a form of joint calling approach, that ensures a consistent motif representation across different sequences and samples, enhancing the reliability and comparability of the results. However, the stability varies with locus sequence complexity. For example, the notch 2 N-terminal like C (NOTCH2NLC) TR locus (Chr1:149,390,803–149,390,842) showed high stability across genomes with one single motif present in all genomes, while the RFC1 TR locus showed high diversity across individuals due to the presence of eight unique motifs in the HPRC genomes. As a result, for RFC1, a larger number of individuals had to be analyzed to accurately capture the extensive motif diversity in the population (Supplemental Fig. 6B).
Profiling clinically relevant loci in the population
TRs are known to have a wide variability in motif sequence, motif size, and repeat size across populations, suggesting the importance of examining multiple samples from different populations collectively. For example, a pathogenic repeat in the gene brain expressed associated with NEDD4 1 (BEAN1) (Chr16:66,490,397–66,490,466) is associated with an autosomal dominant neurological disorder, Spinocerebellar Ataxia Type 31 (SCA31), and coincides with a specific GAATG repeat insertion found solely in the Japanese population (Ishikawa and Nagai 2019). We applied MotifScope to this locus using 47 genome assemblies from the HPRC, and identified a cluster of African alleles with distinct motif structure, with A, T, and ATT insertions in the TAAAA repeat (in red box in Fig. 5A). Additionally, two expanded alleles were observed: a 2.47 kb expansion with CAATA motifs in an Admixed American allele and a 0.99 kb TAAAA expansion in an East Asian allele. Notably, we observed expansions with CAATA and TAAAA motifs (Fig. 5B) as well as another motif TAACA that was found in a South Asian individual (Fig. 5A).
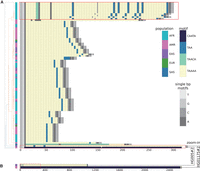
BEAN1 repeat in HPRC samples. For A, the leftmost panel presents the clustering of assembly sequences from the HPRC sample (n = 47) with 10 bp flanking the repeat, along with the population origin of the alleles in the adjacent column. The color code, denoting population origin, is in the second-to-right color bar (SAS: South Asian; EUR: European; EAS: East Asian; AMR: Admixed American; AFR: African). The right panel visually represents repeat composition, with distinct colors signifying different motifs. The two expanded alleles are truncated in this figure. The full motif compositions of these two alleles are shown in B.
We also applied MotifScope to a pathogenic repeat recently identified in the gene fibroblast growth factor 14 (FGF14) (Chr13:102,161,575–102,161,726) (Supplemental Fig. 7; Pellerin et al. 2023; Rafehi et al. 2023). We used the HPRC assemblies as well as the otter-assembled allele sequences of 246 Dutch AD patients and 238 Dutch centenarians (see Methods; Supplemental Fig. 8). The expansion of this repeat has been associated with an autosomal dominant adult-onset ataxia SCA27B. Despite sharing the same GAA repeat backbone, 10 different repeat structures were identified across all evaluated individuals. Similar to the GRCh38 human reference genome, the majority of the alleles carried a nonexpanded GAA repeat. However, the other assemblies carried different insertions inside the GAA repeat: insertions of single As; an GAAGAG repeat insertion; an GAG repeat insertion immediately followed by a GAGAAG repeat insertion; an [(GAA)1(CAG)2] repeat insertion; a GAGAAG repeat insertion; insertions of different single nucleotides in slightly expanded GAA repeat; and an East Asian individual with an 1.85 kb expansion composed of an [(GAG)1(GAA)4] repeat, an [(GCA)2(GAA)2] repeat, an [(GCA)1(GAA)2] repeat, CAGAAG repeats, and CAG repeat insertions (Supplemental Fig. 7). Notably, MotifScope revealed that all African individuals had the same starting sequence of the repeat as the nonexpanded GAA alleles, whereas individuals of other ancestry that did not carry the pure nonexpanded GAA allele had a different starting sequence (Supplemental Fig. 7). This case illustrates that the motifs and motif structures of TRs can be highly variable in the population and MotifScope is able to identify them and cluster sequences with the same motif structure together.
MotifScope was also applied to TRs in the ataxin 8 (ATXN8) gene (Chr13:70,139,383–70,139,428), where interruptions in the CAG repeat with CCG repeat are believed to increase pathogenicity and are more likely to cause Spinocerebellar Ataxia Type 8 (SCA8) (Koob et al. 1999). However, no such interruptions were found in the Dutch centenarians, AD patients, or HPRC individuals (Supplemental Fig. 9). Similarly, in the ataxin 2 (ATXN2) TR (Chr12:111,598,950–111,599,019), where interruptions in the CAG repeat with CAA motifs are associated with Spinocerebellar Ataxia Type 2 (SCA2), no reported pathogenic interruption patterns were detected in these samples (Supplemental Fig. 10; Charles et al. 2007).
We also applied MotifScope to an intronic repeat in gene ABCA7 (Chr19:1,049,437–1,050,066) on HG002, where the motif size is 25 bp (Supplemental Fig. 11). MotifScope was able to identify 24 different motifs in this sequence including the four single nucleotides. However, a substantial portion of the sequence was annotated with single-nucleotide motifs: 21.4% and 25.3% for the two allele sequences, respectively.
Analyzing TRs at the read level
Whereas MotifScope can jointly analyze multiple individuals, it can also jointly characterize and visualize all mapped sequencing reads that span a TR region, which can unveil somatic and technical variations. For instance, in Figure 6A, we display all spanning reads from the blood of a Dutch CANVAS patient on the RFC1 repeat, revealing a motif structure characterized by (GACAG)n(GACAA)n(AAAAG)n. However, these reads varied in size, ranging from 7.0 kb to 9.5 kb, and contained different SNVs at different positions. These reads showed varying copies of different motifs suggesting the presence of somatic mutation and/or technical errors (Supplemental Fig. 12A).
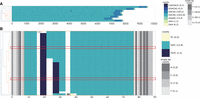
Motif characterization with MotifScope on long reads. (A) Motif characterization of the spanning reads of the RFC1 repeat in the blood of a CANVAS patient. The sequences contain the RFC1 repeat and 10 bp sequences flanking both sides of this region. (B) Motif characterization of the spanning reads of forensic loci D3S135. The two assembly alleles of the HG002 genome are highlighted in red boxes and the repeat is highlighted in the orange box with 10 bp flanking this region. In each figure, the clustering of the sequences is shown in the left panel, the right panel shows the motif composition of the repeat, with distinct motifs represented in different colors, as indicated by the color bar on the right side of the figure. The number following the motif on the color bar indicates the average number of occurrences of the motif per read.
Another example is shown in Figure 6B, where we analyzed all reads as well as the phased assemblies for the forensic locus D3S1358 (Chr3:45,540,738–45,540,802) in HG002, using the MSA feature provided by MotifScope. Our analysis revealed that 14 out of 31 reads matched one assembly allele, (TCTA)1(TCTG)2(TCTA)12, 12 out of 31 reads matched the other assembled allele, (TCTA)1(TCTG)1(TCTA)14, while 5 out of 31 reads did not exactly match to any of the two assembled alleles. Of these five reads, three reads contained a whole motif gain or loss compared to the two assemblies, one read contained a single C deletion, and one read contained a C insertion in addition to a whole TCTA motif loss. This is also evident from the count of different motifs across different reads (Supplemental Fig. 12B). This analysis allows one to assess somatic stability and/or the propensity for technical sequencing errors in a region. Additionally, we randomly selected 8, 15, and 30 reads from forensic locus D2S1338 (Supplemental Fig. 13). AAGG, CAGG, and AGG motifs were consistently identified by MotifScope with similar counts per read. Additional AGGA motifs were discovered in one of the 15 and 30 reads, which contained single-nucleotide deletion. This suggests that the influence of sequencing coverage on motif discovery is minimal.
MotifScope can also be applied to reads sequenced with Oxford Nanopore Technologies. We applied MotifScope to the forensic locus D8S1179 using HG002 reads sequenced with PacBio Sequel II, PacBio Revio, Nanopore R9.4.1 (simplex) chemistry, and Nanopore duplex, in reference motifs guided mode, as this locus has well-defined known motifs. As shown in Supplemental Figure 14, MotifScope produced similar results for reads from PacBio Sequel II, PacBio Revio, and Nanopore duplex, while Nanopore R9.4.1 chemistry displayed less repeat purity. This likely reflects the lower sequencing quality of Nanopore R9.4.1 chemistry compared to the other technologies. Local assemblies from these technologies consistently showed the same motif compositions.
Discussion
Advancements in long-read sequencing technologies have significantly enhanced our ability to explore TRs within the human genome. These variants have emerged as crucial elements in genetic studies due to their implications in evolution and diseases (Perry et al. 2008; Weischenfeldt et al. 2013). They often display multiallelic patterns in the human population, and therefore, a detailed examination of the composition of these repeats is important for unraveling their functional implications and evolutionary history. To address the complexities of TRs, we developed MotifScope, a tool designed to characterize and visually represent TR compositions. In a benchmarking study against existing tools, MotifScope outperformed by identifying more motifs and more accurately reflecting sequence composition.
By performing motif discovery de novo, MotifScope does not have to rely on a predetermined set of motifs to characterize TR sequences, which makes it able to detect motifs that are rare or unseen in the general population. This is especially important in a clinical setting, as studies have shown that motif alterations in repeats can be the cause of disease. For instance, in CANVAS patients, pathogenic alleles of the RFC1 repeat have been identified as expansions of GACAG and AAGGG sequences, rather than the AAAAG repeat observed in the GRCh38 human reference genome (Cortese et al. 2019; Scriba et al. 2020; van de Pol et al. 2023). Similarly, in five familial adult myoclonic epilepsy (FAME) subtypes, expanded TTTCA segments were found inside the TTTTA repeat in sterile alpha motif domain containing 12 (SAMD12), StAR related lipid transfer domain containing 7 (STARD7), membrane associated ring-CH-type finger 6 (MARCHF6), trinucleotide repeat containing adaptor 6A (TNRC6A) and Rap guanine nucleotide exchange factor 2 (RAPGEF2) genes (Ishiura et al. 2018; Corbett et al. 2019; Florian et al. 2019; Bennett et al. 2020). Given that both the original and efficient set of motifs from vamos were constructed with the HPRC and Human Structural Variation Consortium (HGSVC) samples, some of these pathogenic motifs are not represented in the default motif sets (Fig. 3A; Ren et al. 2023). Consequently, there is a risk of overlooking these rare, biologically relevant motifs, which could have significant implications for disease diagnosis and understanding.
MotifScope also offers the capability for joint motif analysis across samples. Similar to vamos, which utilizes motifs present in the HPRC and HGSVC samples, MotifScope thereby uses sequence information from other samples to enrich motif characterization (Fig. 4). This allows it to correctly annotate single occurrences of a motif in a haplotype, by leveraging information from other genomes in which the motif is expanded, highlighting patterns that reflect evolutionary proximity. Furthermore, joint analysis in combination with motif canonicalization ensures consistent annotation of motifs across all sequences. This prevents the occurrence of different motifs and/or cyclic shifted forms of the same motifs in different sequences, enhancing comparison across sequences.
Such comparisons will also highlight population differences. Several studies have demonstrated that while overall patterns of repeat variations are highly similar across populations, there are notable exceptions with population-specific patterns (Ziaei Jam et al. 2023). For instance, common CAG expansions in an intronic repeat within the gene carbonic anhydrase 10 (CA10) were found predominantly in African individuals, and the motif usage of an intronic repeat in gene PCBP3 showed substantial differences across modern superpopulations (Course et al. 2021; Ziaei Jam et al. 2023). Another example is the BEAN1 repeat, which showed population-specific pathogenic expansions (Fig. 5; Ishikawa and Nagai 2019). The ataxia SCA31, found exclusively in the Japanese population, aligns with a specific GAATG repeat insertion in this repeat, which is also exclusive to the Japanese population, suggesting a strong founder effect. This insertion ranges 2.5–3.8 kb in size and was found to inversely correlate with disease age of onset. Our analysis of the FGF14 repeat in the HPRC samples, Dutch AD patients, and Dutch cognitively healthy centenarians (Supplemental Fig. 7) also revealed highly population-specific patterns and revealed that only certain haplotype clusters showed expansions.
Repeat sequences might also differ at the read level. Somatic instabilities within TRs have been widely observed, including in forensic STRs (Fig. 6B) and pathological conditions such as cancers and repeat expansion disorders like Huntington's disease, myotonic dystrophy type 1 and CANVAS (Fig. 6A), contributing to variability at the individual read level (Veitch et al. 2007; dos Santos et al. 2012; Ciosi et al. 2019; Chintalaphani et al. 2021; Monckton 2021). Simultaneously, despite the high accuracy of long-read sequencing technologies, such as PacBio HiFi sequencing with an error rate lower than 0.05% and Oxford Nanopore duplex with an error rate lower than 0.09%, errors can still occur during sequencing, leading to variations between individual reads (Tesi et al. 2024). This can make it challenging to separate somatic from technical variation. However, we find that variations in reads also include whole-motif gain or loss in addition to SNVs. Given that homopolymer errors (i.e., indels) are the primary source of long-read sequencing systematic errors when using the PacBio technology, whole-motif gain and loss might be reflective of somatic variations (Au et al. 2012). With MotifScope, we can examine these different classes of variations between reads while annotating them with a consistent motif set. MotifScope also uses grayscale to color single-nucleotide motifs to visually separate them from multibase motifs.
Alignment of sequences can further facilitate repeat sequence comparisons. However, traditional alignment methods may struggle to accurately align highly repetitive sequences, in which motif gains and losses occur, while only subtle differences can be used as alignment markers. Standard alignment approaches therefore do not always yield the most meaningful results. Here, we addressed this issue by aligning sequences based on their motif composition, performing alignment on so-called “motif sequences,” which lead to a more biologically relevant alignment. This approach also enhances the visual representation and comparability of repeat loci, particularly for analyzing differences between reads (Fig. 6B) in complex loci and identifying variations among haplotypes with similar motif structures.
One element that sets MotifScope apart is that motifs are annotated exactly as they appear in the input sequences, providing a precise representation of repeat structures. In contrast, other tools usually allow for some flexibility in describing repeat sequences. This leads to simpler and more condensed motif patterns, but can also hide crucial information. In particular, small variations can highlight evolutionary proximity and expansion locations. Moreover, small variations can also be highly relevant in clinical settings, in which certain repeats have been found to only have pathogenic effects when the repeat is pure, i.e., is not interrupted by small sequence variations. One example is an intronic repeat in gene FGF14. Expansion of this repeat (>250 repeats) can cause adult-onset ataxia SCA50/ATX-FGF14 (Rafehi et al. 2023). Previous studies have found that only individuals carrying pure GAA repeat expansions developed the disease, and it was hypothesized that it is because these long GAA repeats are known to form secondary structures, inhibiting the transcription of the gene (Rafehi et al. 2023). With MotifScope, the structure of the repeat can be easily checked and visualized. As shown in Supplemental Figure 7, three individuals had >250 GAA repeats, yet they all have SNVs within the repeat sequence, and none were given ataxia diagnosis. However, it is important to note that these samples have low coverage, with 3, 2, and 1 reads supporting the assemblies, respectively, suggesting the possibility of sequencing errors affecting both the purity and size of the repeats.
Due to biological variations in TRs and technical errors introduced during sequencing, TR sequences often deviate from a perfect repetition of a single motif. Despite these variations, k-mers that form longer continuous sequence stretches are still more likely to explain more of the sequences and are consequently the frequently observed motifs. While this approach is effective for active repeats in which motif copies have not substantially diverged, it can also present a limitation in analyzing highly diverged sequences (Supplemental Fig. 3). Exact motif discovery approaches can struggle with sequences in which repeats are hard to identify due to the buildup of mutations. This is more likely to occur in VNTRs with large motifs. For instance, Supplemental Figure 11 shows an example of an intronic ABCA7 repeat, where the sequence is highly complex. While the concatenation of results from MotifScope produces the true sequence, these additional motifs do not enhance the interpretation of the repeat's structure, as much of the sequence is labeled with single nucleotides. MotifScope prioritizes motifs that can annotate long consecutive sequences, but when nearby motifs are all different, it fails to identify them. In such cases, tools like uTR, which allows distances to the true sequence, or vamos, which utilizes an established motif set to decompose repeat sequences, may be more suitable alternatives. Future iterations of MotifScope might therefore benefit from incorporating a more robust initial repeat structure identification step. Exact motif descriptions of highly complex repeats in combination with the here proposed motif color mapping could thereby reveal both the high-level structure of the repeat locus as well as more recent patterns of motif divergence.
With the continued adoption of long-read data in research and clinical settings, characterization and visualization of TRs will remain an active research area, as TRs constitute a major source of biological variation (Jeffreys et al. 1985). MotifScope offers a new unique angle to this, which might lead to new insights into motif structure and pathogenic mechanisms. Joint motif analysis thereby facilitates sequence comparisons, which are leveraged in assembly-pileups for comparative analyses of motif structures across individuals, as well as read-pileups for analyzing somatic differences within an individual. The joint analysis of TR motif structures might also open up new avenues for case/control studies to not only take into account repeat length but also repeat structure in a biologically meaningful manner. This could shed new light on the biological and phenotypical impact of TRs.
Methods
MotifScope aims to characterize and visualize motif organization of TRs. It is designed to specifically target TRs that are variable among individuals. Although it works best with genome assemblies, it can be applied to any collection of genomic sequences (e.g., individual long reads) from different technologies (e.g., PacBio, Nanopore).
It should be noted that MotifScope operates on sequences provided by the user, and therefore, it is important for users to provide the target sequences for analysis. MotifScope is primarily for long-read sequencing data as long-read sequencing enables the characterization of the majority of TR sequences. However, it is worth noting that the tool accepts FASTA-formatted files as input, allowing for the analysis of various sequence data types, including reference sequences, genome assemblies, long reads, and potentially short-read sequencing data. It can be run in three modes: assembly mode (by providing sample information) for assessing variations between individuals, reads mode for comparing (somatic) variations within sequencing reads obtained from a single individual, and single-sequence mode (by disabling sequence clustering) for analyzing the motif structure of a single sequence. Optionally, motif discovery can be guided by providing an expected set of motifs (in a TSV file). Finally, MotifScope can be used for performing MSA based on motif composition. MotifScope always outputs the motif composition in a FASTA-formatted file (including each motif and the relative number of copies). Additionally, it provides a tab-separated file reporting the fraction of the sequence covered by each motif. Optionally, MotifScope can generate a visual representation of motif composition across sequences.
Algorithm
Motif discovery and annotation
MotifScope identifies repeat motifs across a given set of input sequences S = {s1, s2, …, sn} based on highly occurring k-mers (see Supplemental Algorithm 1 for implementation details). Due to the repetitive nature of TRs, a single k-mer kx in a tandemly repeated sequence can yield a set of distinct k-mers (Kx) as the repeat motif can be circularly permuted, for example for k-mer kAAG, KAAG = {“AAG,” “AGA,” “GAA”}.
k-mer frequencies for varying lengths of k (the user can specify the maximum size of k-mer to screen) are first computed across all sequences. To that end, input sequences are concatenated into a string (scombined = “{s1}${s2}$…${sn},” and a suffix array and LCP array is computed for scombined using the libsais library (available at https://github.com/IlyaGrebnov/libsais) (Nong et al. 2011). We iteratively walk through the suffix array, while keeping a set of active k-mers and their count. At each position i in the suffix array, the count of active k-mers with a size ≤ LCP[i] is increased by one. Active k-mers with a size > LCP[i] are stored in a result list, accompanied by the count with which they occurred in the sequence scombined.
k-mers that can be represented as repetition of shorter sequences are excluded, e.g., AGAG is removed because it can be viewed as (AG)2. Also, k-mers are not considered if they contain the sequence separation symbol “$” or occur only once.
The final list of these k-mers is then sorted based on k × count so that k-mers that can mask longer sequences will be considered first. Given the set of sorted k-mers K = {ka, kb, kc, …}, MotifScope then iteratively identifies and annotates them across the input sequences (Algorithm 1). In brief, for each iteration, a k-mer kj is selected as the candidate motif, and the maximum continuous masked sequence length lmcs (kj) is determined. This is done for all k-mers, until k × count of the next k-mer is smaller than the maximum value of lmcs that has already been observed for previously considered k-mers. The k-mer with the largest lmcs value is subsequently selected, and masked from the sequence. The masking operation is detailed in Supplemental Algorithm 2.
Motif annotation.

The unmasked sequences are then used as the input for the subsequent iteration to discover the next candidate motif. For the ith iteration (where i > 1), an additional step is taken to provide a canonicalized description of candidate motifs (Supplemental Algorithm 3). For example, if the set of already selected k-mers M = {“TGAGA”}, the next candidate motif, m2, is canonicalized toward TGAGC instead of one of its cyclical rotations GAGCT, AGCTG, GCTGA, or CTGAG. This aims to ensure that these motifs are in a comparable representation, enabling clearer comparison between them. To achieve this, MotifScope considers all cyclical rotations of candidate k-mer mi, and selects the rotation that produces the maximum sum of pairwise alignment scores compared to all previously identified candidate motifs in M. This k-mer is then considered canonicalized and is added to M.
This k-mer selection process stops when the longest remaining sequence ≤1 bp in length or the length of the identified motif is 1 bp. All single occurrences of previously identified candidate motifs in the remaining sequences are then tagged with the corresponding motif as well. The bases that remain uncharacterized are then tagged with the single nucleotide at that position. In this way, each base in the sequences is assigned to one motif.
Clustering and alignment
To effectively compare the patterns of motifs in sequences and to enable further downstream analysis, MotifScope clusters sequences based on their motif organization and length. Hierarchical clustering is subsequently performed on the pairwise distance matrix of these motif sequences.
By default, sequences are first translated into motif sequences, in which each unique motif is assigned a character. Nucleotide sequences are then translated into a “motif sequence” with these characters according to the motif assignments. Next, edit distances between pairs of sequences are calculated using the Levenshtein algorithm.
Alternatively, full MSA is performed. This allows for an aligned representation in the figure, and also provides clustering distances. MotifScope makes use of the partial order alignment algorithm and dual affine gap penalties, as implemented in abPOA library (Gao et al. 2021). MSA can both be performed at the nucleotide level, as well at the level of motif sequences. For motif sequences, in which individual letters represent the motif occurrences, match and mismatch costs are set according to pairwise alignment scores of the motif sequences.
Dimensional reduction of motifs to a color map
MotifScope accurately represents the underlying sequences, and uses a color-based visualization to display TR composition. It supports reflecting motif similarity in a color spectrum such that similar colors correspond to similar motifs. This is achieved by projecting the pairwise alignment score matrix of motifs into a 1D space using Uniform Manifold Approximation and Projection (UMAP) or multidimensional scaling (MDS). It also supports using random RGB colors to represent motifs. Single-nucleotide motifs are colored with grayscale to ensure they are well-separated from multinucleotide motifs.
Benchmarking
We benchmarked MotifScope's ability to identify motifs and accurately represent TR sequences in the context of recently developed methods: uTR, vamos, and TRF. To do so, we used the HG002 genome assembly, and an overlapping set of 5486 TRs between the PacBio repeat catalog (version 0.3.0, available at https://github.com/PacificBiosciences/trgt/tree/main/repeats) and the vamos repeat catalog (based on the exact same start and end coordinates). We also randomly sampled 5000 repeats from the vamos VNTR catalog and compared the performances between these four tools on this set of repeats.
For vamos, several motif databases have been made available by the authors: “vamos original,” which uses motifs identified in samples from the HPRC and HGSVC; and “vamos efficient,” in which rare motifs have been replaced with more common ones while ensuring a bounded total replacement cost (compression strength q = 0.2) (Ren et al. 2023).
The number of motifs discovered was calculated for the 5486 TRs on HG002. MotifScope, uTR, and vamos generate a single representation that can consist of multiple motifs for each sequence: in these cases, all the motifs included in the representation were included. For TRF, which can produce multiple representations each with a different motif, all the motifs reported were included. For the comparisons, all motifs were corrected for cyclic shifts and all motifs were represented with the shortest unit possible: for example, AGAG would be represented as AG.
We calculated the edit distance for MotifScope, uTR, and vamos, between the concatenation of the motif representation of the repeat and the true sequence of the repeat. For example, if a tool annotated a TR as (AGG)3, then the relative motif-derived sequence would be AGG AGG AGG. This sequence was compared to the true underlying sequence. Edit distance was then normalized by the length of the true repeat sequence. Because MotifScope annotates TR sequences through exact matching, the edit distance for MotifScope is always 0 by definition. In the case of TRF, where multiple results were sometimes provided for a single repeat, for each characterization, the edit distances were calculated using the result from TRF (i.e., motif * copy number) and the true underlying sequence of the characterization. These distances were further normalized by dividing by the length of the corresponding parts of the true repeat sequence. The average of these values was used to represent the normalized edit distance for TRF. For the motif overlap between MotifScope and the other tools, the fraction of intersected motifs between MotifScope and another tool over the total number of motifs found by the other tool (A / Method X in Fig. 2C), and the fraction of intersected motifs between MotifScope and another tool over the total number of motifs found by MotifScope (A / MotifScope in Fig. 2C) were calculated for each locus.
Sequencing data
Public sequencing data: Individual PacBio HiFi reads as well as publicly available whole-genome assemblies of the paternal and maternal haplotypes of the HG002 genome were used for benchmarking and read-level analysis (Wang et al. 2022). PacBio-based whole-genome assemblies of the publicly available HPRC samples were also used to assess repeats across individuals (Wang et al. 2022; Liao et al. 2023).
CANVAS patients: The PacBio HiFi sequencing data of a blood sample of a Dutch CANVAS patient was additionally used to evaluate the performance of different tools in a clinical setting based on the RFC1 repeat (van de Pol et al. 2023). The genome of the Dutch CANVAS patient was assembled with hifiasm (version 0.16). In addition, PacBio HiFi sequencing data of a blood sample of another Dutch CANVAS patient was used to show read variability (van de Pol et al. 2023).
Dutch AD patients and cognitively healthy centenarians: PacBio HiFi sequencing data of 246 AD patients and 238 Dutch cognitively healthy centenarians were additionally used for multigenome comparisons of TR motifs. The sequencing data are available through Alzheimer Genetics Hub (https://www.alzheimergenetics.org/). Formal data requests can be submitted via the contact form at https://alzheimergenetics.org/contact/. Additional information about the sequencing and data processing can be found in Salazar et al. (2023). Targeted local assembly of the regions of interest was done on these genomes using otter on HiFi reads (available at https://github.com/holstegelab/otter) (Tesi et al. 2024).
Software availability
MotifScope has been written in Python (version ≥ 3.10). The analyses were done with MotifScope version 1.0.0. The code, documentation, example files, a conda environment, a packaged Docker image, and scripts used in this manuscript are publicly available at GitHub (https://github.com/holstegelab/MotifScope) and as Supplemental Code. Additionally, MotifScope is also available as a web server at https://motifscope.holstegelab.eu.
Competing interest statement
H.H. has a collaboration contract with Muna Therapeutics, PacBio, Neurimmune, and Alchemab. She serves on the scientific advisory boards of Muna Therapeutics and is an external advisor for Retromer Therapeutics.
Acknowledgments
The authors are grateful to all study participants, their family members, the participating medical staff, general practitioners, pharmacists, and all laboratory personnel involved in patient diagnosis, blood collection, blood biobanking, DNA preparation, and sequencing. Part of the work in this manuscript was carried out on the Cartesius supercomputer, which is embedded in the Dutch national e-infrastructure with the support of SURF Cooperative. Computing hours were granted to H.H. by the Dutch Research Council (“100plus”: project# vuh15226, 15318, 17232, and 2020.030; “Role of VNTRs in AD”; project# 2022.31, “Alzheimer's Genetics Hub” project# 2022.38). This work is supported by a VIDI grant from the Dutch Scientific Counsel (#NWO 09150172010083) and a public–private partnership with TU Delft and PacBio, receiving funding from ZonMW and Health∼Holland, Topsector Life Sciences & Health (PPP-allowance), and by Alzheimer Nederland WE.03-2018-07. H.H. and S.v.d.L. are recipients of ABOARD, a public–private partnership receiving funding from ZonMW (#73305095007) and Health∼Holland, Topsector Life Sciences & Health (PPP-allowance; #LSHM20106). S.v.d.L. is the recipient of ZonMW funding (#733050512). H.H. was supported by the Hans und Ilse Breuer Stiftung (2020), Dioraphte 16020404 (2014), and the HorstingStuit Foundation (2018). Acquisition of the PacBio Sequel II long-read sequencing machine was supported by the ADORE Foundation (2022).
Research of Alzheimer center Amsterdam is part of the neurodegeneration research program of Amsterdam Neuroscience. Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting Steun Alzheimercentrum Amsterdam. The clinical database structure was developed with funding from Stichting Dioraphte.
Author contributions: Conceived the study: H.H.; wrote the manuscript: Y.Z., A.S., M.H., N.T., and H.H.; patient selection: N.T., H.H., and E-J.K.; patient blood collection and sequencing: S.W., J.K., L.K., and E-J.K.; data management: N.T., M.H., and S.v.d.L.; bioinformatic analysis: Y.Z. and M.H.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279278.124.
-
Freely available online through the Genome Research Open Access option.
- Received March 8, 2024.
- Accepted October 31, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








