
Deciphering the largest disease-associated transcript isoforms in the human neural retina with advanced long-read sequencing approaches
- Merel Stemerdink1,2,
- Tabea Riepe3,4,
- Nick Zomer4,
- Renee Salz3,
- Michael Kwint4,
- Jaap Oostrik1,
- Raoul Timmermans4,
- Barbara Ferrari5,
- Stefano Ferrari5,
- Alfredo Dueñas Rey6,7,
- Emma Delanote6,7,
- Suzanne E. de Bruijn1,4,
- Hannie Kremer1,2,4,
- Susanne Roosing4,
- Frauke Coppieters6,7,8,
- Alexander Hoischen4,9,
- Frans P.M. Cremers4,
- Peter A.C. ‘t Hoen3,
- Erwin van Wijk1,10 and
- Erik de Vrieze1,10
- 1Department of Otorhinolaryngology, Radboud University Medical Center, Nijmegen 6525 GA, The Netherlands;
- 2Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Center, Nijmegen 6525 GA, The Netherlands;
- 3Department of Medical BioSciences, Radboud University Medical Center, Nijmegen 6525 GA, The Netherlands;
- 4Department of Human Genetics, Radboud University Medical Center, Nijmegen 6525 GA, The Netherlands;
- 5Fondazione Banca degli Occhi del Veneto, Zelarino, Venice 30174, Italy;
- 6Center for Medical Genetics, Ghent University Hospital, Ghent 9000, Belgium;
- 7Department of Biomolecular Medicine, Ghent University, Ghent 9000, Belgium;
- 8Department of Pharmaceutics, Ghent University, Ghent 9000, Belgium;
- 9Department of Internal Medicine and Radboud Center for Infectious Diseases (RCI), Radboud University Medical Center, Nijmegen 6525 GA, The Netherlands
-
↵10 These authors contributed equally to this work.
Abstract
Sequencing technologies have long limited the comprehensive investigation of large transcripts associated with inherited retinal diseases (IRDs) like Usher syndrome, which involves 11 associated genes with transcripts up to 19.6 kb. To address this, we used PacBio long-read mRNA isoform sequencing (Iso-Seq) following standard library preparation and an optimized workflow to enrich for long transcripts in the human neural retina. While our workflow achieved sequencing of transcripts up to 15 kb, this was insufficient for Usher syndrome–associated genes USH2A and ADGRV1, with transcripts of 18.9 kb and 19.6 kb, respectively. To overcome this, we employed the Samplix Xdrop System for indirect target enrichment of cDNA, a technique typically used for genomic DNA capture. This method facilitated the successful capture and sequencing of ADGRV1 transcripts as well as full-length 18.9 kb USH2A transcripts. By combining algorithmic analysis with detailed manual curation of sequenced reads, we identified novel isoforms characterized by an alternative 5′ transcription start site, the inclusion of previously unannotated exons, or alternative splicing events across the 11 Usher syndrome–associated genes. These findings have significant implications for genetic diagnostics and therapeutic development. The analysis applied here on Usher syndrome–associated transcripts exemplifies a valuable approach that can be extended to explore the transcriptomic complexity of other IRD-associated genes in the complete transcriptome data set generated within this study. Additionally, we demonstrate the adaptability of the Samplix Xdrop System for capturing cDNA, and the optimized methodologies described can be expanded to facilitate the enrichment of large transcripts from various tissues of interest.
The human retina is a complex multicellular tissue that plays a crucial role in visual function by converting light into the electrical signals that are interpreted by the brain. Understanding the molecular composition of the human retina, particularly the wide variety of transcript isoforms expressed there, is essential for comprehending disease mechanisms and designing effective treatment strategies for inherited retinal diseases (IRDs) (Braun et al. 2013). A key factor contributing to the diversity of transcript isoforms expressed in the retina is alternative splicing, a biological process that allows a single gene to encode multiple transcript isoforms leading to tissue-specific differences in gene expression and function. This process involves the use of alternative transcription initiation and termination sites, intron retention, exon skipping, and alternative splice donor and acceptor sites. The retina is a highly specialized tissue that is known to be enriched for these tissue-specific alternative splicing events (Cao et al. 2011; Liu and Zack 2013). While previous studies using RNA short-read sequencing provided valuable insights, they often fall short in fully characterizing the diverse array of transcript isoforms present in the human retina (Murphy et al. 2016; Sarantopoulou et al. 2021; Ciampi et al. 2022; Ruiz-Ceja et al. 2023).
The Pacific Biosciences (PacBio) long-read mRNA isoform sequencing (Iso-Seq) technology (Wang et al. 2016) provides deeper insights into the transcriptome complexity caused by alternative splicing events. This technology eliminates the need for de novo transcript assembly, and consequently provides greater certainty in the identification of alternative splicing events, as it can generate full-length transcripts up to 10 kb in length. While the length of the average human transcript is ∼2 kb, many transcripts associated with IRDs are considerably longer, and are, therefore, at (or beyond) the limit of what can be reliably investigated using PacBio Iso-Seq technology. The longest two annotated transcripts of IRD-associated genes are USH2A (18.9 kb) and ADGRV1 (19.6 kb), both associated with Usher syndrome, which is an autosomal recessively inherited disorder characterized by the combination of sensorineural hearing loss and the progressive loss of visual function due to retinitis pigmentosa (RP). The disorder is clinically and genetically diverse, with 11 associated genes that have been identified (MYO7A [Weil et al. 1995], USH1C [Verpy et al. 2000], CDH23 [Bolz et al. 2001], PCDH15 [Ahmed et al. 2001], SANS [Weil et al. 2003], CIB2 [Riazuddin et al. 2012], USH2A [Eudy et al. 1998], ADGRV1 [Weston et al. 2004], WHRN [Ebermann et al. 2007], CLRN1 [Adato et al. 2002], and ARSG [Abad-Morales et al. 2020]). With the exception of CIB2, the full-length isoforms of the Usher syndrome–associated genes surpass the average human transcript length of 2 kb, with five of them exceeding the mean transcript length of RETNET genes (4.7 kb). Although the association of CIB2 with Usher syndrome type 1J (USH1J) has been called into question (Booth et al. 2018), functional studies leave room for the involvement of CIB2 in Usher syndrome (Sethna et al. 2021; Linnert et al. 2023). Therefore, CIB2 is included in our analyses as these results could add to the discussion of whether it qualifies as an Usher syndrome–associated gene.
Obtaining a comprehensive understanding of the Usher syndrome–associated transcript isoforms in the human retina is crucial, as it facilitates the development of therapeutic strategies and enables accurate classification of genetic variants linked to the disorder. To this end, we aimed to provide an overview of the Usher syndrome–associated transcript isoforms in the human neural retina, using the PacBio long-read mRNA Iso-Seq data set from our previous study (Riepe et al. 2024), and supplementing it with two additional data sets aimed at capturing full-length reads from the longest known transcript isoforms. Our previously generated data set was obtained using the standard PacBio Iso-Seq workflow, optimized for transcripts centered ∼2 kb—the average human transcript length—making it suitable for genome-wide studies as we performed in Riepe et al. (2024). However, as nearly all Usher syndrome–associated genes have transcripts exceeding 2 kb, we aimed to generate an additional Iso-Seq data set using an optimized PacBio long transcript workflow to enable the sequencing of larger transcripts up to >10 kb. Despite our efforts, this workflow remained insufficient to enrich for the longest USH2A (18.9 kb) and ADGRV1 (19.6 kb) transcript isoforms. In an attempt to further enhance the capture of full-length transcripts for these two genes, we also employed an “indirect targeted enrichment” approach using the Samplix Xdrop System (Madsen et al. 2020), followed by PacBio long-read sequencing. By integrating the data from these three sequencing workflows and employing a combined strategy of algorithmic analysis and manual curation, we aimed to obtain a comprehensive overview of the Usher syndrome–associated transcript isoforms present in the human neural retina.
Results
Three human neural retina samples were used for PacBio long-read mRNA Iso-Seq, to gain an overview of the Usher syndrome–associated transcript isoforms expressed in the human retina. By integrating the data from three distinct sequencing workflows and combining algorithmic analysis with manual curation (Fig. 1), we obtained a comprehensive overview of the landscape of Usher syndrome–associated transcript isoforms in the human neural retina. Table 1 summarizes which previously annotated Usher syndrome–associated transcripts were identified in the human retina by IsoQuant analysis. Additionally, it highlights frequently observed novel transcript isoforms and events identified by IsoQuant, and validated by Oxford Nanopore Technologies (ONT) long-read mRNA sequencing of independent retina samples. A detailed overview of minor events observed following the manual curation of sequenced reads using the BAM files in the Integrative Genomics Viewer (IGV) is presented in Supplemental Table S1. The findings for MYO7A (Fig. 3), WHRN (Fig. 4), USH2A (Fig. 5), and ADGRV1 (Fig. 6) illustrate the different types of observations in the data set, such as the identification of novel, previously unidentified isoforms, novel coding exons, or regions sensitive to pseudoexon (PE) inclusions. For the remaining Usher syndrome–associated genes, an overview of the algorithmic output and manual curation results has been generated and presented in Supplemental Figures S1–S8.
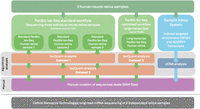
Overview of the sequencing workflows and subsequent analyses. The figure illustrates the sequencing workflows and subsequent analysis performed on RNA extracted from three human neural retina samples. The workflows include PacBio long-read mRNA Iso-Seq using both the standard and an optimized long transcript workflow. The analysis was carried out in three distinct data sets: data set 1 comprised the standard workflow samples analyzed with IsoQuant, data set 2 involved a combined analysis of the reads obtained with standard and optimized long transcript workflows, and data set 3 focused solely on reads obtained with from the long transcript workflow. Additionally, an “indirect targeted enrichment” of transcripts for the USH2A and ADGRV1 genes was achieved using the Samplix Xdrop System, followed by PacBio long-read sequencing and cDNA analysis. All reads mapping to Usher syndrome–associated transcript isoforms were manually curated using BAM files in the IGV. An independent ONT long-read sequencing data set of three independent retina samples was used to validate findings.
Identified Usher syndrome–associated retinal transcript isoforms and prevalent events observed across the majority of transcripts
Exploring Usher syndrome–associated transcript isoforms through Iso-Seq PacBio standard and optimized long transcript workflows
We previously generated a PacBio long-read mRNA Iso-Seq data set from three human neural retina samples (data set 1) for which we conducted a genome-wide integration with proteomic and genomic data to construct the proteogenomic atlas presented in Riepe et al. (2024). Transcripts identified in this data set had an average read length of 2.6 kb. Because this is shorter than most Usher syndrome–associated genes, we optimized the PacBio long transcript workflow to enrich for larger transcript sizes (data sets 2 and 3). Supplemental Table S2 illustrates the successful enrichment for long transcripts as the average subread length was increased while a similar amount of reads was retrieved with both workflows. The optimized PacBio long transcript workflow produced HiFi (CCS3) reads of over 15 kb with an average size of 4.7 kb (Fig. 2A). Transcripts for all 11 Usher syndrome–associated genes were identified in the samples prepared according to both the PacBio standard and long transcript workflow, and the optimized long transcript workflow yielded an increased number of reads from Usher genes with transcripts that exceed the average GENCODE transcript length of 2.4 kb (Fig. 2B; Supplemental Fig. S9). The limited overlap in read length distribution between the data sets underscores the complementary nature of the standard and long transcript workflows (Fig. 2A).
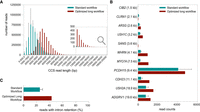
Exploring the Usher syndrome–associated transcript isoform landscape in the human neural retina using PacBio long-read mRNA Iso-Seq. (A) The size distribution of sequenced transcripts derived from the standard workflow (blue) and optimized long workflow (red) data sets. For the standard workflow data set, the mean size distribution across the three sequenced samples is depicted ± standard deviation (SD). (B) Comparison of Usher syndrome–associated transcript coverage between the standard workflow and optimized long workflow data set. The Usher genes are arranged in order from smallest to largest coding sequence, with the coding sequence length of the largest known transcript for each gene provided in brackets. For the standard workflow data set, the mean ± SD transcript length across the three sequenced samples is presented. (C) Quantification of the percentage of reads displaying intron retention in standard workflow samples 1–3 (mean of 3 samples ± SD) versus long workflow sample 4.
In Riepe et al. (2024), the IsoQuant algorithm was used to analyze and categorize the PacBio Iso-Seq standard workflow samples, resulting in data set 1. A second IsoQuant analysis was conducted on the combined reads from the three PacBio standard workflow samples and the retina sample prepared following the optimized long transcript workflow, resulting in the IsoQuant data set 2. However, the incorporation of the PacBio long transcript workflow sample in the combined IsoQuant analysis negatively impacted the number of identified unique full splice match (FSM) transcripts. The increase in the unique novel in catalog (NIC) and novel not in catalog (NNIC) transcripts suggests that many reads are reallocated in the transcript models predicted by IsoQuant. (Supplemental Fig. S10). The long transcript workflow results in an enrichment for intron retention events (Fig. 2C), which we suspect causes the observed difference between the IsoQuant transcript models. This is exemplified in Supplemental Figure S11, showing the IsoQuant transcript models for MYO7A based on data sets 1 and 2, of which only those proposed based on data set 1 were supported by the raw reads. To reliably identify the largest possible transcripts, we reanalyzed the data from the long transcript workflow separately from the standard workflow data (data set 3). We furthermore lowered the consensus read requirement to 0 (CCS0) and manually curated the reads in IGV.
Iso-Seq identifies a novel predominant MYO7A transcript isoform with an alternative 5′ transcription start site
The MYO7A gene encodes the unconventional myosin VIIa motor protein, which is expressed in the outer hair cells of the cochlea (Hasson et al. 1995), the retinal pigmented epithelium (RPE), and in the photoreceptor cells (Liu et al. 1997; Udovichenko et al. 2002). Recent literature describes two human MYO7A transcript isoforms that are expressed in the RPE and the neural retina (Fig. 3A,B), which differ in the length of exon 35 (Gilmore et al. 2023). IsoQuant analysis confirmed the presence of both isoforms in the human retina (Fig. 3A), with the transcript harboring the short form of exon 35 being the most abundant (4.81 ± 4.04 Transcripts Per Million [TPM] for ENST00000458637.6 vs. 0.88 ± 0.86 TPM for ENST00000409709.9) (Fig. 3C), consistent with the findings of Gilmore et al. (2023). Moreover, IsoQuant also identified novel transcript isoforms (transcript20052.Chr11.nic; transript20055.Chr11.nic) characterized by an alternative 5′ transcription start site (TSS) coupled with an extended 3′ untranslated region (UTR), which have not been previously reported in humans. Notably, a similar transcript isoform with a comparable TSS has been identified in mice (Li et al. 2020). Manual curation of sequenced transcripts using the BAM files in IGV revealed substantial read support for this alternative TSS across both the standard and long transcript workflow samples. The use of this alternative TSS was confirmed by ONT long-read mRNA sequencing on three independent retina samples (Supplemental Fig. S12). Protein domain analysis using SMART (Fig. 3D; Letunic et al. 2021) and structural modeling with AlphaFold2 (Fig. 3E; Jumper et al. 2021) revealed differences between the canonical MYO7A isoform and the protein isoform encoded by transcripts with the alternative TSS (transcript20052.Chr11.nic). The canonical isoform encodes a protein of 2215 amino acids, whereas transcript20052.Chr11.nic is predicted to encode a protein of 2245 amino acids, with a larger N-terminal tail and a predicted low complexity region. To quantify the relative expression of these two transcript isoforms, we performed a qPCR using primers specifically targeting the alternative 5′ sequence, canonical 5′ sequence, and canonical 3′ sequence. The results suggest that transcripts produced from the alternative TSS are the most abundant MYO7A transcript isoform in the human neural retina (Fig. 3F). Further manual curation of sequencing reads uncovered skipping or inclusion of certain novel exons, which are summarized in Supplemental Table S1. These splicing events are incidental occurrences, except for the previously reported 5′ truncation of exon 35 and the observed retention of introns 30 and 37 that is found in ∼25% of the sequenced transcripts.
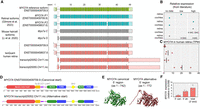
MYO7A transcripts identified by IsoQuant analysis compared to known isoforms from the literature. (A) The GENCODE reference transcript is depicted at the top in green, followed by the known human MYO7A transcript isoforms in blue (Gilmore et al. 2023) and the murine isoforms in gray (Li et al. 2020). The MYO7A IsoQuant transcripts are depicted in red. The light green, blue, gray, and red colors indicate the UTR and the dark green, blue, gray, and red colors indicate the open reading frame (ORF) of each transcript. Differences between the IsoQuant transcript isoforms and the GENCODE reference transcript are highlighted in gray boxes. (B) Relative expression of MYO7A isoforms based on literature in either the retina or the cochlea. (C) The TPM (based on data set 1) for each IsoQuant isoform are presented for the three individual samples. (D) The predicted 2D protein domain architecture of the MYO7A protein isoforms with the canonical 5′ start and the alternative 5′ start from transcript20052.Chr11.nic. The bar below the 2D protein structures displays the amino acid positions. (IQ) isoleucine–glutamine motif, (CC1) coiled-coil domain, (LowC) low complexity region, (MyTH4) myosin tail homology 4, (SH3) SRC homology 3 domain. (E) AlphaFold2 3D protein predictions of the MYO7A protein isoforms, modeled from the 5′ start to the end of the Myosin motor head domain. (F) RT-qPCR analysis of the relative expression of the MYO7A canonical 5′ start site, the alternative 5′ start, and the 3′ end is shown. The locations of the primers for this RT-qPCR are indicated with the arrows on top of the IsoQuant isoforms in Figure 3A.
Iso-Seq reveals a novel coding exon and WHRN retina-specific transcript isoforms
The existing knowledge of WHRN transcript isoforms, encoding whirlin proteins, has predominantly been derived from research on mutant mouse models (Mburu et al. 2003; Belyantseva et al. 2005; Ebrahim et al. 2016). Studies by Mburu et al. (2003) and Belyantseva et al. (2005) have suggested the presence of human retinal WHRN transcript isoforms based on cDNA clones, of which only the full-length isoform (ENST00000362057.4) and a C-terminal isoform (ENST0000674048.1) were verified in the human retina using RT-PCR (Fig. 4A,B; van Wijk et al. 2006). IsoQuant analysis confirmed the expression of the full-length reference isoform of the WHRN gene in the human retina but did not detect the previously reported C-terminal isoform (ENST0000674048.1). However, the IsoQuant analysis did identify an alternative C-terminal WHRN transcript isoform starting with a distinct noncoding exon (ENST00000265134.10), although its expression is relatively low (0.3 ± 0.5 TPM) (Fig. 4C). An N-terminal WHRN isoform was proposed based on the murine Whrn transcripts, but not yet experimentally validated in humans (Mburu et al. 2003; Belyantseva et al. 2005). IsoQuant indeed reveals the presence of the N-terminal transcript isoform ENST00000374057.3 in 2 out of the 3 retinal samples.
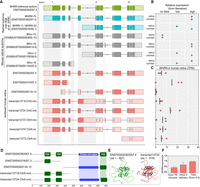
WHRN transcript isoforms identified by IsoQuant analysis compared to known isoforms from the literature. (A) The GENCODE reference transcript is depicted at the top in green, followed by human WHRN transcript isoforms from literature in blue (van Wijk et al. 2006) and the murine transcript isoforms in gray (Mburu et al. 2003; Belyantseva et al. 2005; Ebrahim et al. 2016). The WHRN IsoQuant transcripts are depicted in red. The light green, blue, gray, and red colors indicate the UTR and the dark green, blue, gray, and red colors indicate the ORF of each transcript. Differences between the IsoQuant transcripts and the GENCODE reference transcript are highlighted in gray boxes. (B) Relative expression of WHRN isoforms based on literature in either the retina or the cochlea. (C) The TPM (based on data set 1) for each IsoQuant transcript isoform are presented for the three individual samples. (D) The predicted 2D protein domain architecture of the encoded WHRN protein isoforms. Light blue and green boxes highlight the difference between the WHRN reference isoform and the protein isoform encoded by exon 7B-containing transcript13724.Chr9.nic. (E) AlphaFold2 3D protein predictions of two WHRN isoforms; reference isoform ENST00000362057.4 in green and transcript13724.Chr9.nic in red, with the alpha helix encoded by the novel exon 7B highlighted in blue. (F) RT-qPCR analysis of the expression of the WHRN transcripts containing exon 7B, and WHRN transcripts with intron 4 retention, relative to all WHRN transcripts containing exons 8–9.
Multiple IsoQuant transcript isoforms exhibited intron 4 retention, introducing a premature stop codon, and therefore these transcripts are predicted to encode a truncated protein containing only the first two PDZ domains (Fig. 4D). Manual curation of sequenced reads also confirmed intron 4 retention in the majority of the sequenced reads. Similarly, intron 4 retention was observed in ONT long-read mRNA sequencing data obtained from three independent retinal samples. Quantification of intron 4-containing transcripts using qPCR indicates that 70% of the expressed WHRN transcripts exhibited retention of intron 4 (Fig. 4F). In the absence of a universally present WHRN transcript region, we included a primer pair targeting WHRN exons 8–9 to amplify a segment present in nearly all isoforms, which we designated as “total” WHRN.
Finally, two novel exons were identified within intron 7: designated exons 7A and 7B. While exon 7A was exclusively observed as a noncoding exon and always co-occurred with exon 7B, exon 7B was additionally detected as an independent protein-coding exon in transcript13724.Chr9.nic. Manual inspection of the sequencing data also revealed the widespread presence of exon 7B in the Iso-Seq transcripts, which was further corroborated in an independent ONT long-read mRNA sequencing data set. This 33-nt-long exon 7B is not present in the GENCODE reference annotations for the human WHRN gene but does resemble a corresponding 33-nt exon found in the mouse Whrn consensus sequence. Structural modeling with AlphaFold2 revealed that this novel exon 7B encodes an additional alpha helix of 11 amino acids in the center of the protein (Fig. 4E). A comparison of the TPM levels of the WHRN reference transcript (5.5 ± 3.5 TPM for ENST00000362057.4) with those of the exon 7B-containing transcript (13.6 ± 8.9 TPM for transcript13724.Chr9.nnic), suggests that the latter may represent the predominant retinal isoform. Quantitative PCR confirms that the inclusion of exon 7B occurs in the majority of WHRN transcripts (Fig. 4F). Because biallelic pathogenic variants in WHRN can cause USH2D, we queried whole-genome sequencing (WGS) data of our in-house cohort of unsolved IRD patients (de Bruijn et al. 2023) for rare variants in exon 7B, but found no candidate pathogenic variants in this exon.
Identification of regions prone to PE inclusion and the successful enrichment of full-length USH2A isoform B transcripts using Samplix Xdrop Sort
The USH2A gene gives rise to two distinct transcripts and protein isoforms. Isoform A is the shorter variant, composed of 1546 amino acids encoded by a 21-exon mRNA transcript (Eudy et al. 1998). Conversely, isoform B represents the larger isoform, composed of 5202 amino acids and encoded by a transcript consisting of 72 exons (Fig. 5A,B; van Wijk et al. 2004). IsoQuant analysis confirmed the presence of isoform A transcripts (Fig. 5A). Additionally, our IsoQuant analysis revealed transcripts (transcript51429.Chr1.nnic and transcript51430.Chr1.nnic) that demonstrate significant diversity in the 5′ region. This entails both the use of alternative TSS that are slightly upstream of the annotated TSS, and the use of alternative splice sites in exons 1 and 2. The latter leading to the inclusion of additional amino acids at the 5′ end of the encoded ORF, while maintaining the same reading frame and sharing the same 3′ UTR as isoform A. Notably, this diversity in TSS was also observed in the independent ONT long-read mRNA sequencing data set.

USH2A transcript isoforms were identified by IsoQuant analysis, manual curation, and Samplix Xdrop targeted enrichment. (A) The GENCODE reference transcript is depicted at the top in green, followed by the known human USH2A transcript isoforms in blue (van Wijk et al. 2004). The USH2A IsoQuant transcripts are depicted in red. The light green, blue, and red colors indicate the UTR and the dark green, blue, and red colors indicate the ORF of each transcript. Differences between the IsoQuant transcript isoforms and the GENCODE reference transcript are highlighted in gray boxes. (B) Relative expression of USH2A isoforms based on literature in either the retina or the cochlea. (C) The TPM (based on data set 1) for each IsoQuant transcript are presented for the three individual samples. (D) Proposed USH2A transcript isoforms based on manual curation and Samplix Xdrop targeted enrichment. The GENCODE reference transcript is depicted in green, followed by the proposed USH2A transcript isoforms and events based on manual curation of BAM files using the IGV in red, and the proposed transcript isoform following the Samplix Xdrop targeted enrichment in orange. The light green, red, and orange colors indicate the UTR and the dark green, red, and orange colors indicate the ORF of each transcript. Differences between the proposed transcript isoforms and the GENCODE reference transcript are highlighted in gray boxes. The overview of sporadic incorporation of cryptic exons indicates the presence of PE8 and PE20 as previously described by Reurink et al. (2023). Additionally, locations, where cryptic exons are occasionally incorporated at sites that are not yet associated with deep-intronic pathogenic variants, are indicated with black arrows.
IsoQuant analysis revealed transcript51485.Chr1.nic, spanning exons 1–15, as the most abundant transcript (15.35 ± 2.85 TPM) (Fig. 5C). Manual inspection of the BAM files in IGV confirmed substantial read support for this alternative isoform. However, the presence of a 12-adenine stretch at the 3′ end of this transcript suggests possible internal priming of the oligo(dT) primer. Additionally, its shorter length relative to known usherin isoforms may have resulted in preferential amplification and loading on the PacBio SMRT-cells, potentially explaining its higher TPM values, therefore, raising uncertainty about whether it represents a bona fide transcript isoform or an experimental artifact. Additional N-terminal USH2A transcripts were identified that spanned different exon ranges, including exons 1–9 (transcript 51552.Chr1.nnic), exons 1–26 (transcript51403.Chr1.nic), and exons 1–3 (transcript51634.Chr1.nic), as well as a transcript covering exons 16–21 (transcript 51591.Chr1.nic) (Fig. 5A). The authenticity of these transcripts can be questioned as the PacBio standard workflow returned numerous partial reads, as can be observed in BAM files, and was not able to capture the full-length USH2A transcripts encoding isoform B.
Although also unable to capture the full-length USH2A transcripts encoding isoform B, the value of the optimized long transcript workflow is clearly demonstrated through the manual examination of the sequenced reads (Fig. 5D). While tiling of reads resulting from the PacBio standard workflow samples did not fully cover the complete USH2A isoform B transcript, the reads from the sample prepared following the optimized long transcript workflow together were able to cover all 72 exons of USH2A. These data did not reveal any evidence of natural exon skipping (NES); a phenomenon where specific exons are excluded from the mature transcripts in healthy tissues, as observed for the ABCA4 gene by Tomkiewicz (2024). Furthermore, numerous reads support the presence of isoform A transcripts and the observed variation in the 5′ region. While individual reads do not clearly indicate whether this 5′ variation is specific to isoform A, we did identify reads extending beyond exon 21 that are linked to the variation in the 5′ region, suggesting it may also occur in the larger isoform B transcripts.
Manual curation of sequenced transcripts furthermore revealed sporadic inclusion of intronic sequences, for example, the inclusion of 87 bp of intron 20 that was previously identified as pseudoexon 20 (PE20) by Reurink et al. (2023). In addition to the previously reported USH2A PE8 and PE20, our investigation also revealed the occasional incorporation of other uncharacterized cryptic exons. These cryptic exons are denoted by arrows in the overview provided in Figure 5D, with genomic positions provided in Supplemental Table S3. As demonstrated by Reurink et al. (2023), pathogenic deep-intronic variants can induce the inclusion of PEs harboring an in-frame stop codon across all USH2A transcripts. This prompted us to evaluate WGS data from our in-house cohort of unsolved IRD patients (de Bruijn et al. 2023). However, we did not identify any candidate pathogenic deep-intronic variants surrounding the identified sporadic cryptic exons.
Since both the PacBio and ONT long-read sequencing approaches failed to sequence the full-length USH2A isoform B transcripts, we employed a targeted enrichment approach on cDNA using the Samplix Xdrop Sort technology in an effort to capture these USH2A transcripts. The Samplix Xdrop Sort is a technique designed for single-molecule enrichment of genomic DNA. For the first time, we here demonstrated the utility of this approach to capture cDNA as well. By using three detection sequences targeting either 5′—middle and 3′ sites of the longest known transcript encoding usherin isoform B, we were able to enrich for USH2A transcripts, as evidenced by the qPCR results (Supplemental Table S4). The enriched pool of transcripts was then subjected to HiFi long-read sequencing and subsequent cDNA analysis. Manual curation of the obtained sequencing reads revealed that the detection sequence targeting exons 30–31 enabled us to capture and sequence full-length transcripts encoding usherin isoform B for the first time. Although the cDNA amplification approach associated with this method yields shorter reads, they are tiled along the full-length USH2A transcript encoding isoform B with an average coverage of 73 reads for each of the 72 exons. Because all these reads are from a single capture region and therefore must be connected to this region, this finding provides conclusive evidence for the presence of full-length USH2A transcripts encoding isoform B in the human retina (Fig. 5D). Similar to the reads obtained with the PacBio and ONT workflows, this capture-based sequencing approach also does not indicate the presence of alternative splicing events such as NES.
The 19.6 kb ADGRV1 transcript defies the limits of long-read mRNA sequencing approaches
With the largest annotated transcript spanning 19.6 kb, ADGRV1—previously known as MASS1, VLGR1, and GPR98—is the largest Usher syndrome–associated gene, and variants in this gene are responsible for Usher syndrome type 2C (Weston et al. 2004). Three distinct human ADGRV1 transcript isoforms have been previously reported: VLGR1a, VLGR1b, and VLGR1c (Supplemental Fig. S6). VLGR1b represents the transcript encoding the longest isoform, composed of 90 exons and encoding a protein of 6306 amino acids. Similar challenges to those encountered with the large coding sequence of USH2A were observed for ADGRV1, which prevented the PacBio standard and long transcript workflows and ONT long-read mRNA sequencing, from sequencing the VLGR1b transcript. Furthermore, the IsoQuant analysis did also not identify any transcript isoforms corresponding to the shorter VLGR1a and VLGR1c transcripts, despite their smaller sizes not posing a barrier to sequencing (Supplemental Fig. S6). Manual curation of the sequenced reads revealed that the 5′ extension of exon 65, which defines the start of the VLGR1a transcript, was present in reads across all sequenced samples. Notably, only the sample prepared following the PacBio long transcript workflow contained reads that perfectly spanned from the 5′ extension of exon 65 to exon 90, demonstrating the presence of the VLGR1a transcript in the human retina. Additionally, manual curation of the sequenced transcripts revealed that approximately half of the reads contained the 83 bp truncation at the 3′ end of exon 31, as also observed in the N-terminal VLGR1c transcript isoform. While no sequencing reads spanned exon 1 to exon 31, the observed utilization of the splice site resulting in the 83 bp truncation of exon 31 suggests the presence of the VLGR1c transcripts in the human retina (Fig. 6). Further manual inspection of the PacBio Iso-Seq data, and independent ONT long-read mRNA sequencing data revealed that nearly half of the reads contained an in-frame novel exon situated in intron 39, which has been designated as exon 39A (33 bp in size). We again queried the WGS data of our in-house cohort of unsolved IRD patients to identify whether pathogenic variants could be present in exon 39A, but found no candidate pathogenic variants in this exon.
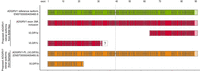
ADGRV1 proposed transcript isoforms from manual curation and Samplix Xdrop targeted enrichment. The GENCODE reference transcript is depicted at the top in green, followed by the ADGRV1 proposed retinal transcript isoforms and events based on manual curation of BAM files using the IGV in red, and proposed transcript isoforms following the Samplix Xdrop targeted enrichment in orange. The light green, red, and orange colors indicate the UTR and the dark green, red, and orange colors indicate the ORF of each transcript. Differences between the proposed transcript isoforms and the GENCODE reference transcript are highlighted in a gray box.
In an effort to capture full-length ADGRV1 (19.6 kb) transcripts encoding the longest isoform VLGR1b, we employed the targeted enrichment approach using the Samplix Xdrop Sort system, by using three detecting sequences targeting the 5′, middle, and 3′ regions of VLGR1b transcripts. qPCR results confirmed the enrichment of ADGRV1 transcripts (Supplemental Table S4). While we cannot conclude that the detection sequences were able to capture the full-length isoform, manual curation of sequenced reads revealed that the 5′ detection sequence (targeting exons 16–17) provided the best coverage, spanning exons 3–77 and 80–90, with an average coverage of 818 reads covering the sequenced exons. This suggests the existence of the full-length ADGRV1 transcript isoform encoding VLGR1b in the human retina. Although the absence of reads mapping against exons 78–79 in the 5′ enriched sample may hint at NES, no reads were found that indicated that exon 77 is immediately followed by exon 80. Furthermore, we did not observe any indication for the skipping of exons 78–79 in the data obtained with the PacBio standard- and long transcript workflows. Finally, data obtained with Samplix Xdrop indicate the presence of an even shorter N-terminal transcript isoform that is similar to murine Vlgr1e (Supplemental Fig. S6).
Discussion
Given the limitations of current sequencing approaches to characterize the largest transcripts expressed in the human neural retina, we conducted PacBio long-read RNA sequencing following the standard library preparation and an optimized workflow to enrich for long transcripts in the human neural retina. Both the standard- and the optimized workflow revealed several novel findings for Usher syndrome–associated transcripts. However, these workflows were insufficient for sequencing the longest two Usher syndrome–associated genes USH2A and ADGRV1, with transcripts of 18.9 kb and 19.6 kb, respectively. Therefore, we employed the Samplix Xdrop System for indirect targeted enrichment of these transcripts, which enabled the successful capture and sequencing of ADGRV1 transcripts as well as full-length 18.9 kb USH2A transcripts.
Our focus on the identification of Usher syndrome–associated transcripts in the human retina enabled a more detailed examination compared to genome-wide studies that rely heavily on advanced tools and algorithms to manage large-scale data. By narrowing our scope to Usher genes, we were able to sift through novel transcripts with greater precision through a combination of algorithmic and manual analysis. For the algorithmic analysis of sequencing data, we used the IsoQuant algorithm, which was shown to have the lowest rate of false positive isoforms compared to alternatives like SQANTI3 and TALON (Prjibelski et al. 2023; Pardo-Palacios et al. 2024). However, integrating the long transcript workflow data set into a combined IsoQuant analysis with the standard workflow data sets posed challenges in isoform classification. This was likely due to the enrichment of intron-retaining transcripts in the long transcript data set. We, therefore, reanalyzed the long transcript data set separately, and combined the IsoQuant analysis with manual curation of sequenced reads to reliably identify the largest possible transcripts.
Our integrated strategy, combining algorithmic and manual analysis, uncovered novel isoforms and alternative splicing events across the 11 Usher syndrome–associated genes, which we highlighted for MYO7A, WHRN, USH2A, and ADGRV1. For instance, we discovered a novel predominant MYO7A transcript isoform. Previous research using a targeted PCR approach had only identified two retinal MYO7A transcript isoforms differing in exon 35 length, with the truncated exon 35 variant being the predominant form (Gilmore et al. 2023). This PCR-based targeted methodology is incapable of identifying isoforms characterized by potentially novel transcription initiation and termination sites. In contrast, the PacBio Iso-Seq allowed us to identify a novel MYO7A transcript with an alternative 5′ TSS. While Gilmore et al. (2023) have suggested that both previously known MYO7A transcript isoforms should be considered when designing gene therapies, our findings indicate that the isoforms with a novel 5′ TSS were the most predominant in the human retina. Notably, the variation in exon 35 length as described by Gilmore et al. is also found in this novel isoform. While we do not dispute the relevance of this variation for therapy development, it might be even more important to incorporate the novel TSS isoform we identified in the design of retinal gene augmentation therapies. Furthermore, the novel, previously unannotated MYO7A 5′ TSS should be incorporated into diagnostic screening pipelines as it may harbor pathogenic variants of potential clinical significance.
Additionally, sporadic intron retention events were observed across all 11 Usher syndrome–associated genes, likely indicating ongoing splicing in these transcripts. The high frequency of introns 30 and 37 retention in the MYO7A transcripts, and the intron 4 retention in WHRN are particularly interesting. While intron splicing does not follow a strict 5′–3′ order or depend on intron length (Pandya-Jones and Black 2009; Singh and Padgett 2009), Gazzoli et al. (2016) demonstrated that inefficient multistep splicing of introns can lead to the formation of “exon blocks”—groups of consecutive exons that tend to be spliced together as a unit. The observed retention of introns 30 and 37 in the MYO7A transcripts may indicate the creation of exon blocks encompassing exons 31–37. The interplay between intron retention and exon blocks may be relevant for the design of antisense oligonucleotide (ASO) therapies as for example proposed for the DMD exon block 45–55 to achieve exon skipping with fewer ASO molecules (Gazzoli et al. 2016). Although multiexon skipping does not seem particularly feasible for transcripts encoding myosin motor proteins such as MYO7A, our findings illustrate the value of including partially spliced transcripts in the analysis of Iso-Seq results. With several ASO-based splicing modulation therapies under development for IRDs, some also based on multiple exon skipping (Schellens et al. 2023), our data set could help uncover exon blocks in other retinal disease-associated genes.
Another notable instance of intron retention was observed in WHRN transcripts, where nearly half of the sequenced transcripts exhibited retention of intron 4. While intron retention is generally thought to disrupt protein production due to the introduction of premature stop codons that trigger nonsense-mediated decay (NMD), Boutz et al. (2015) described a class of intron-retaining transcripts that are polyadenylated, sequestered in the nucleus, and resistant to NMD. These retained introns may undergo subsequent splicing and export to the cytoplasm for translation, potentially enabling cells to rapidly adapt to environmental changes or stress. Although we cannot confirm the nuclear origin of WHRN intron 4 retaining transcripts in our data set, their amplification with an oligo(dT) primer indicates they are polyadenylated mature mRNAs. Alternatively, these intron 4-retaining transcripts may be translated into a truncated whirlin protein, containing only the first two PDZ domains, comparable to the proposed murine N-terminal truncated protein isoforms (Mburu et al. 2003; Belyantseva et al. 2005; Mathur et al. 2015). Unfortunately, antibodies to detect the N-terminus of whirlin are not available to determine if this transcript is translated. Nevertheless, the high prevalence of these transcripts suggests their functional relevance, either through encoding a truncated protein or via mechanisms akin to those described by Boutz et al. (2015).
The PacBio Iso-Seq standard and long transcript workflows were unable to sequence transcripts encoding the currently known largest isoforms of usherin and ADGRV1, but did identify the presence of USH2A transcripts encoding usherin isoform A, and several shorter ADGRV1 transcripts analogous to murine Vlgr1d and Vlgr1e that all exceed the average GENCODE transcript length of 2.4 kb. Moreover, the long transcript workflow provided improved coverage across the largest isoforms of these genes. Manual examination of the USH2A reads revealed the sporadic inclusion of intronic sequences, which we have termed cryptic exons. The occurrence of these cryptic exons may be a result of ongoing splicing, as previous studies using ultra-deep sequencing have demonstrated that recursive splicing can take place in which introns are spliced out in multiple steps which can lead to the generation of a cryptic exon as an intermediate product (Gazzoli et al. 2016). Furthermore, it has been shown that cryptic exons can mark regions susceptible to pathogenic PE-inducing variants (Braun et al. 2013). For example, the sporadic inclusion of 87 bp of USH2A intron 20, previously identified as PE20, was also observed in our data set. Reurink et al. (2023) showed that the c.4397-3890A > G pathogenic variant resulted in consistent inclusion of PE20, containing an in-frame stop codon (p.Ala1465_Ala1466ins*5), across all USH2A transcripts. This example illustrates how deep-intronic variants can lead to the permanent inclusion of potentially pathogenic PEs. An ASO-based splicing correction therapy was developed to target this aberrant PE20 inclusion. This underscores the diagnostic potential of a detailed Iso-Seq data analysis, which could inform the development of targeted diagnostic panels and therapeutic strategies.
Despite using our PacBio long transcript workflow, we were unable to sequence the currently known largest transcript isoforms of USH2A and ADGRV1. We chose to use PacBio Iso-Seq for our study due to its reputation for offering higher sequencing accuracy compared to ONT (Tvedte et al. 2021). While the ONT platform presents advantages, such as adaptive sampling for target enrichment and direct RNA or cDNA sequencing, which may improve transcript capture in future studies, an independent ONT data set from human retina samples also failed to capture the full-length transcripts for USH2A and ADGRV1, highlighting the current challenges faced by both sequencing platforms. Considering these challenges, we used the Samplix Xdrop Sort system for an “indirect target enrichment” (Madsen et al. 2020) on human retina cDNA to enrich for USH2A and ADGRV1 transcripts. This facilitated the successful capture and sequencing of ADGRV1 transcripts as well as cDNA molecules of USH2A encoding the largest isoform B, thereby demonstrating the presence of this complete cDNA molecule (18.9 kb) for the first time. In addition to its original purpose of genomic DNA enrichment, this marks the first demonstration of applying the Samplix Xdrop Sort system to enrich cDNA samples. However, a limitation of this technique is that the captured cDNA molecules require an amplification step using multiple displacement amplification (MDA), which introduces branched transcripts. Consequently, an enzymatic digestion step is necessary before sequencing. Ideally, the workflow would enable full-length amplification of the captured molecules followed by PacBio long-read sequencing, but this remains an area for future improvement.
Collectively, our findings underscore the importance of employing integrated sequencing and analysis approaches to capture the full complexity of the transcriptome in specialized tissues like the human neural retina. Through a comprehensive analysis of Usher syndrome–associated transcript isoforms in the human retina, we revealed a more intricate isoform landscape than previously understood. This analysis led to the discovery of novel isoforms and splicing events, with significant implications for diagnostics and therapeutic development. While functional studies are necessary to validate the transcript isoforms and elucidate their roles in retinal physiology and pathology, our work highlights the power of combining algorithmic and detailed manual analyses to achieve a deeper understanding at the individual gene level. We, therefore, advocate for the utilization of the human neural retina sequencing data sets we generated here for similar comprehensive studies, as these data sets have the potential to yield valuable insights into other genes and significantly advance our understanding of complex transcriptomic landscapes.
Methods
Tissue collection for PacBio Iso-Seq long-read mRNA sequencing
Human donor eyes, deemed unsuitable for corneal transplantation, were used in this study. Written consent was obtained from the donors’ next of kin in accordance with the guidelines of the Italian National Transplant Centre (Centro Nazionale Trapianti, Rome, Italy). The procurement and processing of these tissues followed Italian law and adhered to the principles of the Declaration of Helsinki and the guidelines of the European Eye Bank Association. Three human neural retinal samples were obtained from nonvisually impaired individuals through the Fondazione Banca degli Occhi del Veneto (Venice, Italy). Before this study, we performed WGS on DNA isolated from these retinal samples to screen for and exclude any known genetic variants associated with inherited retinal disorders. The eyes were enucleated within 2–12 h postmortem, and the retinal extraction followed established protocols (Niyadurupola et al. 2011; Osborne et al. 2016). After cornea removal, the eyeball was dissected at the ora serrata; the iris, lens, and vitreous body were removed before carefully detaching the retina from the sclera and retinal pigment epithelium (RPE) by cutting at the optic nerve head. Subsequently, retinal samples were snap-frozen in cryovials using liquid nitrogen and shipped on dry ice. Detailed information about the donors is presented in Supplemental Table S5.
RNA isolation and PacBio Iso-Seq library preparation following the standard workflow
To preserve the integrity of long mRNA molecules, all samples were handled with care during RNA isolation and library preparation, avoiding vortexing or vigorous pipetting to prevent shearing of mRNA molecules. Total RNA was extracted from the human neural retina samples by adding 500 µL of TRIzol reagent to each sample, which was then homogenized in a 2 mL tube containing a sterile glass bead using a TissueLyser (Qiagen, Aarhus, Denmark) in two cycles at 30 Hz for 30 sec. Following a 5 min incubation at room temperature (RT), 100 µL chloroform was added to the samples which were then mixed and incubated for another 3 min at RT before being centrifuged at 12,000g for 15 min. The resulting aqueous phase was collected and mixed with 1 µL glycogen (5 µg/µL) and an equal volume of isopropanol. This mixture was incubated at 20°C for 75 min followed by centrifugation at 12,000g for 30 min at 4°C. Subsequently, the supernatant was removed and the remaining RNA pellet was dissolved in MQ water and further purified and DNase treated using the Nucleospin RNA Clean-up Kit (Macherey-Nagel, Düren, Germany) according to the manufacturer's instructions. The total isolated RNA quantity was measured with a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). Additionally, RNA integrity number (RIN) values were determined using a 2100 Bioanalyzer (Aligent Technologies, Santa Clara, CA, USA). The RIN values of all three samples exceeded 7.0 (Supplemental Table S5), and 300 ng of RNA input was used to generate Iso-Seq SMRTbell libraries following the Iso-Seq-Express-Template-Preparation protocol PN 101-763-800 version 2.0 (Pacific Biosciences, CA, USA). These libraries were prepared using the standard workflow, suitable for samples with a predominant transcript size of ∼2 kb, and the SMRTbell library binding kit 2.1 (Pacific Biosciences). The samples were not labeled with a barcode, and 500 ng of cDNA was used for the subsequent steps in the procedure. The quantification and assessment of the SMRTbell library for each sample were conducted using Qubit (Thermo Fisher Scientific) and an Agilent Bioanalyzer 2100 employing HS RNA screentape. The on-plate loading concentration of the final Iso-Seq SMRTbell libraries was set at 80 pM, and sequencing was carried out on a Sequel IIe system (Pacific Biosciences) with a movie time of 24 h.
PacBio Iso-Seq library preparation following adjusted workflow optimized for large transcripts
In total, 300 ng of RNA sample 2 was used to generate the Iso-Seq SMRTbell library using the “Iso-Seq-Express-Template-Preparation” protocol PN 101-763-800 version 2.0 (Pacific Biosciences). The library was prepared using the PacBio long transcript workflow suitable for samples with a transcript size larger than 3 kb using the SMRTbell library binding kit 2.1. To further enhance the enrichment for large transcript sizes, an additional size selection step was included using diluted AMPure PB beads. This was performed after the standard purification of amplified cDNA for long transcripts (pages 6–7 of protocol PN 101-763-800 version 2) and after performing an additional five PCR cycles as outlined in Appendix 1 of PN 101-763-800 (page 11). Next, instead of the recommended ProNex bead purification (page 12), the additional size selection was carried out using a 3.3× ratio of diluted AMPure PB beads according to the PacBio Procedure “Using AMPure PB Beads for Size Selection” protocol PN 101-854-900 version 2.0 (Pacific Biosciences). The concentration and size distribution of the resulting SMRTbell library was evaluated using Qubit (Thermo Fisher Scientific) and an Agilent Bioanalyzer 2100 with HS RNA screentape. The on-plate loading concentration of the final Iso-Seq SMRTbell libraries was set at 120 pM, and sequencing was carried out on a Sequel IIe system (Pacific Biosciences) with a movie time of 30 h.
PacBio long-read Iso-Seq data analysis
Four PacBio long-read RNA Iso-Seq samples (three samples prepared following the PacBio standard workflow, 1 sample prepared following the optimized PacBio long transcript workflow) were analyzed with IsoQuant (Prjibelski et al. 2023). Three separate data sets were created (Fig. 1): the first data set included the IsoQuant analysis of the three standard workflow samples, like we previously performed in Riepe et al. (2024). The second data set combined the analysis of PacBio standard workflow retina samples 1, 2, and 3 with the retina sample prepared following the optimized long transcript workflow. These two data set used CCS3 reads. The third data set consists only of the long transcript workflow sample, analyzed with less strict filtering settings and using CCS0 reads under the assumption that the longest transcripts might not reach CCS3 as frequently as an average-sized transcript. The sequencing data resulting from the PacBio Iso-Seq approaches that were used for IsoQuant analyses are available via the European Genome-phenome Archive (EGA; https://ega-archive.org) with the identifier EGAD50000000720. Detailed properties of the IsoQuant analyses can be found in the Supplemental Code document.
Data analysis and transcript visualization
RStudio software (v4.3.2) (R Core Team 2021) was used to obtain read and transcript counts from the IsoQuant output files. ggtranscript package (Gustavsson et al. 2022) was used to visualize transcripts from the GTF output files. ORFs were predicted with SnapGene. Details and R scripts are posted on GitHub (https://github.com/erikdevrieze/USH_retina), and can be found in the Supplemental Code document.
Targeted Iso-Seq for USH2A and ADGRV1 transcripts using the Samplix Xdrop Sort
The Samplix Xdrop Sort (Samplix ApS, Birkerød, Denmark) was employed for a targeted Iso-Seq analysis, enriched for USH2A and ADGRV1 transcripts. Of each retina sample, 300 ng of RNA was used for cDNA synthesis using the NEBNext Single Cell/Low Input cDNA Synthesis kit (New England Biolabs). For this purpose, the “Preparing Iso-Seq libraries using SMRTbell prep kit 3.0” protocol 102-396-000 version 2.0 (Pacific Biosciences) was followed until the cDNA amplification step. The synthesized cDNA of all three samples was pooled and the final DNA concentration of the resulting pool was determined using the Qubit with the single-stranded DNA kit (Thermo Fisher Scientific) according to the standard workflow. To perform the indirect target enrichment, single cDNA molecules were packaged in double emulsion (DE) droplets together with detection sequence primers using the Samplix Xdrop Sort. The detection sequence primers were designed to target a 100–150 bp region located in either the 5′, middle or 3′ region of full-length USH2A (ENST00000307340.8) and ADGRV1 (ENST00000405460.9) transcripts, respectively, as to increase the likelihood of capturing full-length transcripts as well as known and novel shorter isoforms. Primer sequences and targets are provided in Supplemental Table S6.
The encapsulation process was performed separately for each of the six dPCR primer sets to ensure the targeting of a single region during enrichment. Samples were run in technical duplicates, and two positive controls for amplification and sorting provided by Samplix were included on each Xdrop DE20 Cartridge (Samplix ApS). The input concentration was 2.8 ng of pooled cDNA, to which 20 µL of DE PCR mix (Samplix ApS), 0.8 µL of the respective forward and reverse dPCR primers, and a volume of nuclease-free water was added, resulting in a sample mix with a total volume of 40 µL per sample. For the positive controls, the Samplix positive control DNA and dPCR primers were employed. To ensure proper DE droplet production, the sample and additional reagents were loaded onto the Xdrop DE20 Cartridge in the following order: Firstly, 300 µL of DE PCR buffer (Samplix ApS) (diluted in a 1:1 ratio using nuclease-free water) was loaded into well #A of the cartridge. Next, 40 µL of diluted DE PCR buffer was loaded on the shelf in well #D. Subsequently, 40 µL of the sample mix was pipetted into well #C. Lastly, 100 µL of DE Droplet Oil (Samplix ApS) was loaded into well #B. The cartridge was sealed using a gasket and placed into the Xdrop Sort, after which the DE20 droplet production program was run. Afterward, the DE20 droplets were collected from well #D and dispensed into four equal aliquots per sample in PCR-tubes, which were then placed in a thermal cycler in which the following program was run: 30°C for 5 sec, 94°C for 3 min, 40 cycles of 94°C for 3 sec followed by 65°C for 30 sec, ending in a hold at 4°C. Amplification takes place exclusively in droplets containing cDNA molecules in which the detection sequence targeted by the dPCR primers is present. The encapsulated DNA can be stained using an intercalating dye, resulting in a stronger fluorescent signal in the DE20 droplets containing a target transcript, which enables the sorting of these droplets using the Xdrop Sort. To do so, the four aliquots of each sample were pooled again and the aqueous phase was removed before adding 1 mL of DE staining buffer (Samplix ApS). Samples were incubated for a duration of 15 min at room temperature in the absence of light. An Xdrop DE20 Sort Cartridge (Samplix ApS) was sealed with the accompanying sorting foil, ensuring a tight seal around all wells. To prevent residual fluorescence from neighboring lanes, the sorting process was performed in two separate steps: first sorting of the uneven lanes took place, followed by a second sorting run for the even lanes, taking along one of the previously mentioned positive controls in each step. Using the provided lane opener, the lanes to be used in the sorting run were punched open and the cartridge was loaded with the sample and additional reagents in the following order: 5 µL of Xdrop Blank Oil Droplets (Samplix ApS) were loaded in well #Out, 600 µL of DE Sorting Buffer (Samplix ApS) (diluted in a 1:1 ratio using nuclease-free water) was loaded in well #B1, 300 µL of diluted DE Sorting Buffer was loaded in well #B2 and lastly 300 µL of sample in staining buffer was pipetted in well #In. The cartridge was sealed using a gasket and left to settle in the Xdrop Sort for 5 min, after which the Sorting program was run for the selected lanes. Thresholds for detection and sorting were determined based on the displayed signal of droplets and background fluorescence. As these values differed per sample, the guidelines for picking a threshold as established in the Xdrop Sort Manual (Samplix ApS) were consulted. Directly after sorting, droplets were removed from the #Out well of the cartridge. After incubation for 5 min at room temperature, the aqueous phase was removed in order to start the process of breaking the droplets to prepare the DNA for the subsequent MDA. The droplets were washed twice using 200 µL of Droplet Sorting Wash Buffer (Samplix ApS), after which all the wash buffer was carefully removed. Subsequently, 20 µL of Droplet Break Solution (Samplix ApS) was added as well as 1 µL of Droplet Break Color (Samplix ApS). After vortexing, the clear break solution phase could be removed, leaving the colored aqueous phase containing the enriched DNA.
Since the DNA amount after sorting was insufficient for direct sequencing, the captured transcripts were amplified using MDA, employing non-sequence-specific primers. Additionally, the Xdrop Sort was used to package single transcripts in single emulsion (SE) droplets to avoid bias toward smaller transcripts. For this purpose, 10 µL of enriched DNA from each sample was transferred to separate PCR-tubes. Furthermore, 1 pg of unenriched cDNA dissolved in 10 µL nuclease-free water was taken along as a positive control, as well as a nontemplate control. To each of the samples, 1 µL of SE MDA enzyme (Samplix ApS), 4 µL of SE MDA mix (5×) (Samplix ApS), and 5 µL of nuclease-free water were added on ice. Subsequently, 20 µL of each sample was loaded onto the Xdrop SE85 Cartridge (Samplix ApS) according to the manufacturer's instructions, after which 75 µL of SE Droplet Oil (Samplix ApS) was administered to each inlet well of the cartridge. The cartridge was then placed into the Xdrop Sort and the SE droplet production program was run, after which droplets were collected in PCR-tubes. All but 1–2 mm of droplet oil was removed from the bottom of the PCR-tubes, which were then placed in a thermal cycler to be incubated at 30°C for 16 h, 65°C for 10 min, and 4°C until the release of DNA from the droplets. To break the SE droplets, 20 µL of Droplet Break Solution is added to each of the tubes, together with 1 µL of Droplet Break Color. After vortexing, the clear break solution phase could be removed, leaving the colored aqueous phase containing the enriched and amplified DNA.
Enrichment validation was performed using qPCR to compare enriched samples with the original, unenriched cDNA pool. The amplified 1 pg of unenriched cDNA and nontemplate control from the MDA served as positive and negative controls, respectively. qPCR primers, designed to target 100–125 bp regions close to the targeted regions of the dPCR primers, were used in technical replicates to negate bias from unintended target capture. Primer compositions and targeted exons are detailed in Supplemental Table S6. Each qPCR reaction contained 1 µL of template DNA, 10 µL of GoTaq 2x Master Mix (Promega Corporation), 1 µL of forward and reverse qPCR primers, and 7 µL of nuclease-free water. Reactions were run on a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific) with the following program: 50°C for 1 sec, 95°C for 10 min, 40 cycles of 95°C for 15 sec, and 60°C for 30 sec, then 95°C for 15 sec, 60°C for 1 min, and finally 95°C for 15 sec.
PacBio library preparation and sequencing—adjusted workflow for Samplix-enriched transcripts
To remove the branched structures present in the amplified transcripts generated through the MDA process, an enzymatic digestion was performed. To do so, the enriched samples were diluted with nuclease-free water to a total volume of 25.5 µL. To each sample, 3 µL NEBuffer 2 (New England Biolabs) and 1.5 µL of T7 Endonuclease (New England Biolabs) were added before incubation at 37°C for 15 min in a thermal cycler. Afterward, 20 µL of TE buffer (pH8) was added to each of the samples and a custom bead cleanup was performed. To do so, 40 µL of AMPure PB beads were resuspended and pelleted on a magnet. After the supernatant had been completely removed the beads were washed with nuclease-free water twice and resuspended in an equal volume of the custom bead buffer consisting of the following components: 20 µL of 1 M Tris-HCL (Sigma-Aldrich), 4 µL 0.5 M EDTA pH8 (Thermo Fisher Scientific), 640 µL 5 M NaCl (Thermo Fisher Scientific), 550 µL 40% PEG8000 (Sigma-Aldrich), and 778 µL nuclease-free water. Of this custom bead suspension, 35 µL was added to each sample and incubated at room temperature for 20 min. Subsequently, the samples were pelleted on a magnet and the supernatant was removed. Samples were then washed with 70% ethanol and dried for 30 sec. Afterward, samples were removed from the magnetic rack and resuspended in nuclease-free water before being incubated for 1 min at 50°C and 5 min at room temperature. After spinning the samples down, the beads were pelleted on the magnetic rack until the eluate was clear and the purified DNA could be collected. A total of 500 ng of MDA-amplified and purified DNA was used to create multiplexed SMRTbell amplicon libraries, following the manufacturer's instructions for the SMRTbell prep kit 3.0 102-359-000 (Pacific Biosciences). If the sample had <500 ng of DNA, linearized plasmids not containing the region of interest were added to spike the samples. The samples were then prepared for sequencing using the Sequel IIe Binding Kit 3.2 (Pacific Biosciences), following the recommended protocols from SMRT Link 11.0.0.146107. Finally, 115 µL of the final mix was loaded per well, and long-read sequencing was performed using the Sequel IIe system (Pacific Biosciences). Following sequencing, the reads were processed following a cDNA analysis: subreads were demultiplexed using lima V.2.5.0, and combined to create a consensus sequence using CCS V.6.3.0. These reads were filtered RQ 0.99 to obtain HiFi reads, and HiFi reads were then mapped along the GRCh38 reference genome using pbmm2 V1.8.0 with the –preset Iso-Seq mode. Instead of performing an IsoQuant analysis, we visualized the data in IGV, because despite the enzymatic digestion following the MDA procedure, debranched reads can still contain multiple fragments of the captured transcript and are incompatible with algorithms such as IsoQuant.
Tissue collection for ONT sequencing
For the independent ONT validation data set, the rest material from human donors (n = 3) without any known or clinical evidence of retinal disease was collected from the tissue banks of either Ghent University Hospital or Antwerp University Hospital. This collection adhered to the ethical standards of the Declaration of Helsinki and received approval from the Ethics Committee of Ghent University Hospital (IRB approval B670201837286). Details about the donors can be found in Supplemental Table S7. The eyes were transported in CO2 Independent Medium (Gibco) before dissection. To preserve RNA integrity and minimize the effects of autolysis, retinas were only harvested from eyes with a total postmortem interval of <20 h. Following a visual inspection to ensure no contamination from the RPE, the neural retinas were either processed immediately for total RNA isolation or snap-frozen and stored at −80°C for later use.
RNA isolation and ONT sequencing
Total RNA was extracted from the postmortem adult human neural retina samples using the RNeasy Mini kit (Qiagen), following the manufacturer's instructions. The extracted RNA then underwent DNase treatment (ArcticZymes, Tromsø, Norway) followed by poly(A) capture. Samples of poly(A) mRNA with sufficient quality (RNA Integrity Value, RIN > 8.0) were used for direct-cDNA library preparation using the SQK-DCS109 kit (ONT), with minor modifications to the supplier's protocol. Each prepared library was then loaded onto a FLO-PRO002 flow cell (ONT) and sequenced on an ONT PromethION device for 72 h. Details regarding the number of reads can be found in Supplemental Table S7.
ONT data analysis of selected genes
MinKNOW version 5.1.0 was used to produce FAST5 files, which were subsequently base-called with Guppy version 6.1.5. The reads were then aligned to the Human Reference Genome GRCh38/hg38 using minimap2 (Li 2021) version 2.24 with the -ax splice flags for spliced alignments. Alignment files were converted to BAM format, sorted, and indexed using SAMtools version 1.15 (Li et al. 2009) to produce the data set investigated here.
Relative expression of MYO7A and WHRN isoforms
Quantitative PCR analysis was conducted to determine the relative expression levels of the different MYO7A- and WHRN transcript isoforms identified in the PacBio Iso-Seq data sets across three human neural retina samples. The same RNA used for preparing the PacBio libraries was employed as the template, with 250 ng of RNA being used for cDNA synthesis using the SuperScript IV Reverse Transcriptase kit (Thermo Fisher Scientific 18090200). Quantitative PCR analysis was performed using GoTaq qPCR Master Mix (Promega), following the manufacturer's protocol. For MYO7A, transcript-specific primers were designed and validated to target the 5′ canonical start site, the 5′ alternative start site, and the 3′ end of the identified MYO7A isoforms, as well as the reference gene GUSB. Amplifications were carried out using the QuantStudio 3 Real-Time PCR system (Applied Biosystems, Waltham, MA, USA), with PCR reactions performed in triplicate. Relative gene expression levels compared to the reference gene GUSB were determined using the 2−ΔCt method.
For WHRN, we designed and validated transcript-specific primers to target isoforms with intron 4 retention and those containing exon 7B. In the absence of a universally present WHRN transcript region, we included a primer pair targeting WHRN exons 8–9 to capture a segment present in nearly all isoforms, designated as “total” WHRN. Expression levels of WHRN transcripts were determined using the 2−ΔCt method, and plotted relative to the expression of the exons 8–9 target representing “total WHRN transcripts” set at 1. The primers used are listed in Supplemental Table S8.
Analysis of protein isoforms
To predict the 2D protein domain architecture of encoded proteins in silico analyses were performed using the SMART online tool (Letunic et al. 2021). 3D protein structures were modeled using AlphaFold (Jumper et al. 2021) using the Google Colab notebook (v1.5.3) with standard settings. The 3D structures were visualized with YASARA (Krieger and Vriend 2014), and structural alignments were conducted using the MUSTANG algorithm (Konagurthu et al. 2006).
Data access
The PacBio Iso-Seq data generated for this study, and the ONT long-read mRNA sequencing data used for validation of selected transcripts and events, have been submitted to the European Genome-phenome Archive (EGA; https://ega-archive.org) under accession number EGAD50000000720. Additionally, genome browser tracks of the analyzed PacBio Iso-Seq data can be accessed at https://genome-euro.ucsc.edu/s/tabeariepe/retina_atlas. The original codes have been made publicly accessible through the GitHub repository (https://github.com/cmbi/Neural-Retina-Atlas and https://github.com/erikdevrieze/USH_retina) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This study was financially supported by Stichting UitZicht (2019-16), CUREUsher, and Stichting Ushersyndroom. Purchase of the Samplix Xdrop Sort Instrument was enabled by an internal Radboudumc technology innovation grant (to A.H.). We thank the R&D department of Samplix ApS for their expert guidance in optimizing the research protocol to enable cDNA capturing. Finally, we also thank the Radboud Technology Center Genomics for the library preparation and sequencing of all samples.
Author contributions: M.S.: Investigation, Visualization, Methodology, Project administration, Writing—original draft. T.R.: Formal analysis, Data curation, Writing—review and editing. N.Z.: Investigation, Methodology, Writing—review and editing. R.S.: Formal analysis, Writing—review and editing. M.K.: Investigation, Writing—review and editing. J.O.: Investigation, Writing—review and editing. R.T.: Formal analysis, Writing—review and editing. B.F.: Resources, Writing—review and editing. S.F.: Resources, Writing—review and editing. A.D.R.: Resources, Writing—review and editing. E.D.: Resources, Writing—review and editing. S.E.B.: Formal analysis, Writing—review and editing. H.K.: Funding acquisition, Writing—review and editing. S.R.: Writing—review and editing. F.C.: Resources, Writing—review and editing. A.H.: Funding acquisition, Methodology, Writing—review and editing. F.P.M.C.: Conceptualization, Funding acquisition, Writing—review and editing. P.A.C.’t H.: Conceptualization, Funding acquisition, Writing—review and editing. E.W.: Funding acquisition, Supervision, Conceptualization, Writing—review and editing. E.V.: Funding acquisition, Conceptualization, Methodology, Supervision, Writing—review and editing.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280060.124.
- Received September 24, 2024.
- Accepted February 11, 2025.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








