
Heterogeneous and novel transcript expression in single cells of patient-derived clear cell renal cell carcinoma organoids
- Tülay Karakulak1,2,3,7,
- Natalia Zajac3,4,7,
- Hella Anna Bolck2,5,
- Anna Bratus-Neuenschwander4,
- Qin Zhang4,
- Weihong Qi3,4,
- Debleena Basu2,
- Tamara Carrasco Oltra4,
- Hubert Rehrauer3,4,
- Christian von Mering1,3,
- Holger Moch2 and
- Abdullah Kahraman3,6
- 1Department of Molecular Life Sciences, University of Zurich, 8057 Zurich, Switzerland;
- 2Department of Pathology and Molecular Pathology, University of Zurich and University Hospital Zurich, 8091 Zurich, Switzerland;
- 3Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland;
- 4Functional Genomics Center Zurich, ETH, 8057 Zurich, Switzerland;
- 5Centre for AI, School of Engineering, Zurich University of Applied Sciences (ZHAW), 8400 Winterthur, Switzerland;
- 6School for Life Sciences, Institute for Chemistry and Bioanalytics, University of Applied Sciences Northwestern Switzerland, 4132 Muttenz, Switzerland
-
↵7 These authors are joint first authors and contributed equally to this work.
Abstract
Splicing is often dysregulated in cancer, leading to alterations in the expression of canonical and alternatively spliced isoforms. We used the multiplexed arrays sequencing (MAS-seq) protocol of PacBio to sequence full-length transcripts in patient-derived organoid (PDO) cells of clear cell renal cell carcinoma (ccRCC). The sequencing revealed a heterogeneous dysregulation of splicing across 2599 single ccRCC cells. The majority of novel transcripts could be removed with stringent filtering criteria. The remaining 31,531 transcripts (36.6% of the 86,182 detected transcripts on average) were previously uncharacterized. In contrast to known transcripts, many of the novel transcripts have cell-specific expression. Novel transcripts common to ccRCC cells belong to genes involved in ccRCC-related pathways, such as hypoxia and oxidative phosphorylation. A novel transcript of the ccRCC-related gene nicotinamide N-methyltransferase is validated using PCR. Moreover, >50% of novel transcripts possess a predicted complete protein-coding open reading frame. An analysis of the most dominant transcript-switching events between ccRCC and non-ccRCC cells shows many switching events that are cell- and sample-specific, underscoring the heterogeneity of alternative splicing events in ccRCC. Overall, our study elucidates the intricate transcriptomic architecture of ccRCC, underlying its aggressive phenotype and providing insights into its molecular complexity.
Alternative splicing is a pivotal mechanism by which eukaryotic cells enhance their transcriptomic and proteomic diversity (Graveley 2001). By allowing a single gene to encode multiple RNA variants, alternative splicing contributes significantly to cellular complexity, tissue specificity (Xu 2002), and organismal adaptability (Verta and Jacobs 2022; Marasco and Kornblihtt 2023). In the context of human disease, notably cancer, dysregulation of alternative splicing events can lead to the expression of oncogenic isoforms, influencing tumor initiation, progression, and resistance to therapy (Sciarrillo et al. 2020; Bradley and Anczuków 2023). Despite its recognized importance, the comprehensive characterization of alternative splicing at the resolution of individual cells remains a formidable challenge, primarily due to the limitations of conventional sequencing technologies in capturing the full spectrum of splicing events.
Recent advances in single-cell RNA sequencing (scRNA-seq) have revolutionized our understanding of cellular heterogeneity in complex tissues and tumoral environments, revealing unprecedented insights into the transcriptomic variations that define cell types, states, and functions (Cha and Lee 2020; Travaglini et al. 2020; Li et al. 2022; Dondi et al. 2023; Yao et al. 2023). However, most single-cell studies have relied on short-read sequencing technologies, which, despite their high throughput, fall short of accurately resolving complex splice variants due to their limited read lengths. Long-read sequencing technologies offer a promising solution to these limitations. With the ability to generate reads that span entire transcript isoforms, long-read sequencing enables the direct observation of splicing patterns and the identification of novel isoforms that would be missed or misassembled by short-read technologies (Bolisetty et al. 2015; Byrne et al. 2017; Amarasinghe et al. 2020). However, long-read sequencing was inappropriate for single-cell transcriptome measurements due to the initial lower throughput and high sequencing errors. With the recent advances in sequencing chemistries and transcript concatenation protocols, the restrictions could be overcome, enabling the measurement of transcripts in full length at single-cell resolution.
Using derivatives of this new technology, the research community has begun to investigate the transcriptome of various samples at single-cell resolution. For example, Shiau et al. (2023) identified a distinct combination of isoforms in tumor and neighboring stroma/immune cells in a kidney tumor, as well as cell-type-specific mutations like VEGFA mutations in tumor cells and HLA-A mutations in immune cells. In a recent study, Dondi et al. (2023) identified over 52,000 novel transcripts in five ovarian cancer samples that had not been reported previously. They discovered cell-specific transcript and polyadenylation site usages and were able to locate a gene fusion event that would have been missed using short-read sequencing.
Clear cell renal cell carcinoma (ccRCC) is the most prevalent form of kidney cancer, characterized by its heterogeneous cellular composition and a complex genetic landscape (Hsieh et al. 2017; Turajlic et al. 2018a). A hallmark of ccRCC is the loss of the von Hippel–Lindau tumor suppressor (VHL) gene through genetic (point mutations, indels, and 3p25 loss) and/or epigenetic (promoter methylation) mechanisms. Loss of the VHL gene can lead to the stabilization of hypoxia-inducible factors (HIFs) and subsequent activation of a hypoxic response even in oxygenated tissue microenvironments. The uncontrolled activation of transcriptional targets that regulate angiogenesis, metabolic pathways, apoptosis, and other processes can drive tumor progression and survival while accelerating clonal evolution and subclonal diversification (Turajlic et al. 2018b).
Here, we have applied Pacific Biosciences' (PacBio's) new multiplexed arrays sequencing (MAS-ISO-seq) protocol (Al'Khafaji et al. 2024) to probe full-length transcriptomic profiles of single cells in patient-derived kidney organoids from four individuals with ccRCC. Cancer-derived organoids serve as an ideal starting material. They closely mirror the biological features of the original tumors, including genetic intratumor heterogeneity (ITH), and provide a renewable source of living cells for analysis (Bolck et al. 2021). Applying long-read scRNA-seq to patient-derived organoids (PDOs) can reveal important insights into the transcriptional landscapes of these important translational models.
Despite extensive research highlighting the role of alternative splicing in ccRCC development and treatment response (Zhang et al. 2021a; Wang et al. 2022), the transcriptome landscape of ccRCC at the single-cell resolution remains unexplored. Given the well-known genetic heterogeneity and complexity of the tumor microenvironment in ccRCC (Turajlic et al. 2018a,b), understanding these processes at the single-cell resolution could reveal critical insight for ccRCC biology. For example, recent single-cell studies have suggested VCAM1-positive renal proximal tubule cells (PTC) to be the likely origin of ccRCC (Zhang et al. 2021b; Schreibing and Kramann 2022), which is consistent with the hypothesis that ccRCC is derived from the proximal tubules. Also, ccRCC tumors were found to detain many CD8+ T cells and macrophages in immune checkpoint inhibition responsive and resistant samples, respectively (Krishna et al. 2021).
Here, for the first time, we explored the transcriptome landscape of ccRCC samples and one matched-normal PDOs in single-cell resolution using single-cell long-read sequencing technology. We aimed to understand the heterogeneity within cells and between samples at the alternative splicing level and identify isoform-switching events in ccRCC cells that could pave the way for novel therapeutic strategies.
Results
Full-length single-cell sequencing reveals transcript diversity and the cell heterogeneity of known and novel transcripts
To discern the transcriptome diversity in ccRCC, we have applied full-length single-cell sequencing using the MAS-seq protocol (Al'Khafaji et al. 2024) on a PacBio Sequel IIe instrument to five PDO samples (Fig. 1A). The PDOs were established from fresh tissue samples obtained from four individuals with ccRCC (Fig. 1B). We included one PDO generated from matching normal kidney tissue from sample ccRCC2. All ccRCC-derived organoids carried a VHL mutation, a hallmark of ccRCC (Table 1). To sequence the single-cell transcriptomes as deeply as possible, we loaded transcript molecules of as few cells as possible on the flow cell. With 29.4 to 58.8 million segmented reads per sample, we sequenced 310–1091 cells and obtained a total of 216,926–346,107 transcripts. The average sequencing depth ranged from 21,499 to 96,620 reads per cell (Table 2). Calculation of the number of unique genes and transcripts and their UMI counts per cell revealed that the ccRCC4 PDO with the highest number of cells had the lowest number of transcripts, genes, and UMI per cell (Table 2; Fig. 1C; Supplemental Fig. 1A).
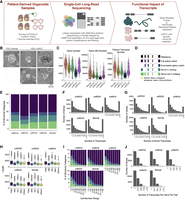
Transcript landscape and cell heterogeneity in Normal and ccRCC-PDOs. (A) Schematic design of the project showing how PDO samples are established, sequenced using single-cell long-read sequencing, and functionally characterized (illustrations were created by BioRender [https://www.biorender.com]). (B) The bright-field representative images of our organoids. The dotted line marks the matched pair. Scale bar, 50 μm. (C) Distribution of the number of genes, UMI counts, and filtered transcript numbers across samples. (D) SQANTI3 transcript categories. (E) The proportion of transcript categories found across four ccRCC-PDO and one normal PDO (see C for the color code). (F) The number of identified transcripts per gene in each PDO. The x-axis denotes the number of transcripts per gene, categorized into bins (1, 2, 3, 4, 5, 6, 7, 8, 9, and ≥10), while the y-axis represents the number of genes. The height of each bar reflects the count of genes that express the corresponding number of transcripts. (G) The number of identified novel transcripts per gene per cell in each PDO. The x-axis shows the number of transcripts detected per gene per cell, categorized into different bins, while the y-axis denotes the total number of genes, with the height of each bar reflecting the count of genes that express the corresponding number of transcripts per cell. (H) Distribution of transcript lengths for each structural category across samples. (I) Proportional distribution of identified transcripts’ structural categories across cell number ranges. (J) Number of transcripts per gene per cell across samples, categorized into bins (1, 2–3, 4–6, 7–9, and ≥10).
Clinical data of PDO samples
The number of HiFi reads, segmented reads, cells, genes, and transcripts, and their structural categories before filtering
The Iso-Seq pipeline in SMRTLink classified transcripts into four categories according to the SQANTI3 classification (Pardo-Palacios et al. 2024a). Based on the alignment profile of exon coordinates of transcripts to the reference transcriptome, SQANTI3 categorized the transcripts as full splice match (FSM), incomplete-splice match (ISM), novel in catalog (NIC), and novel not in catalog (NNC) (Fig. 1D). FSM transcripts perfectly align with reference transcripts at their junctions; ISM transcripts have fewer exons at the 5′ or 3′ ends, while the rest of the internal junctions align with the reference transcript junctions. The novel transcript categories NIC or NNC are made of new combinations of known splice junctions or have at least one new donor or acceptor site, respectively. In addition, SQANTI3 subcategorizes isoforms based on their 5′ and 3′ ends (Pardo-Palacios et al. 2024a). We grouped the remaining SQANTI3 transcripts, namely antisense, genic intron, genic genomic, and intergenic, into a single category called “Other.” Filtering based on the CAGE peak, 3′ and 5′ support, and TSS ratio left on average 86,182 isoforms, of which 31,531 (36.6%) were novel (Table 3). While 37.2% of the transcripts were identified as ISM before filtering (Table 2), this number reduced to 9.31% (Table 3), showing that many ISM isoforms have missing 3′ or 5′ support and are prone to degradation. Filtered isoforms had, on average, 53.7% FSM followed by 36.6% novel transcripts, of which 17.1% and 19.4% were identified as NIC and NNC, respectively (Fig. 1E; Table 3).
The number of cells, genes, and transcripts and their structural categories after filtering
FSM transcripts mainly consisted of the alternative 3′ end subcategory, while ISM transcripts had primarily 5′ ends (Supplemental Fig. 1B). FSM and NIC transcripts tended to be the longest and to have similar lengths on average, with 1338 bp length (t-test P-value = 0.53) (Fig. 1H), while the ISM transcripts showed shorter lengths compared to FSM and NIC (t-test, P-value < 2.2 × 10−16). Across five samples, we detected more than 10 transcripts for the subset of genes, ranging from 11.4% in ccRCC3 to 27.3% in ccRCC2 (Fig. 1F). With ∼8%, the highest percentage of genes with at least 10 or more novel isoforms were detected in ccRCC2 and Normal samples. (Fig. 1G). Even though many genes expressed more than one transcript in a sample, most genes were found to express a single transcript per cell across all samples (Fig. 1J). The result could reflect low transcript diversity across most genes or an artifactual loss of lowly abundant transcripts due to 10 × bead capture or insufficient coverage. On average, ∼55% of transcripts found in only one or two cells were novel, while 93% of the transcripts found in more than 150 cells (representing 14.8% to 44.9% of cells across samples) were FSM (Fig. 1I).
Both known and novel transcripts tended to have a higher expression within a cell if they were also expressed in many cells (Supplemental Fig. 1C,D). The highest UMI counts were found for FSM transcripts compared to other categories (Supplemental Fig. 1E). Of the novel isoforms, 63.3% were found only within one cell of a sample. However, there were some exceptions; for example, the novel transcripts of the genes glyceraldehyde 3-phosphate dehydrogenase (GAPDH), pyruvate kinase (PKM), angiopoietin-like 4 (ANGPTL4), nicotinamide N-methyltransferase (NNMT), and karyopherin subunit alpha 2 (KPNA2) were expressed in at least 30% of the cells of one or more samples (see Supplemental File 1 for the complete list). Among those, GAPDH and PKM are known to be involved in glycolysis, and it is reported that enzymes having a role in glycolysis are upregulated in the occurrence of VHL-deficient ccRCC due to the upregulation in hypoxia inducible factor 1 subunit alpha (HIF1A) (Miranda-Poma et al. 2023). ANGPTL4 is another hypoxia-inducible gene, and its expression has been shown as a potential diagnostic marker for ccRCC (Verine et al. 2010). KPNA2 is overexpressed in many cancers (Sun et al. 2021), including ccRCC, and its knockdown has been shown to inhibit kidney tumor proliferation (Zheng et al. 2021). NNMT is another gene overexpressed in ccRCC, and it was previously characterized as a promising drug target for ccRCC (Reustle et al. 2022). Our findings suggest that those novel transcripts expressed more broadly across cells might play an important role in the pathogenesis of ccRCC.
More than 50% of novel transcripts have translation capability
We predicted the open reading frame (ORF) using TransDecoder (https://github.com/TransDecoder/TransDecoder) to assess whether the novel transcripts are protein-coding. Based on the occurrence of start and stop codons and coding regions, TransDecoder assigned transcripts into varying sub-ORF categories, including 3′ partial (transcripts with missing stop codons), 5′ partial (transcripts with missing start codons), internal (transcripts that miss both start and stop codons), and complete transcripts (including all necessary parts to code a protein) (Fig. 2B). For about 77.9% of the NIC transcripts, we could predict an ORF (Fig. 2A). The highest ORF prediction rate was found with transcripts combining known junctions (Supplemental Fig. 2A). Despite applying our stringent filtering criteria, ISM transcripts remained with the lowest proportion of complete ORFs (Fig. 2C). Transcripts commonly expressed in a sample were significantly more likely to have complete ORFs as compared to cell-specific transcripts (Cochran–Armitage test for trend test: P < 2.2 × 10−16) (Fig. 2D). To understand whether the predicted protein isoforms form a stable protein structure that could hint toward a biological function, we predicted intrinsically disordered regions (IDRs) for all isoforms with complete ORFs using iupred2 (Mészáros et al. 2018). The calculations demonstrated that ISM transcripts had the highest proportion of disordered residues (Wilcoxon rank-sum test, P.adj: ISM-FSM: 8 × 10−13, ISM-NIC: 2.20 × 10−18, ISM-NNC P.adj value: 4 × 10−21) (Fig. 2E) and NNC and NIC transcripts with intron retention showed a higher disordered score than those with a new splice site (Wilcox rank-sum test P.adj value < 2 × 10−16), a combination of known junctions (Wilcoxon rank-sum test, P.adj value: 1.7 × 10−14) or known splice sites (Wilcoxon rank-sum test, P.adj value < 2 × 10−16). Among FSM subcategories, the mono-exon transcripts were predicted to have the highest disorder score (Supplemental Fig. 2B). On the other hand, transcripts with a new combination of a splice site from the NIC category showed the least proportion of disordered regions (Fig. 2F).
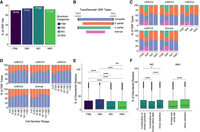
Distribution of ORF categories and intrinsically disordered protein predictions. (A) Percentage of ORF hits across different structural categories in all data sets. For the percentage of ORF hits across isoform subcategories, see Supplemental Figure 2A. (B) ORF types predicted by TransDecoder. (C) The fraction of different ORF types across data sets in each structural category. Each color represents various ORF types. (D) Distribution of ORF types in novel transcripts as a function of cell number range across data sets. The x-axis categorizes the cell number range, while the y-axis shows the proportion of each ORF type (see B for the legends). (E) Comparison of disordered scores for the protein sequences of complete-ORF transcripts across structural categories. (F) Comparison of disordered scores for the protein sequence of NIC and NNC showing complete ORFs across different substructural categories. For the percentage of disordered region scores across FSM subcategories, see Supplemental Figure 2B.
Genes expressing ccRCC cell–specific transcripts participate in ccRCC-related pathways
To evaluate the cell types in our samples, we examined the expression of a ccRCC-specific marker (CA9), a kidney proximal tubule marker (GGT1), and an epithelial cell marker (EPCAM). ccRCC marker CA9 was predominantly expressed in PDO cells from samples ccRCC2, ccRCC4, and ccRCC5 (Fig. 3A; Supplemental Fig. 3B,D,E). As ccRCC originates from the PTC, we also found that nearly all CA9-expressing cells also expressed the PTC marker GGT1 (Supplemental Fig. 3). EPCAM was expressed predominantly in the normal sample and in the PDO cells of ccRCC3 (Supplemental Fig. 3A,C). In addition, we annotated the cells with a manually curated list of genes. Most CA9-expressing cells were found as ccRCC or cells undergoing epithelial-mesenchymal transition (EMT) (Supplemental Fig. 4). The transcript expression profile of ccRCC3 stood out compared to the other ccRCC organoids, as we could not detect CA9 expression in this organoid sample. ccRCC3 had P25L at the VHL mutations (Table 1). This variant was previously described as a polymorphic likely benign mutation, which could explain the VHL-positive-like expression profile of ccRCC3 (Rothberg 2001; Nickerson et al. 2008) lacking overexpression of the VHL–HIF pathway.
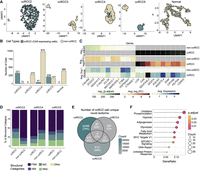
Categorizing cells as ccRCC and non-ccRCC in PDOs. (A) UMAP plot of the ccRCC marker CA9 expression in cells across all PDOs, with darker colors indicating CA9 expression levels. (B) The graph shows the number of ccRCC and non-ccRCC cells in each PDO categorized based on their CA9 expression. (C) The heatmap shows the differential gene expression between ccRCC and non-ccRCC cells. (D) The proportion of expressed transcripts’ structural categories across ccRCC and non-ccRCC cells. (E) Overlap of novel isoforms unique to ccRCC cells only. (F) Overrepresentation hallmark analysis of genes expressing common novel transcripts explicitly in ccRCC cells.
Moreover, to explore the gene and transcript diversity between typical ccRCC and non-ccRCC cells, we categorized cells based on their CA9 expression. CA9 expression results from HIF upregulation due to VHL inactivation (Tostain et al. 2010). ccRCC2, ccRCC4, and ccRCC5 samples contained 321 (96.1%), 41 (4%), and 209 (57%) ccRCC cells with CA9 expression, respectively (Fig. 3B). Differential gene expression analysis between ccRCC and non-ccRCC cells revealed upregulation of several ccRCC-related genes in ccRCC cells, including NDUFA4 mitochondrial complex associated like 2 (NDUFA4L2), lysyl oxidase (LOX), vascular endothelial growth factor A (VEGFA), ANGPTL4, and egl-9 family hypoxia inducible factor 3 (EGLN3) (Fig. 3C). Each of these genes is known to have a role in the progression of ccRCC through various mechanisms. NDUFA4L2 and EGLN3 are critical for the adaptation of ccRCC cells to hypoxic conditions (Wang et al. 2017; Tamukong et al. 2022), VEGFA is a key factor for new blood vessel formations, essential for tumor metastasis, and LOX contributes to ccRCC progression by increasing the stiffness of the collagen matrix, which in turn, facilitates the cellular migration (Di Stefano et al. 2016).
We explored the number of overlapping transcripts to understand the intertumor heterogeneity of alternative splicing between patients. As the PacBio Iso-Seq pipeline assigns transcript IDs randomly, we matched the transcripts based on their exon-boundaries as described before by Healey et al. (2022). Using the Tama tool, we could detect 256,088 unique isoforms across all samples (Fig. 4A), of which 11,283 (from 4746 genes) were shared among all samples. Transcripts that were not unique to a sample were found to be expressed in more cells (Fig. 4B, Wilcoxon test, P-value < 2.2 × 10−16). A comparison of the number of all matched transcripts revealed the highest similarities between Normal:ccRCC5 (Jaccard similarity index: 0.23) and ccRCC2:ccRCC5 PDOs (Jaccard similarity index: 0.23) followed by Normal:ccRCC2 (Jaccard similarity index: 0.22) (Supplemental Fig. 5).
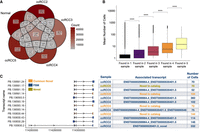
Shared transcripts across samples. (A) The number of overlapping transcripts across all samples. (B) Box plots showing the mean number of cells a transcript was found in a sample on average (Wilcoxon test, P < 2.2 × 10−16). (C) The top four transcripts of NNMT are based on the number of cells across ccRCC2, ccRCC4, and ccRCC5 and their exon structures (left panel). The commonly found novel transcripts from ccRCC2, ccRCC4, and ccRCC5 are depicted in orange, FSM transcripts are shown in blue, and a distinct novel transcript from ccRCC4 is highlighted in yellow. The table next to the transcript structures lists the SQANTI3 categories, aligned reference transcripts, and the number of cells in which the transcripts were identified (right panel).
The comparison of transcripts found explicitly in CA9+ or CA9− cells in each sample revealed 1364 transcripts commonly detected in ccRCC cells of ccRCC2 and ccRCC5 PDOs, 48 transcripts between ccRCC4 and ccRCC5, and 100 between ccRCC2 and ccRCC4 (Supplemental Fig. 6A).
Next, we explored the splicing diversity between ccRCC and non-ccRCC cells. No preference was found for the number of novel transcripts in ccRCC and non-ccRCC cells (Fig. 3D). Explicitly expressed transcripts in ccRCC and non-ccRCC cells showed a diverse structural category pattern (Supplemental Fig. 6B). Nevertheless, we observed 748 novel transcripts from 582 genes commonly found in ccRCC cells (Fig. 3E; see Supplemental File 2 for a complete list of ccRCC-specific novel transcripts) that were primarily associated with ccRCC-relevant pathways, including hypoxia signaling, glycolysis, and oxidative phosphorylation (Fig. 3F). For example, the genes expressing the highest number of common novel isoforms in CA9-expressing cells in the ccRCC5 and ccRCC2 were the NDUFA4L2 gene with 17 novel transcripts, and ANGPTL4 with 11 novel transcripts. Both genes play a role in ccRCC progression, as mentioned earlier.
One of the most frequently found novel transcripts belonged to the NNMT gene. It was categorized as NIC having a combination of known junctions between three exons (Fig. 4C).
PCR validation experiments
We selected two frequently found novel transcripts for PCR validation experiments associated with NNMT and TMEM91. We validated the commonly found novel transcript of NNMT using PCR (see Fig. 4C). The novel transcript of NNMT differed from the commonly found FSM isoform by the presence of 36 nt at the end of exon 2 (Fig. 5A). ORF prediction showed that the candidate novel isoform contained a stop codon at the beginning of the unique sequence. The novel isoform of NNMT has 121 amino acids, corresponding to part of the catalytic domain, NNMT_PNMT_TEMT, compared to the canonical NNMT protein sequence (PDB ID: 3ROD [Peng et al. 2011]) (Fig. 5B). Thus, the novel transcript lacks the complete binding pocket and likely any enzymatic activity. We successfully detected the unique region of the novel transcript using PCR and Sanger sequencing (Fig. 5D,E, NNMT novel; Supplemental Fig. 7B).

NNMT novel transcripts. (A) Reads that align with the NNMT novel transcript. (B) Alphafold3 structure of the NNMT novel isoform aligned to the canonical isoform of NNMT (PDB ID: 3ROD). (C) Representation of the primer design strategy to validate and sequence the novel and canonical transcripts of NNMT. In the novel isoform, the dotted box indicates the position of the unique sequence at the 3′ end of exon 2. (D) Agarose gel (2%) electrophoresis image of PCR validation of different NNMT transcripts. All lanes are marked with corresponding tumor samples and product names. The canonical transcript is amplified with canonical forward primer and reverse primer 1 (see orange arrow). The novel transcript is amplified with a novel forward primer and reverse primer 2 (see green arrow). (E) Sanger sequencing result of NNMT novel transcript. Reverse sequencing confirmed the unique sequence at the novel transcript's 3′ end of exon 2. The splice junction between exon 2 and exon 3 is marked.
Additionally, we could detect the FSM's unique region (NNMT Canonical). It should be noted that there is a known isoform of NNMT in the Ensembl database, ENST00000545255, which contains only two exons and shares 100% sequence similarity with the identified region of novel isoform in Sanger sequencing. However, the ENST00000545255 isoform was not identified in our data, which supports the conclusion that the identified isoform is indeed novel. Note that validating a novel transcript of TMEM91 gave nonconclusive results (see Supplemental Material; Supplemental Fig. 7B).
Most dominant transcript-switching events in ccRCC cells
As alternatively spliced transcripts can have different exons, they may result in different protein domains, disrupt protein interactions, or form interaction with new protein partners. Previous research has shown that most protein-coding genes have one most dominant transcript (MDT) expressed at a significantly higher level than any other transcript of the same gene. These dominant transcripts can be tissue-specific (Gonzàlez-Porta et al. 2013; Ezkurdia et al. 2015; Tung et al. 2022). We previously demonstrated that these MDTs switch during malignant transition in cancer, including in ccRCC (Kahraman et al. 2020). To explore variations in MDT profiles between ccRCC and non-ccRCC cells, we analyzed MDT distribution and their switches between ccRCC and non-ccRCC cells across three PDOs, ccRCC2, ccRCC4, and ccRCC5. The highest number of MDT genes was found for ccRCC5 non-ccRCC cells (Fig. 6A). We identified 7986 unique cancer-specific MDTs (cMDTs) in 571 single cells, ranging between one and 48 switches per cell (Fig. 6B). Most of these switches were found only in one cell in a sample (78.1% in ccRCC2, 99.6% in ccRCC4, 88.4% in ccRCC5, Supplemental File 3). The cMDT was found in three samples (Fig. 6C, left panel). However, 73 genes expressed different cMDT across all CA9+ samples (Fig. 6C, right panel), while 764 genes showed a cMDT in at least two ccRCC samples. Over-representation analysis of these genes revealed functional roles in RNA and mRNA splicing pathways, ubiquitin-dependent protein catabolic process, regulation of mRNA metabolic process, and mitochondrial translation (Fig. 6D). The most frequently found cMDT that was expressed in 115 cells of ccRCC2 was APH1A (Fig. 6E). The APH1A gene encodes for the transmembrane protein Aph-1. This protein is a part of the γ-secretase complex, having a role in the cleavage of various transmembrane proteins, including proteins associated with cancer, such as NOTCH, ERBB4, CD44, VEGFR, etc. (Song et al. 2023). The ccRCC MDT aligns to ENST00000369109.8 with an alternative 5′ end. It had seven exons and encoded 265 amino acids long protein. The non-ccRCC MDT mapped to ENST00000360244.8 with an alternative 5′ end consisting of six exons and encoding for 247 amino acid long protein (Fig. 6E). On GTEx, both isoforms were found to be expressed in high abundance; ENST00000369109.8 is the most abundant isoform in Kidney Medulla, while ENST00000360244.8 is the most abundant transcript in Kidney Cortex (https://www.gtexportal.org). The expression of both isoforms in the cells expressing ENST00000369109 as cMDT and in the normal cells are shown in Figure 6F.
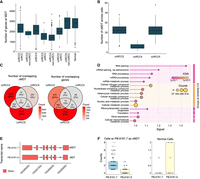
MDTs and MDT switch between ccRCC and non-ccRCC cells. (A) Distribution of the number of MDTs in PDOs. (B) Distribution of the number of cMDT in ccRCC2, ccRCC4, and ccRCC5 PDOs. (C) The number of overlapping isoforms (left panel) and genes (right panel) showing transcript-switching events across three data sets. (D) GO term enrichment analysis of genes commonly shows transcript-switching events in ccRCC2, ccRCC5, and ccRCC4 PDO data sets. (E) Exon structures of ccRCC (PB.8161.7) and non-ccRCC (PB.8161.6) MDTs. (F) UMI counts of cMDT and MDT APH1A transcripts in ccRCC and non-ccRCC cells.
In ccRCC5 PDO cells, the most frequently found cMDT belonged to the gene TMEM161B divergent transcript (TMEM161B-DT), a long noncoding RNA. Higher expression of TMEM161B-DT has been associated with malignancy of glioma cells (Chen et al. 2021), while it was found to be downregulated in oesophageal squamous cell carcinoma (Shi et al. 2021). Here, we identified a cMDT of TMEM161B-DT in 71 ccRCC cells of ccRCC5 PDO (Supplemental File 3). The cMDT had three exons, mapped to ENST00000665319.2 with an alternative 3′ end. In non-ccRCC cells of ccRCC5, we identified diverse MDTs classified as FSM, NIC, or ISM, having three to four exons.
In addition to cMDT analysis, we also applied differential splicing analysis using acorde (Arzalluz-Luque et al. 2022), which revealed three upregulated transcripts in non-ccRCC cells within the ccRCC2 sample that matched MDTs in the Normal sample (Supplemental Table 3).
Discussion
The recent advent of single-cell long-read sequencing technologies provides a unique opportunity to gain insight into intra- and intertumor heterogeneity of tumors and to discover potential novel predictive biomarkers. To reveal the heterogeneity in ccRCC, we utilized PacBio's MAS-ISO-seq single-cell long-read sequencing protocol. We generated a comprehensive catalog of known and novel transcripts for one normal and four ccRCC PDOs. PDOs with the highest number of sequenced cells had the least number of detected transcripts per gene per cell.
Here, we uncovered, on average, 86,182 transcripts across PDOs, of which 36.6% were novel. To interpret the biological impact of these transcripts, we investigated the prevalence across cells and their protein-coding capability. Our analysis revealed that, as expected, conserved well-characterized known transcripts were more widely expressed across cells and samples. In contrast, on average, 63.3% of novel transcripts were found only in one cell. The low detection rate of novel or rare isoforms in our study could potentially be of a technical nature caused by the throughput limitations of PacBio (Al'Khafaji et al. 2024) or by a low bead capture rate of 10x Genomics (Ortolano 2024). Additionally, rare and novel isoforms could have been filtered out as artifacts by the Iso-Seq workflow (Zajac et al. 2024). While the latter would affect any application using 10x Genomics, we tried to address the low throughput of PacBio flow cells by concatenating transcripts using the MAS-seq protocol. We indeed managed to increase the sequencing depth on average by a factor of ∼16, which enabled us to detect transcript expressions for most Illumina-detected genes (Zajac et al. 2024). Thus, the MAS-seq protocol seems to allow the detection of highly abundant transcripts in single cells. Since the selection of transcripts for concatenation and sequencing is stochastic and we detected, on average, only 1.12 transcripts per gene per cell (see Fig. 1J), we believe many of the reported transcripts are the most dominant gene transcript in a cell. It remains to be seen if further improvements to the throughput of single-cell long-read sequencing technologies will increase the number of detected transcripts per gene per cell and reveal additional insights into the heterogeneity of cancer-specific transcript expression.
ccRCC cells tended to have unique novel transcripts in ccRCC-related pathways (e.g., for oxidative phosphorylation, hypoxia, and glycolysis), proposing a contribution to ccRCC cancer progression. Our most dominant switch analysis between ccRCC and non-ccRCC cells revealed many cell-specific and sample-specific switching events. The genes were often part of the mRNA-splicing pathway, highlighting their pivotal role in alternative splicing regulation.
Our study provides insight into the complex and under-explored functional diversity of cells driven by splicing changes in ccRCC. Where possible, we meticulously addressed the issue of potential artifacts and biases introduced by sample processing or data analysis. Despite our efforts to minimize artifacts, some limitations might remain, like artifacts in the PCR amplification, which is an essential step in the MAS-ISO-seq library protocol. Another issue could be the difficulty delineating the actual isoform architecture disguised by transcript degradation, fragmentation, or incompleteness. Iso-Seq addresses the issue via heuristically merging isoforms with differing internal and external junctions. However, the parameters might need to be adjusted based on cell, sample, or tissue types. Lastly, Iso-Seq works on a per-sample basis and provides arbitrary isoform IDs for each sample, prohibiting the matching of identical transcripts between samples. The tool “Tama merge” used in this work can merge identical transcripts based on exon coordinates. Still, it does not consider the sequence identity between the matched transcripts, which can mask some of the isoforms’ sequence diversity. Other tools like Bambu can perform multisample isoform discovery and quantification (Chen et al. 2023) but tend to report fewer novel transcripts (Pardo-Palacios et al. 2024b). The inconsistency in merging identical transcripts raises the question about isoform collapsing parameters, read correction methods, and the extent of evidence required for novel transcript calling. Thus, single-cell long-read sequencing projects like the one presented here should validate their most important findings using molecular validation assays. This aligns with the recent benchmarking studies, the “Long-read RNA-Seq Genome Annotation Assessment Project” (Pardo-Palacios et al. 2024b).
The presented study revealed hundreds of thousands of novel transcripts, of which only the minority are commonly expressed in single or multiple patients, highlighting the intra- and intertumor heterogeneity of ccRCC. The discovery of frequently found novel transcripts provides insights into cancer progression and a new avenue for discovering potential novel biomarkers or therapeutic targets. The functional role of the commonly expressed novel transcripts remains to be further explored and validated.
Methods
Generation and characterization of ccRCC patient–derived organoid samples
Patient tissue samples were provided by the Department of Pathology and Molecular Pathology at University Hospital Zürich. The tissues were collected and biobanked according to previously described procedures (Bolck et al. 2019). The study was approved by the local Ethics Committee (BASEC# 201 9-01 959) and in agreement with the Swiss Human Research Act (Swiss Human Research Act). All patients gave written consent. Organoids were established as previously described (Bolck et al. 2021) (for details, see Supplemental Materials).
Full-length single-cell isoform sequencing and data processing of PDO cells via MAS-ISO-seq
Cell culture media was removed to obtain single-cell suspension, and PDOs from one well of a ULA 6-well plate were collected in ice-cold Cell Recovery Solution (Corning) and incubated for 1 h at 4°C to resolve the Matrigel. Subsequently, PDOs were dissociated with TrypLE by incubation on a thermal shaker set to 37°C, 300 rpm. Every 2 min, the samples were picked up and mechanically dissociated by pipetting up and down. The progress of dissociation was evaluated under a microscope using a small fraction of the cells and trypan blue. After dissociation, PBS, supplemented with 20% FBS, was added to stop the reaction. Samples were centrifuged at 1000g for 5 min, and the supernatant was aspirated. The pellet was washed once in 1× PBS with 0.04% BSA and filtered through a 70 µm strainer. Finally, cells were counted and diluted to the target cell concentration using PBS with 0.04% BSA. Cell viability and concentration were determined using a LUNA-FX7 Automated Cell Counter (Logos).
Generation of full-length cDNAs with 10x Genomics platform and PacBio MAS-seq library preparation and sequencing
10x Genomics Chromium platform was used to analyze the dissociated organoid cells (Zheng et al. 2017). We targeted the recovery of 700 cells per library preparation to have a greater sequencing depth using the PacBio platform. Library preparation was conducted following the 10x Genomics Single Cell 3′ Reagent Kits v3.1 (Dual Index) User Guide. The single-cell full-length cDNAs were directed for single-cell MAS-seq library preparation using the MAS-seq 10x Single Cell 3′ kit (PacBio). Template switch oligo (TSO) priming artifacts generated during 10x cDNA synthesis were removed in the PCR step with a modified PCR primer (MAS capture primer Fwd) to incorporate a biotin tag into desired cDNA products, followed by capture with streptavidin-coated MAS beads. TSO artifact-free cDNA was then further directed to incorporate programmable segmentation adapter sequences in 16 parallel PCR reactions/samples followed by directional assembly of amplified cDNA segments into a linear array. The obtained 10–15 kb fragments were subjected to DNA damage repair and nuclease treatment. The quality and quantity of the single-cell MAS-seq libraries were assessed with Qubit 1× dsDNA High Sensitivity Kit (Thermo Fisher Scientific) and pulse-field capillary electrophoresis system Femto Pulse (Agilent), respectively. Each single-cell MAS-seq library was used to prepare the sequencing DNA-polymerase complex using 3.2 binding chemistry and further sequenced on a single 8 M SMRT cell (PacBio), on Sequel IIe sequencer (PacBio) yielding in ∼2 M HiFi reads and ∼30 M segmented reads per sample. Please see Supplemental Material for short-read sequencing.
SMRTLink Iso-Seq pipeline
We utilized the “Read Segmentation and Iso-Seq workflow” from SMRTLink version 11.1 to process our long-read sequencing data. For two specific samples, Normal and ccRCC2, we combined the data from three SMRTcells to enhance coverage. Within the pipeline, the HiFi reads were converted into segmented reads using the Skera tool, followed by processing with the Iso-Seq for removal of cDNA primers and barcode and UMI tags, reorientation, trimming of poly(A) tails, cell barcode correction, real cell identification, and PCR deduplication via clustering by UMI and cell barcodes. The reads were then aligned to the human genome (GRCh38.p13) using pbmm2. We verified the presence of the selected 10x cell barcodes using the GenomicAlignments R package (Lawrence et al. 2013).
Full-length single-cell data analysis
Isoform filtering
After mapping, isoforms were collapsed into a unique set of transcripts with Iso-Seq using the default options, setting –max-fuzzy-junction to 5 bp, –max-5p-diff to 1000 bp, –max-3p-diff to 100 bp, –min-aln-coverage to 0.99, –min-aln-identity to 0.95, –max-batch-mem 4096, and –split-group-size to 100. In addition, at the Iso-Seq collapse step, reads that mapped chimerically or mapped with low identity were filtered out. The pigeon make-seurat function was run on the remaining reads to generate the gene count matrices. Subsequently, the pigeon was used to classify the unique isoforms into SQANTI3 classification categories (Pardo-Palacios et al. 2024a). After isoform classification, pigeon filtered out intrapriming (with accidental priming of adenine stretches in the genomic position downstream from the 3′ end), RT switching (reverse transcriptase template switching), and low coverage/noncanonical isoforms (having noncanonical splice junctions).
In addition to the pigeon-based filtering, we manually filtered transcripts based on their transcription start site (TSS) ratio, their distance to the gene's TSS and transcription termination site (TTS), and their distance to the gene's CAGE peak (data available through SMRTLink v11.1, also available here: https://reftss.riken.jp/datafiles/3.3/human/). We calculated the TSS ratio using Illumina short reads as an input to SQANTI3's stand-alone sqanti3_qc.py function (run with default parameters and skipping ORF prediction) and discarded any 5′ end-degraded transcripts. BAM files used as input from SQANTI3 QC were generated by mapping Illumina short reads to the human reference genome (GRCh38.p13) as part of the CellRanger (v7.2.0) workflow. We used different filtering criteria for each SQANTI category: FSM isoforms: TSS ratio > 1; ISM, NNC, NIC, and other isoforms: TSS ratio > 1, distance to CAGE peak ≤ 50 bp, and distance to the gene's TTS and TSS ≤ 50 bp. All other isoforms were discarded for downstream analysis.
The isoform count matrices were generated with the pigeon make-seurat function on the filtered isoforms with default parameters. Reads mapping to mitochondrial and ribosomal genes were not retained during isoform and gene count matrix generation. Additionally, we only kept the cells with mitochondrial content <30% for the downstream analysis. All subsequent analyses were conducted using the high-confidence filtered isoforms.
Transcript types and their prevalence across cells
We calculated the percentage of structural categories and their length in each sample using filtered scisoseq_classification.filtered_lite_classification.txt files. We then checked transcript prevalence across varying cell number ranges and the number of transcripts per gene and cell using R version 4.2.1 (R Core Team 2021).
Functional annotation of long-read sequencing transcripts
ORFs were identified on long-read transcript sequences listed in FASTA files from the Iso-Seq collapse function using TransDecoder v5.7.1 (https://github.com/TransDecoder/TransDecoder). The TransDecoder.LongOrfs function predicted all possible ORFs with a length ≥100 nt. To calculate protein sequences from the predicted ORFs, an extensive human reference database containing 226,259 canonical and alternatively spliced isoform protein sequences was generated using UniProt (release date: 2023-11). The predicted ORFs were aligned to this database via BLASTP (Camacho et al. 2009), setting the e-value to 1 × 10−5. In addition, hmmscan v3.4 was applied to predict potential Pfam domains using the Pfam database (release date: 2023-09-12) with a maximum e-value of 1 × 10−10. The results from hmmscan (Eddy 2011) and BLASTP were used to predict the final ORFs using the TransDecoder. Predict function. We then selected one ORF for each transcript based on the highest score assigned by TransDecoder. We applied iupred2a (Mészáros et al. 2018) on the transcripts having complete ORFs to predict their IDRs. A residue was annotated as ordered or disordered if its iupred2a score was below or above 0.5, respectively. We calculated the percentage of disordered residues for each transcript and assigned a percentage disordered score.
Transcript matching among samples
Due to Iso-Seq assigning transcript IDs randomly, we first converted all sqanti_classification.filtered_lite.gff files to BED format using bedparse's gtf2bed function. A “geneID;TranscriptID” column was added to the BED file (Healey et al. 2022). Tama's tama_merge.py function combined all transcript IDs among samples using their exon and junction coordinates. Mismatches up to 50 and 100 nt from the 5′ and 3′ ends, respectively, and mismatches of 5 nt from any exon junction were accepted. The similarities of the samples were calculated in R using the Jaccard similarity matrix, i.e., the number of overlapping transcript IDs divided by the total number of transcripts found in two samples. The heatmaps were visualized using the pheatmap function (https://CRAN.R-project.org/package=pheatmap) in R (R Core Team 2021), and the number of overlapping transcripts was plotted by UpsetR's upset function (Conway et al. 2017).
Cell-type annotations
Seurat (Hao et al. 2024) was used for quality control and integration of the samples using the output files of the Iso-Seq make-seurat function. For gene-level analysis, each sample was normalized by the SCTransform function (Hafemeister and Satija 2019). Three thousand features were selected using SelectIntegrationFeatures, and anchors for integration were identified with FindIntegrationAnchors. The samples were integrated with the IntegrateData function using the SCT normalization. Subsequently, the PCA and UMAP analyses were performed using the RunPCA and RunUMAP functions, respectively. Markers for each cluster were defined using the PrepSCTFindMarkers and FindAllMarkers functions. To categorize the cells in each PDO, we analyzed the samples separately. SCT normalized gene expression matrices were scaled, and the cells were classified into two categories using the scGate R package (Andreatta et al. 2022) by defining the CA9 as a ccRCC positive marker. The other cells were assigned as non-ccRCC. We used the SCpubr R package to visualize marker expressions and clusters (Blanco-Carmona 2022). Genes expressing ccRCC-specific novel transcripts in ccRCC cells of ccRCC2, ccRCC4, and ccRCC5 were analyzed using ClusterProfiler's enricher function (Yu et al. 2012). For the analysis, a hallmark gene set from MSigDB was used as the background gene set (Liberzon et al. 2015). An overrepresentation analysis was performed, setting pValueCutoff = 0.05, qvalueCutoff = 0.1, and pAdjustMethod = BH (Benjamini-Hochberg). In addition, we annotated cells using manual curation of ccRCC and kidney-related markers (see Supplemental Material) using sc-Type (Ianevski et al. 2022).
Most dominant transcripts switch between ccRCC and non-ccRCC cells
We used transcript UMI counts to assess MDTs and cMDTs in our ccRCC samples. Each MDT was required to have at least two times higher UMI counts than the second most abundant transcript (Kahraman et al. 2020). Orphan transcripts of genes were automatically counted as MDT. cMDT were computed based on the comparison of MDTs between ccRCC and non-ccRCC cells using the following strict rules:
-
- cMDT are unique to ccRCC cells.
-
- A distinct MDT of the same gene exists for at least 20% of non-ccRCC cells.
-
- If an MDT and potential cMDT were mapped to the same transcripts within the sample, cMDT was discarded.
-
- UMI counts of MDTs in ccRCC cells should be higher than the mean of the MDTs’ UMI count in non-ccRCC cells.
A cMDT was identified when an MDT switch event fulfilled all criteria. STRINGdb (Szklarczyk et al. 2025) was used for the enrichment analysis of genes showing MDT switches between ccRCC2, ccRCC4, and ccRCC5 PDOs. The human gene list was used as a background for the enrichment analysis. ggVennDiagram R package (Gao et al. 2021) generated a Venn diagram of overlapping cancer-specific MDTs among samples. Exon structures of the transcripts were generated with the ggtranscript R package (Gustavsson et al. 2022).
In addition, a differential isoform expression analysis was performed between ccRCC and non-ccRCC cells in each sample using the acorde software (Arzalluz-Luque et al. 2022). The software calculates cell-level weights for each isoform using ZinBWaVE R package (Risso et al. 2018), followed by performing differential expression with DESeq2 (Love et al. 2014) and edgeR (Chen et al. 2016).
Visualization of NNMT reads and structural modeling
The protein structure of the novel NNMT isoform was modeled using AlphaFold3 (Abramson et al. 2024) based on its ORF sequence. The structure was rendered with PyMOL (https://www.pymol.org). Sequence reads were visualized using Gviz (Hahne and Ivanek 2016) and GenomicRanges (Lawrence et al. 2013).
PCR validations
To validate the isoforms using PCR, we targeted two novel isoforms of two genes, NNMT and TMEM91. We selected these novel isoforms based on their frequency and presence across samples. The novel isoform of NNMT was classified as NIC by SQANTI3. It was found in all samples (IDs: PB.100830.44 in ccRCC2, PB.139561.14 in ccRCC5, PB.136593.16 in ccRCC4, PB.130901.11 in Normal). The novel isoform of TMEM91 was classified as NNC by SQANTI3. The transcript was identified predominantly in CA9 expressing ccRCC cells of ccRCC2 and ccRCC5 PDOs.
For the PCR experiment, total RNA was isolated directly from corresponding frozen tissue samples of ccRCC2, ccRCC3, ccRCC4, and ccRCC5 using Maxwell RSC simplyRNA Tissue (Promega AS1340). Following the manufacturer's protocol, 500 ng RNA was used to synthesize cDNA by qScript cDNA Synthesis Kit (Quanta bio 95048-100). Synthesized cDNA was used as a template for PCR amplification. To capture novel and canonical isoforms, we designed three types of primers against the novel transcripts (Fig. 5C):
-
- Common_primer: targeting sequences shared in both canonical and novel isoforms
-
- Canonical_primer: targeting sequences unique to canonical isoform
-
- Novel_primer: targeting sequences unique to novel isoform
The details of the primer design are mentioned in Supplemental Material. All the primers were designed using Primer3 software (Untergasser et al. 2012) and synthesized by Microsynth AG. The details of primer sequences and primer pair combinations are listed in Supplemental Tables 1 and 2, respectively. PCR amplification was performed using AmpliTaq Gold DNA Polymerase (Applied Biosystems 4311806) following the manufacturer's protocol. The amplified products were subjected to agarose gel electrophoresis (2%) and visualized with GelRed (Biotium 41003-1). The PCR products were further purified by the MinElute PCR Purification Kit (Qiangen 28006) and validated by Sanger sequencing at Microsynth AG.
Data access
The raw single-cell long-read RNA sequencing data generated in this study have been submitted to the European Nucleotide Archive (ENA: https://www.ebi.ac.uk/ena) under the accession number PRJEB73513. The codes used to generate the figures can be found at Zenodo (https://zenodo.org/records/14697181) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Mark Robinson from UZH for valuable discussions about single-cell data analysis. We also would like to thank Harini Lakshminarayanan and Adriana Von Teichmann from the Department of Pathology and Molecular Pathology of USZ for their technical assistance while preparing PDO samples for the single-cell analysis. This work was funded by Krebsliga Zurich and the EMDO Foundation.
Author contributions: A.K. and T.K. conceptualized the study, T.K. and N.Z. conducted data analysis and interpretation, and created visualizations. H.A.B. generated and characterized the organoids. A.B.N., Q.Z., and T.C.O. performed full-length single-cell isoform and short-read sequencing experiments. N.Z. and W.Q. processed the sequencing data using SMRTLink. H.A.B. and D.B. carried out PCR validation experiments. H.R., C.v.M., H.M., and A.K. provided funding and conceptualized the study. T.K., H.A.B., N.Z., and A.K. critically interpreted the results, contextualized the findings, and shaped the study's narrative. A.K. was actively involved in all aspects of the study. All authors contributed to the manuscript writing. T.K., N.Z., H.A.B., and A.K. revised and edited the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.279345.124.
-
Freely available online through the Genome Research Open Access option.
- Received March 15, 2024.
- Accepted February 20, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








