
Assessing DNA methylation detection for primary human tissue using Nanopore sequencing
- Rylee Genner1,2,9,
- Stuart Akeson3,9,
- Melissa Meredith4,9,
- Pilar Alvarez Jerez1,5,
- Laksh Malik1,
- Breeana Baker1,
- Abigail Miano-Burkhardt6,
- CARD-long-read Team1,
- Benedict Paten4,
- Kimberley J. Billingsley1,6,10,
- Cornelis Blauwendraat1,6,10 and
- Miten Jain1,3,7,8,10
- 1Center for Alzheimer's and Related Dementias, National Institute on Aging and National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA;
- 2Department of Biology, Johns Hopkins University, Baltimore, Maryland 21218, USA;
- 3Department of Bioengineering, Northeastern University, Boston, Massachusetts 02115, USA;
- 4Department of Biomolecular Engineering, University of California Santa Cruz, Santa Cruz, California 95064, USA;
- 5Department of Neurodegenerative Disease, UCL Queen Square Institute of Neurology, University College London, London WC1N 3BG, United Kingdom;
- 6Laboratory of Neurogenetics, National Institute on Aging, Bethesda, Maryland 20892, USA;
- 7Department of Physics, Northeastern University, Boston, Massachusetts 02115, USA;
- 8Khoury College of Computer Sciences, Northeastern University, Boston, Massachusetts 02115, USA
Abstract
DNA methylation most commonly occurs as 5-methylcytosine (5mC) in the human genome and has been associated with human diseases. Recent developments in single-molecule sequencing technologies (Oxford Nanopore Technologies [ONT] and Pacific Biosciences [PacBio]) have enabled readouts of long, native DNA molecules, including cytosine methylation. ONT recently upgraded their Nanopore sequencing chemistry and kits from the R9 to the R10 version, which yielded increased accuracy and sequencing throughput. However, the effects on methylation detection have not yet been documented. Here, we performed a series of computational analyses to characterize differences in Nanopore-based 5mC detection between the ONT R9 and R10 chemistries. We compared 5mC calls in R9 and R10 for three human genome data sets: a cell line, a frontal cortex brain sample, and a blood sample. We performed an in-depth analysis on CpG islands and homopolymer regions, and documented high concordance for methylation detection among sequencing technologies. The strongest correlation was observed between Nanopore R10 and Illumina bisulfite technologies for cell line–derived data sets. Subtle differences in methylation data sets between technologies can impact analysis tools such as differential methylation calling software. Our findings show that comparisons can be drawn between methylation data from different Nanopore chemistries using guided hypotheses. This work will facilitate comparison among Nanopore data cohorts derived using different chemistries from large-scale sequencing efforts, such as the NIH CARD Long Read Initiative.
DNA methylation is an epigenetic mechanism that most commonly involves the addition of a methyl group to the fifth carbon of a cytosine residue to form a 5mC (5-methylcytosine) complex. This reversible reaction is catalyzed by DNA methyltransferases (DNMTs) and is positively correlated with the recruitment of histone proteins that compact the DNA structure, making it inaccessible to the transcription machinery. As a result, transcription levels can undergo repression leading to a silencing effect of the associated gene. The dominant form of DNA methylation in terminally differentiated human cells occurs proximal to a guanosine residue, and the resulting loci are often referred to as CpG sites. These CpG sites cluster within promoters to form high methylation frequency regions known as CpG islands (Bird 1986). Methylation differences can impact transcription levels. Methylation patterns can also be inherited and play an important role in cellular processes such as embryo development (Monk et al. 1987; Li et al. 2018), genomic imprinting (Li et al. 1993; Suzuki et al. 2007; Court et al. 2014), X-Chromosome inactivation (Sharp et al. 2011), and transcription repression (Moore et al. 2013). As a result, variations in DNA methylation have been associated with human diseases such as aging, neurodegeneration, and cancer (Lunnon et al. 2014; Maschietto et al. 2017; Gasparoni et al. 2018; Smith et al. 2018, 2019; Altuna et al. 2019; Lardenoije et al. 2019; Semick et al. 2019; Wei et al. 2020).
The quality and throughput of Oxford Nanopore Technologies (ONT) long-read sequencing has rapidly improved over the past decade. The median alignment identity has increased from 85% in 2014 (R9 ONT and chemistry) to 99.7%+ in 2023 (R10 Nanopore Duplex chemistry). Throughput has also increased to the point where sequencing a 30×+ genome is routinely achievable using a single PromethION flow cell (Kolmogorov et al. 2023). While these improvements present promising opportunities for future genomics studies, they pose certain challenges for ongoing, multiyear sequencing projects. The NIH Intramural Center for Alzheimer's and Related Dementias (CARD) is in the process of sequencing thousands of brain tissue samples from donors with and without Alzheimer's disease (AD). The first sample cohort was sequenced in 2023 using ONT R9 chemistry and kits. However, all subsequent samples are being sequenced with the updated R10 chemistry and kits.
The combination of a new Nanopore flow cell configuration, updated chemistry, and improved basecalling software algorithms have yielded key improvements in accuracy, especially in homopolymer-rich regions (Sereika et al. 2022; Kolmogorov et al. 2023). This resulted in documentable improvements in resolving haplotypes, structural variants (SVs), and single nucleotide variants (SNVs). Additionally, a custom R10 pore-specific methylation calling model was released. ONT stated that the training for the methylation detection model—which identifies changes in the ionic current created by modified and unmodified bases moving through the pore—was more comprehensive and robust with R10 chemistry (Ni et al. 2023b). While some of the improvements between the R9 and R10 chemistry-derived data have been published (Kolmogorov et al. 2023), differences in methylation detection have not been thoroughly documented by the academic community.
In this work, we computationally evaluated methylation calls for a cell line, a brain sample, and a blood sample that were sequenced using both R9 and R10 ONT chemistries. We also used Pacific Biosciences (PacBio) long-read sequencing and Illumina bisulfite sequencing data for the cell line to benchmark methylation calls using orthogonal data. This work has been largely motivated by the fact that some of the CARD samples have been sequenced with R9 chemistry and others with R10. Given that many of these samples are limited in quantity, it is important to validate how differences in chemistry and sequencing modalities could affect methylation calling and downstream analyses. Such validations could be used for assessing and implementing strategies for switching over to newer chemistries as technology improves. It can also help devise analysis strategies for comparing variant calling and methylation calling information from different ONT chemistries.
Results
ONT sequencing improvements with R10
We first compared sequencing data for the established Genome in a Bottle (GIAB) human cell line HG002 (also referred to as GM24385). In our recent work, we sequenced DNA from this cell line using a protocol optimized for the R9.4.1 chemistry and analyzed the data using a custom analysis pipeline for ONT data called NAPU (Kolmogorov et al. 2023). These data had 42× genome coverage and 28 kb read N50. We then sequenced DNA from the same cell line using the R10.4.1 chemistry. The resulting data had 45× genome coverage and 29 kb read N50. We performed benchmarking analyses using these cell line-derived data. Additionally, we sequenced two primary tissue samples (a brain and a blood sample) using both R9 and R10 chemistries to further assess performance.
We basecalled these data using Guppy v6.3.8 and then aligned them relative to the GRCh38 reference genome using minimap2 (Li 2018; see Methods). The median alignment identity was 95.05% for the HG002 R9 data set and 98.72% for the HG002 R10 data set (Supplemental Table S1). We then performed variant calling using DeepVariant and Sniffles2 (Smolka et al. 2024). The R10 data we analyzed had demonstrable improvements in variant calling relative to R9 (Supplemental Tables S2, S3), which was in agreement with what was previously documented (Kolmogorov et al. 2023).
Comparing methylation calls between R9 and R10 ONT data
We extracted methylation information from the aligned data using Modkit (https://github.com/nanoporetech/modkit) with the extended BED file format and collapsed strands (see Methods). This resulted in 99.22% and 99.09% of the ∼29.17 million CpG sites in GRCh38 being represented by the HG002 R9 and R10 data sets, respectively. We then filtered the HG002 data set to only include CpG sites that had a minimum coverage of 20× and maximum coverage of 200×. This filtering resulted in 25,937,319 CpG sites (88.92% of sites represented in GRCh38) in R9 data and 27,021,032 sites (92.63% of sites represented in GRCh38) in R10 data (Supplemental Table S4). Some of the discrepancy may be explained by the difference in overall coverage in the two data sets (Supplemental Fig. S1).
We first compared CpG sites that had been detected in all of the ONT-generated HG002 data sets (R9, R10, and whole-genome bisulfite sequencing [WGBS]). Because previous studies have revealed the questionable quality of some WGBS techniques and data sets (Ji et al. 2014; Olova et al. 2018), we compared the ONT WGBS data set to an extensively validated HG002 WGBS data set generated as part of an epigenomics quality control (EpiQC) study (Foox et al. 2021). Details about the generation of this data set, and links to the files used, are included in the Methods section. Our comparison showed that the ONT bisulfite data set was highly correlated with the EpiQC data set (Pearson r methylation frequency correlation = 0.969, root mean squared error [RMSE] = 9.367) (Supplemental Fig. S2) and had significantly higher coverage than the EpiQC data set (26.03× and 92.95× mean coverage levels for the EpiQC and ONT bisulfite data sets, respectively) (Supplemental Fig. S3). Because of the higher coverage, we continued our analysis with the ONT-generated bisulfite sequencing data set.
For the methylation comparison, we binned the CpG sites based on their bisulfite methylation frequencies (reads with 5mC at the site of interest/total reads at the site of interest) that were classified into a combination of 5% and 10% methylation frequency intervals (see Methods). Methylation proportions at both the low (0%–10% for bisulfite) and high (90%–100% for bisulfite) end of the spectrum saw a significant shift (R10 < R9 for 0%–10% bisulfite, Mann–Whitney U test P < 1.12 × 10−16, R10 > R9 for 90%–100% bisulfite, Mann–Whitney U test P < 1.12 × 10−16) in distributions between R9 and R10 proportions (Fig. 1A).
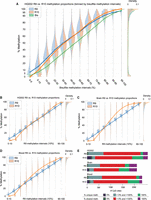
Overall comparison of DNA methylation calls between R9 and R10 data sets across the HG002 cell line, blood, and brain samples. (A) Methylation proportions of R9 (orange) and R10 (blue) data for the HG002 cell line when binned by bisulfite (green) methylation intervals. The portions of R9 and R10 methylation distributions that agree with the bisulfite methylation range are highlighted in green. (B–D) The violin plots underneath show methylation proportions of R9 and R10 data for the HG002 cell line, brain sample, and blood sample binned according to R9 intervals. Each violin plot has lines connecting the median interval points for better visualization of methylation trends. Distributions of CpG site methylation frequencies are depicted on the right side of each panel. (E) Stacked bar charts showing the breakdown of total CpG sites per technology per sample. These sites are further subset into CpG sites with 0% methylation frequency and 100% methylation frequency (Supplemental Tables S5, S6, S8–S11).
We next compared R9 and R10 performance in the HG002 cell line, brain sample, and blood sample. Each CpG site that we identified in our initial filtering step had a coverage of at least 20 reads and had been detected in both the R9 and R10 data sets. We binned the sites based on their R9 methylation proportion and made kernel density estimator (KDE) plots to compare distributions between R9 and R10 methylation proportions in each bin. We selected a 0.1 bin size (10% methylation intervals) to minimize the effect of coverage-based aliasing on our visual analysis. Methylation proportions at both the low (0%–10% for R9) and high (90%–100% for R9) end of the spectrum saw a significant (Mann–Whitney U test P < 1.12 × 10−16 for 0.0–0.1 R9 > all R10, Mann–Whitney U test P < 1.12 × 10−16 for 0.9–1.0 R9 < all R10) shift in distributions between the two chemistries (Fig. 1B–D; Supplemental Fig. S4). The HG002 R9 and R10 data sets shared 25,521,492 CpG sites after filtering. The majority of R9 positions did not have the same complete methylation status in positions where R10 had either 0.0 or 1.0 methylation proportions. In contrast, the majority of R10 positions had 0.0 or 1.0 methylation proportions when R9 achieved 0.0 or 1.0 methylation proportion, respectively (Fig. 1E; Supplemental Tables S5, S6). Relaxing the stringency and counting positions within 0.05 of 0 and 1 as the extremes had the predicted effect of bringing R9 and R10 much closer together by raw counts (Supplemental Table S7).
Characterizing R9 versus R10 methylation differences
Since R10 ONT data have demonstrated an improved resolution of homopolymers compared to R9, we evaluated if this impacted methylation calling. We defined homopolymer regions using the GRCh38 low complexity BED file made available by the Global Alliance for Genomics and Health (GA4GH) Benchmarking Team and the NIST Genome in a Bottle Consortium (https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/genome-stratifications/v3.1/GRCh38/LowComplexity/GRCh38_AllHomopolymers_gt6bp_imperfectgt10bp_slop5.bed.gz). This BED file defines 3,819,657 homopolymeric regions covering 83,977,437 bases. The intersection between homopolymer regions from the HG002 R9 and R10 BEDMethyl files resulted in 296,660 and 314,448 sites with ≥20× and ≤200× coverage, respectively. Of those sites, 290,592 were shared across both chemistries with a Pearson r methylation frequency correlation of 0.966 (RMSE = 10.08). Supplemental Figure S5 shows more positions being identified as 0% or 100% methylated in R10 data relative to R9 data, concordant to what we had documented for the whole genome. We also compared methylation frequency in R9 and R10 data for all cytosines genome-wide, in documented promoter regions, and in CpG islands, and observed a similar pattern (Fig. 2A–F).
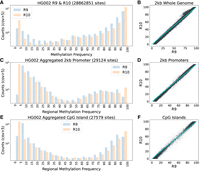
Genome-wide and regional methylation calling trends in HG002 R9 and R10 data sets. (A) Methylation calls for all CpG motifs (strand combined) in HG002 R9 and R10. (B) Scatterplot of 2 kb whole-genome windows with a minimum of 10 CpGs for R9 and R10. The 0–100 diagonal line is overlaid in cyan for reference. (C,D) Average methylation over the 29,124 filtered human promoters with a minimum of 10 CpGs plotted as a histogram and a scatter plot. (E,F) A histogram and scatter plot of the CpG island BED regions with more than 10 CpGs in HG002. All plots are limited to CpG sites with >5× read coverage. The y-axis is log scaled.
We explored the differences in protein coding gene promoter regions (Dreos et al. 2017) in HG002 as measured by R9 and R10. To capture the methylation profile around these gene promoters, we expanded the regions to 2060 bp and calculated average methylation using the BEDTools map function. We then filtered these expanded promoter regions to include those with a minimum of 10 CpGs and a minimum read coverage of 5×. We calculated a Pearson r correlation of 0.998 (RMSE = 4.586) for the 29,124 filtered promoters (Fig. 2C,D). Promoters were generally hypomethylated in HG002. The HG002 R10 data set had 13,073 promoters with <10% methylation while the HG002 R9 data had 8461 promoters in this range. On average, we found a difference of 4.14% in average methylation frequency in these promoters between R9 and R10. In the 27,579 CpG islands passing the CpG quantity and read coverage filters, the Pearson r methylation frequency correlation was 0.998 (RMSE = 5.120) (Fig. 2E,F). The CpG islands had sufficient CpG sites within their boundaries and did not require expansion. Overall, these regional analyses exhibited the same methylation trends that were documented genome-wide.
To further discern the methylation calling differences between R9 and R10, we explored alternative hypotheses. We first examined the impact of strandedness on methylation calling differences and documented no influence (Supplemental Fig. S6). We then tested if the shift in proportion we observed was an artifact due to either sample preparation or the sequencing experiment. To that end, we tested an HG002 Ultra-long data set that was sequenced in a different laboratory on a different PromethION device. This comparison yielded the same observation as we had seen from the data generated at CARD (Supplemental Figs. S7–S11). We tested if variation in genome coverage between the R9 and R10 data sets contributed to methylation calling differences and found that this was not the case (Supplemental Fig. S12). We then tested if the observed difference in methylation proportions could be attributed to the number of CpG motifs in a set window size. We did not notice any discrepancies in either a 100 or 1000 base window (Supplemental Fig. S13). Finally, we examined the covariance of methylation call confidence in reads with at least 20 overlapping CpG sites and noted that R10 had a broader covariance distribution (Supplemental Fig. S14).
We also compared genome-wide CpG site methylation proportions for HG002 data sets generated using the ONT platform (R9 and R10), PacBio platform (HiFi data), and Illumina platform (bisulfite sequencing data; typically considered to be a “gold standard”) (Fig. 3A; Supplemental Fig. S15). We noted that all of the sequencing technologies had good concordance (Pearson r ≥ 0.9, RMSE < 0.2) with fluctuations occurring in the extrema of the methylation proportions (Fig. 3B,C). Based on these comparisons, ONT R10 and Illumina bisulfite sequencing had the highest overall correlation (r = 0.967384 in Illumina mappable regions from BisMap, RMSE = 0.0814881) while ONT R9 and PacBio had the lowest correlation (r = 0.903295, RMSE = 0.153743) (Fig. 3D,E). When comparing R9 and R10, we noted that R9 tended to call the extremes of methylation proportions (0% and 100%) with less frequency than R10. This same phenomenon was observed in varying degrees in each technology that was compared to R10.
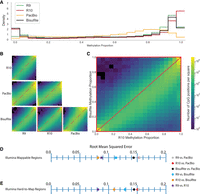
Comparison of HG002 immortalized cell line methylation calling between technologies. (A) Methylation proportion histograms for each technology. (B) Pairwise site-specific CpG methylation proportion comparison between technologies. (C) Site-specific CpG methylation proportion comparison between R10 and bisulfite sequencing. (D) RMSE values for pairwise comparisons between technology in Illumina 150 bp paired-end mappable regions as defined by BisMap. (E) RMSE for pairwise comparisons between technologies in Illumina 150 bp paired-end hard-to-map regions.
The low correlations associated with PacBio data may be due to the fact that the PacBio HG002 reads were processed using Modkit, which was created and optimized for extracting methylation information from Nanopore sequencing data. PacBio's long-read sequencing technology uses a fluorescence-based long-read sequencing approach that is mechanistically different from ONT's voltage-driven approach and comes with its own optimized methylation detection packages. Benchmarking experiments for one such package, called ccsmeth, involved comparing methylation data from HG002 samples that were sequenced/methylation-called by PacBio/ccsmeth, ONT/Deepsignal2 (R9 chemistry), and Illumina bisulfite sequencing/Bismark. Pairwise comparisons of 5mC genome-wide methylation levels revealed a correlation of 0.9287 for ONT/Deepsignal2 and PacBio/ccsmeth (15 kb insert size) and 0.9463 for Illumina/Bismark and PacBio/ccsmeth, which is improved from our correlations of 0.9033 (PacBio/Modkit and ONT/Modkit) and 0.9201 (PacBio/Modkit and Illumina/Bismark). These findings indicate that PacBio methylation calls are more reliable than our data may suggest, especially when paired with optimal processing packages (Cheung et al. 2023; Ni et al. 2023a).
Additionally, we used Integrative Genomics Viewer (IGV) (Robinson et al. 2011) to visualize methylation patterns in the HG002 cell line between ONT methylation calls (for both R9 and R10 data sets) and traditional bisulfite sequencing in constitutively methylated and constitutively unmethylated regions (Supplemental Fig. S16; Edgar et al. 2014). These examples suggest that the methylation detection differences between chemistries do not hinder their ability to draw qualitative conclusions in a conservative hypothesis context.
We phased ONT reads using PEPPER-Margin-DeepVariant (Shafin et al. 2021) and compared R9 and R10 data for haplotype-specific differential methylation using NanoMethPhase (Akbari et al. 2021) which uses the R package DSS (Feng et al. 2014). We applied NanoMethPhase to calculate differentially methylated regions (DMRs) between R9 and R10 haplotype-phased HG002 samples. More DMRs were identified in R10 data than in R9 data, and the number of CpG sites in each DMR was similar (Supplemental Tables S12, S13). The average difference in methylation proportion was higher for R10. This supports our observation that R10 calling the extremes of methylation more frequently than R9 was also contributing to the downstream identification of DMRs.
Methylation comparison for primary human blood and brain tissue samples
We wanted to assess if ONT data derived from human primary tissue exhibited similar patterns between R9 and R10 chemistries. To that end, we sequenced a human brain sample and a human blood sample using the protocol developed at CARD (see Methods). The data from the brain sample had an average genome coverage of 39× (read N50 30 kb) for R9 and 56× (read N50 26 kb) for R10 chemistry. The data from the blood sample had an average genome coverage of 39× (read N50 34 kb) for R9 and 36× (read N50 37 kb) for R10 chemistry, respectively. The median alignment identity for these samples was 95.2% (R9) and 98.52% (R10) for brain-derived and 95.3% (R9) and 98.55% (R10) for blood-derived data (Supplemental Table S1).
We extracted methylation information for the brain and blood samples in the same fashion as HG002 (see Methods). For the brain sample, this resulted in 98.78% and 98.73% of the ∼29.17 million CpG sites in GRCh38 being represented by R9 and R10 data, respectively (Supplemental Table S4). Filtering for 20× coverage or higher resulted in 23,271,407 CpG sites (79.78% of sites represented in GRCh38) in R9 data and 27,742,379 sites (95.11% of sites represented in GRCh38) in R10 data. Of those sites, 23,148,718 overlapped (79.36% of sites represented in GRCh38). For the blood sample, we observed 98.12% and 98.07% of the GRCh38 CpG sites for R9 and R10, respectively. After filtering, R9 had 22,347,084 CpG sites (76.61% of GRCh38 CpG sites) and R10 had 25,723,371 CpG sites (88.18% of GRCh38 CpG sites). Of those sites, 20,977,914 overlapped (71.92% of GRCh38 CpG sites). We observed a similar pattern of methylation proportions across chemistries in the primary tissue samples (Supplemental Fig. S17).
We wanted to assess if there was variation in the identification of haplotype-specific methylation between R9 and R10 data sets. This required a strategy to preserve the phasing information between the two data sets because the assignment of haplotype tags is performed at random by PEPPER-Margin-DeepVariant. To overcome this limitation, we merged the BAM files for R9 and R10 data sets for the HG002 cell line and applied PEPPER-Margin-DeepVariant (using settings for R9 chemistry) to perform the phasing. We then separated the merged and phased R9/R10 haplotagged BAM into phased R9 and R10 BAM files by filtering for the original R9 and R10 read names. This preserved phase 1 and phase 2 haplotag assignments between the two data sets for downstream comparison. We used Modkit to estimate methylation frequencies of the CpG sites and performed differential methylation analysis using the NanoMethPhase DNA module. We used the Methylartist package to visualize haplotype-specific methylation differences associated with a 75 bp deletion on Chromosome 16 in the R9 and R10 HG002 cell line data sets (Fig. 4A), the R10 HG002 cell line, blood, and brain sample data sets (Fig. 4B), and previous R10 HG002, HG02723, and HG00733 GIAB cell line sample data sets (Supplemental Fig. S18; Kolmogorov et al. 2023). We extracted CpG sites that were detected by both R9 and R10 in the DMR of the HG002 cell line (plotted in Fig. 4A) and compared their haplotype-specific methylation frequencies. The methylation frequencies of the 52 CpG sites shared by R9 and R10 in haplotype 1 had an RMSE value of 6.413, and the methylation frequencies of the 113 CpG sites shared R9 and R10 in haplotype 2 has an RMSE value of 5.775. We also visualized haplotype-specific methylation differences in the R10 HG002 cell line, blood, and brain samples in the imprinted GNAS region on Chromosome 20 (Supplemental Fig. S19).
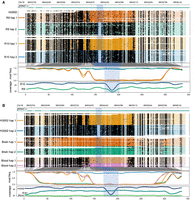
Haplotype-specific methylation differences and similarities between cell, blood, and brain samples. From top to bottom, each plot shows the genome coordinates, labeled gene models (if present), haplotype-aware read mappings with modified bases as black (methylated), or colored (unmethylated) circles, a smoothed methylation fraction plot, and a coverage plot. The highlighted region corresponds to a 75 bp deletion (Chr16:88,534,247–88,534,321) in haplotype 2 of the HG002 cell line that coincides with haplotype-specific methylation. Coordinates across the bottom refer to methylation bins used in the smoothed methylation plot. (A) Haplotype-specific methylation differences and similarities between the R9 and R10 sequenced HG002 cell line. (B) Haplotype-specific methylation differences and similarities between the R10 sequenced cell line, blood, and brain samples.
Comparing methylation calls between Guppy and Dorado basecalling platforms
In 2023, Nanopore released an upgraded basecalling software called Dorado. Key features of this new software included a new architecture that uses graphics processing units (GPUs) instead of central processing units (CPUs) for calling methylated bases. Additionally, the technology switched to a new storage-optimized raw data file format called POD5 (from FAST5 in older iteration). A benchmarking study comparing Guppy v6.4.8 and Dorado v0.2.4 and v0.3.0 on the Amazon Web Services (AWS) platform using an R10 sequenced 30× human genome sample found that Dorado outperformed Guppy in all instance types, including run time and performance with and without methylation calling. More specifically, Dorado outperformed Guppy by a factor of 3.8× with regard to 5-Hydroxymethylcytosine (5hmC) calling (2023).
Many researchers in the genomics field have already begun to transition from Guppy to Dorado, and the brain and blood tissue samples at NIH CARD are now being basecalled with Dorado. This change necessitates the characterization of potential methylation calling differences in these two models, particularly if methylation results from both models are to be combined in analyses. To assess this, we compared an HG002 cell line that had been basecalled with R10 Dorado v0.3.4 (with 5mC and 5hmC modifications called) and R10 Guppy v6.3.8 (with 5mC modifications called). Results revealed that Dorado and Guppy 5mC calls were comparable, particularly at the extremes (Supplemental Fig. S20). We also looked at the proportions of 5mC and 5hmC calls. Almost all of the 5hmC methylation frequencies were in the 0%–20% methylation range, with the majority of calls reporting 0% methylation frequency. This matches previous findings showing that 5hmC levels are generally low in human tissues and are particularly sparse when measured in human cell lines (Supplemental Fig. S21; Li and Liu 2011; Cui et al. 2020). This study was limited to analyzing 5mC calls from the Nanopore data sets because of the presence of orthogonal data sets (e.g., bisulfite sequencing data). We anticipate that our analysis can extend to 5hmC once additional validation is performed using an orthogonal 5hmC truth set for the HG002 cell line.
Discussion
There are several large-scale human genome projects underway in the United States and across the world. One of them is being led by the NIH's CARD and is known as the CARD Long Read Initiative (LRI). Researchers involved with CARD LRI have developed protocols designed to streamline and automate the tissue processing and long-read ONT sequencing of thousands of brain samples from individuals with and without AD. These sequencing data provide a unique opportunity to perform genome-wide, population-scale methylation analyses, and assess methylation levels in poorly resolved genomic regions in the human brain. Like CARD, these large-scale initiatives may want to adopt or incorporate the most up-to-date sequencing methods as they become available. This will result in cohorts of data sequenced with different sequencing technologies, like R9 and R10 for NIH CARD. It is imperative to document the differences in methylation measurements arising due to technology improvements so that they are not misinterpreted as cohort-specific observations.
In this work, we systematically assessed the performance of ONT sequencing for methylation analysis using data sets for cells, blood, and brain tissue from both R9 and R10 chemistries. We also compared ONT methylation detection with other sequencing platforms (ONT, PacBio, and Illumina). These comparisons revealed that the overall differences between R9 and R10 methylation data sets were significant enough that they should be taken into account when comparing data sets across platforms and chemistries. Biologically relevant conclusions for methylation across cohorts sequenced using these two chemistries must account for these differences. We argue that long-read sequencing can be at least an equivalent alternative for methylation to short-read bisulfite sequencing, without requiring any additional sample preparation.
Direct, simultaneous analysis of DNA sequences and their modifications allows for the exploration of elements beyond genomic and structural variation in samples across cell types and tissue. This will be transformative for studying biology and increasing the understanding of disease mechanisms. It is also important to characterize differences across different platforms, between technological improvements, and within different sample types. In the future, we aim to incorporate additional sequencing data sets (such as Hifi and bisulfite) into our methylation analyses of the blood and brain samples. We also plan to compare methylation levels between these two sample types to see if the chemistry-related differences cancel each other out.
Historically, such methylation analyses have focused only on 5mC in CpG contexts. This is especially true of long-read technologies. However, ONT sequencing is now capable of detecting 5mC and 5hmC simultaneously. A comprehensive, genome-wide, and context-agnostic analysis of cytosine modifications in human primary tissue samples will be essential for improving our understanding of basic and disease biology. Our analysis strategy can also extend to other modifications as their informatics inference becomes amenable in sequencing data.
Methods
Sample collection and sequencing
Long-read sequencing data were generated from human blood, brain, and cell line samples. For the blood sample, frozen blood was obtained from the PPMI study (https://www.ppmi-info.org/) of a 56-year-old female donor without known neurological symptoms. For the brain sample, frozen tissue was obtained from the frontal cortex of an 86-year-old male donor without known neurological symptoms at the Banner Sun Health Research Institute (https://www.bannerhealth.com/services/research/about-banner-research/research-programs/brain-and-body-donation-program/tissue-request). The HG002 cell line was purchased from Coriell (https://www.coriell.org/): HG002 (Ashkenazi Jewish ancestry GM24385) and cell culture was performed using Epstein–Barr virus (EBV)-transformed B lymphocyte culture in RPMI-1640 medium with 2 mM l-glutamine and 15% FBS at 37°C.
For DNA processing, the blood (Billingsley 2022; Miano-Burkhardt 2023), brain (Billingsley et al. 2022; Baker 2023), and cell line (Alvarez Jerez 2023; Cogan 2023) protocols are explained in detail and are publicly available on protocols.io. In brief, DNA was extracted using either the Nanobind Tissue Big DNA kit (cell line and brain) or the Nanobind HT 1 mL blood kit (blood) (PacBio). For the cell line and blood samples, the DNA went through a size selection step using a SRE Kit (PacBio SKU-102-208-300) to remove fragments up to 25 kb. The DNA was then sheared to a target size of 30 kb on a Megaruptor3 instrument (Diagenode) with either the DNAFluid + needles at speed 45 for two cycles (cell and brain) or speed 20 for two cycles with the standard shearing kit (blood). For all samples, DNA length was assessed by running 1 μL on a genomic screentape on the TapeStation 4200 (Agilent). DNA concentration was assessed using the dsDNA BR assay on a Qubit fluorometer (Thermo Fisher Scientific). Libraries were constructed using either an SQK-LSK 110 kit (ONT) or SQK-LSK 114 kit (ONT) and were loaded onto R.9.4.1 or R.10.4.1 flow cells, respectively. Each sample was sequenced for a total of 72 h, with roughly one reload every 24 h on a PromethION device per the manufacturer's guidelines (ONT FLO-PRO002).
R9 samples were basecalled using Guppy v6.1.2 (with config file dna_r9.4.1_450bps_modbases_5mc_cg_sup_prom.cfg) and R10 samples were basecalled using Guppy v6.3.8 (with config file dna_r10.4.1_e8.2_400bps_modbases_5mc_cg_sup_prom.cfg). The read batch size and reads per FASTQ were both set to 50,000 and chunks per runner was set to 195 for both R9 and R10. Example commands below:
R9:
guppy_basecaller -i ${FAST5_PATH} -s ${OUT_PATH} -c dna_r9.4.1_450bps_modbases_5mc_cg_sup_prom.cfg -x cuda:all -r ‐‐read_batch_size 50000 -q 50000 ‐‐chunks_per_runner 195 ‐‐bam_out
R10:
guppy_basecaller -i ${FAST5_PATH} -s ${OUT_PATH} -c dna_r10.4.1_e8.2_400bps_modbases_5mc_cg_sup_prom.cfg -x cuda:all -r ‐‐read_batch_size 50000 -q 50000 ‐‐chunks_per_runner 195 ‐‐bam_out
HG002 bisulfite data
HG002 Illumina bisulfite sequencing data were collected from an AWS open data set generated by ONT available here: s3://ont-open-data/gm24385_mod_2021.09/ and described here: https://labs.epi2me.io/gm24385-5mc.
HG002 PacBio HiFi data
HG002 HiFi data are available through the Genome in a Bottle Consortium described here: https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/PacBio_CCS_15kb_20kb_chemistry2/GRCh38/.
CpG site methylation frequency estimation
CpG site methylation frequencies were estimated using Modkit (https://github.com/nanoporetech/modkit), a suite of tools for manipulating ONT modified-base data stored in BAM files. The Modkit pileup command was used with either phased or unphased mapped BAMs as input to create summary counts of modified and unmodified bases in an extended BEDMethyl format—a series of columns detailing the counts of base modifications in each sequencing read over each reference genomic position. Output was restricted to 5mC sites with a CpG dinucleotide in the reference and reported as methylated, unmethylated, or mismatched. Methylation calls were aggregated/collapsed across strands (https://github.com/nanoporetech/modkit).
The Modkit command used to generate BEDMethyl files for samples basecalled with Guppy and Dorado (without 5hmC included) is
modkit pileup ‐‐cpg ‐‐ref ‐‐only-tabs ‐‐ignore h ‐‐combine-strands <IN_BAM> <OUT_BEDMETHYL>
Genome-wide comparison of R9, R10, bisulfite, and HiFi methylation proportions
The unaligned BAM files were aligned to the GRCh38 human reference genome (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz) using a combination of SAMtools (Li et al. 2009) to extract methylation aware FASTQs (-TMm,Ml,MM,ML), minimap2 to align FASTQs to reference genome (-x map-ont) and SAMtools again to sort and index aligned BAM files. Modkit (https://github.com/nanoporetech/modkit) was used to produce BEDMethyl files with collapsed strands from the aligned BAM files. A set of Numpy arrays were created and populated with CpG positions from the reference genome, ratios of modified sites calculated as Modified Calls/(Modified Calls + Nonmodified Calls), and coverage (Modified Calls + Nonmodified Calls) (Li et al. 2009).
The split violin plot for Figure 1A was created by filtering the Modkit BEDMethyl files for CpG sites shared between R9 and R10 ONT technologies and a bisulfite BEDMethyl file. Only CpG sites on the main chromosomes (1–22, X, Y, M) with coverage levels between 20× and 200× for all three data sets were considered. A Pandas dataframe was created with each row featuring a CpG site (defined by genomic coordinates) and its accompanying information. The “pandas.cut” function was used to bin CpG site methylation frequencies into specified intervals ([0–5), [5–10), [10–20), [20–30), [30–40), [40–50), [50–60), [70–80), [90–95), [95–100)) based on the bisulfite data set, with the rightmost edge values included. Split violin plots were used to plot the methylation frequency distributions across each interval, with the R9 distribution on the left in blue and the R10 distribution on the right in orange. The intervals were classified on the x-axis, and the actual distribution of values within those intervals was on the y-axis. A segmented line plot of the median value for each technology at each interval was drawn. A smoothed histogram comparing the distribution of methylation frequencies within each technology (R9 in blue, R10 in orange, and bisulfite in green) was added along the right side of the graph (Fig. 1A). The additional split violin plots were created in the same manner, but were binned by R9 instead of bisulfite (Fig. 1B–D) and by R10 (Supplemental Fig. 4A–C).
RMSE values were calculated for three sets of the data: all CpG sites meeting coverage filtering criteria, all CpG sites meeting coverage filtering criteria in Illumina mappable, 150 bp paired-end reads (Hoffman), and all CpG sites meeting coverage filtering criteria but not present in the Illumina mappable regions.
The heatmaps were created by plotting methylation proportions for each genomic site for one technology on the x-axis and another technology on the y-axis in bins equating to 0.05-sized buckets. Each combination of R9, R10, bisulfite, and HiFi data was plotted.
This process was repeated for R10 and PacBio HiFi Methylation data, and R10 and Illumina bisuflite data. This process was repeated with the added caveat of separating CpG sites by strand of origin, creating a paired violin plot for both strands at each of the R10-binned proportions.
Analysis of phased, differentially methylated regions; R9 versus R10
For each sample, the R9 and R10 GRCh38-mapped BAMs were merged and phased together using PEPPER-MARGIN-Deepvariant with R9 settings (-ont_R9_guppy5_sup flag). This was done to keep phase 1 and phase 2 assignments consistent since they are normally randomly assigned by PEPPER-Margin-DeepVariant. The merged and phased R9/R10 haplotagged BAM was then separated into phased R9 and R10 BAM files by filtering for the original R9 and R10 read names using Picard -FilterSamRead (part of the GATK [DePristo et al. 2011] toolbox).
DMRs were calculated by using NanoMethPhase dma, a Python package built on top of the Bioconductr DSS library. Comparisons between phased haplotypes for the same chemistry (e.g., R10 haplotype 1 vs. R10 haplotype 2) and between chemistries for the same haplotype (e.g., R9 haplotype 1 vs. R10 haplotype 1) were performed for the HG002 cell line data.
Comparisons between DMRs were done with BEDTools intersect using the following command:
bedtools intersect -wao -a {file1} -b {file2} > {outfile}
This produced a BED file from which counts of overlapping bases for each pair of intersecting DMRs between chemistries were calculated. This process needs to be repeated with the inverse orientation of BED files since the overlap calculation is not a symmetric function.
Haplotype-specific DMR visualization
Haplotype-specific methylation differences were visualized using Methylartist, a tool for parsing and plotting methylation patterns from ONT data. Mapped BAM files were used as input and the locus command was used to generate haplotype-aware, smoothed methylation profiles across specified intervals. A coverage plot was added and the raw log-likelihood ratio section was excluded from the final graph https://github.com/adamewing/methylartist. The command used to generate Figure 4 and Supplemental Figure S18 is shown below:
methylartist locus –b <IN_BAM1>,<IN_BAM2> -i chr16:88532537-88536321 –g –-plot_coverage <IN_BAM1>,<IN_BAM2> –-labelgenes –-genes ZFPM1 –-motif CG –-phased –slidingwindowsize 5 –-samplepalette colorblind ‐‐nomask ‐‐coverpallete viridis -–ignore_ps –o <OUT_PREFIX>
Structural variant calling
SVs were called using Sniffles v2.2, a SV caller designed for long-read sequencing data. Methylation-tagged BAMs mapped to GRCh38 were used as the input and the minimum SV length was set to 50 bp.
Software availability
Source code used in the analysis of these data and generation of the figures is available as Supplemental Scripts and at GitHub (https://github.com/NIH-CARD/CARDlongread_meth_R.9vs10).
Data access
Blood and brain: Blood and brain sequencing data generated in this study have been submitted to NCBI's database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap/) under accession number phs003181.v1.p1. The modkit files used for modification analysis are available at Amazon Web Services (AWS): https://s3.amazonaws.com/gtl-public-data/index.html?prefix=R9_R10_methylation_2024/. To preserve patient confidentiality the chromosome and position information have been masked. The positional masking is shared between the Brain and Blood samples, which allows for the reanalysis of this data using the provided code.
HG002 cell line: The HG002 cell line R9 and R10 FASTQ and BAM data generated in this study have been made publicly available through the AnVIL workspace: https://anvil.terra.bio/#workspaces/anvildatastorage/ANVIL_NIA_CARD_Coriell_Cell_Lines_Open. The HG002 Ultra-long BAM data set is publicly available through AWS: https://s3.amazonaws.com/giab-aws/index.html?prefix=WGS/ONT/2022/11_16_22_R1041_HG002_UL_Kit14_400/. The HG002 EpiQC bisulfite sequencing MethylSeq data set used to benchmark the HG002 ONT bisulfite sequencing data set was generated as part of the SEQC2 Epigenomics Quality Control Study (Foox et al. 2021). This study applied six different methylation detection approaches to seven well-characterized human cell lines—including HG002—for a comparative analysis of targeted and genome-wide methylation protocols. Their MethylSeq protocol involved using dual indexing primers to generate MethylSeq libraries with EZ DNA Methylation-Gold kit used for bisulfite conversion. Sequencing was done using Illumina NovaSeq 6000 S4 flow cells with a PE150 read length and yielded 20× genomic coverage, which was the highest coverage of all of the assay-based methylation approaches tested. The MethylSeq libraries and sequencing data were quality checked using a TapeStation 2200 HSD1000 and FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), respectively. The data were mapped to GRCh38 using Bismark (Krueger and Andrews 2011) v.0.23.0 and Bowtie 2 (Langmead and Salzberg 2012). Methylation information was extracted using bismarck_methylation_extractor (default settings), and strand information was merged using MethylDackel mergeContext. Total cytosine conversion in CpG contexts fell within the appropriate range (45%–65% of CpGs) and MethylSeq methylation levels were highly correlated with those from the two other WGBS approaches tested (Pearson r = 0.93 for both) as well as the HG002 long read-sequenced sample (Pearson r = 0.913) (sequenced with ONT R9 chemistry). The MethylSeq replicate with the largest number of reads sequenced (R1) was used to benchmark the HG002 ONT bisulfite sequencing data. This file was submitted to NCBI's BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under Genome in a Bottle project accession number PRJNA646948 (run accession number SRR13051101). Methylome capture of HG002 using Accel-NGS Methyl-Seq has been submitted to NCBI's Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) also under run accession number SRR13051101. MethylSeq BEDMethyl file: https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos3/sra-pub-zq-24/SRR013/13051/SRR13051101/SRR13051101.lite.1>GSM5649480_MethylSeq_HG002_LAB01_REP01.bedGraph.
Competing interest statement
M.J. has received reimbursement for travel, accommodation, and conference fees to speak at events organized by ONT.
Acknowledgments
We thank all of the participants who donated their time and biological samples to be a part of this study. This work used the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). This work was supported in part by the Intramural Research Program of the National Institute on Aging (NIA) (AG000542-01 and AG000538-03). This work was supported by the Center for Alzheimer's and Related Dementias, within the Intramural Research Program of the National Institute on Aging and the National Institute of Neurological Disorders and Stroke, National Institutes of Health, Department of Health and Human Services (ZIAAG000538). We thank members of the North American Brain Expression Consortium (NABEC, phs001300) for providing samples derived from brain tissue. Brain tissue for the NABEC cohort was obtained from the Baltimore Longitudinal Study on Aging at the Johns Hopkins School of Medicine, the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, the Banner Sun Health Research Institute Brain and Body Donation Program, and from the University of Kentucky Alzheimer's Disease Center Brain Bank. Data biospecimens used in the analyses presented in this article were obtained from the Parkinson's Progression Markers Initiative (PPMI) (www.ppmi-info.org/access-dataspecimens/download-data). As such, the investigators within PPMI contributed to the design and implementation of PPMI and/or provided data and collected biospecimens, but did not participate in the analysis or writing of this report. For up-to-date information on the study, visit www.ppmi-info.org. PPMI—a public-private partnership—is funded by The Michael J. Fox Foundation for Parkinson's Research and funding partners, including 4D Pharma, AbbVie Inc., AcureX Therapeutics, Allergan, Amathus Therapeutics, Aligning Science Across Parkinson's (ASAP), Avid Radiopharmaceuticals, Bial Biotech, Biogen, BioLegend, BlueRock Therapeutics, Bristol Myers Squibb, Calico Life Sciences LLC, Celgene Corporation, DaCapo Brainscience, Denali Therapeutics, The Edmond J. Safra Foundation, Eli Lilly and Company, Gain Therapeutics, GE Healthcare, GlaxoSmithKline, Golub Capital, Handl Therapeutics, Insitro, Janssen Pharmaceuticals, Lundbeck, Merck & Co., Inc., Meso Scale Diagnostics, LLC, Neurocrine Biosciences, Pfizer Inc., Piramal Imaging, Prevail Therapeutics, F. Hoffmann-La Roche Ltd and its affiliated company Genentech Inc., Sanofi Genzyme, Servier, Takeda Pharmaceutical Company, Teva Neuroscience, Inc., UCB, Vanqua Bio, Verily Life Sciences, Voyager Therapeutics, Inc., and Yumanity Therapeutics, Inc.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279159.124.
-
Freely available online through the Genome Research Open Access option.
- Received February 19, 2024.
- Accepted February 11, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








