
Ancient dog introgression into the Iberian wolf genome may have facilitated adaptation to human-dominated landscapes
- Diana Lobo1,2,3,
- Hernán E. Morales4,5,
- Cock Van Oosterhout6,
- José Vicente López-Bao7,
- Pedro Silva1,3,
- Luis Llaneza8,
- Carolina Pacheco1,2,3,
- Diana Castro1,3,
- Germán Hernández-Alonso4,
- George Pacheco4,
- John Archer1,3,
- M. Thomas P. Gilbert4,9,
- Nuno Ferrand1,2,3,10 and
- Raquel Godinho1,2,3,10
- 1CIBIO, Centro de Investigação em Biodiversidade e Recursos Genéticos, InBIO Laboratório Associado, Campus de Vairão, Universidade do Porto, 4485-661 Vairão, Portugal;
- 2Departamento de Biologia, Faculdade de Ciências, Universidade do Porto, 4169-007 Porto, Portugal;
- 3BIOPOLIS, Program in Genomics, Biodiversity and Land Planning, CIBIO, Campus de Vairão, 4485-661 Vairão, Portugal;
- 4Center for Evolutionary Hologenomics, The GLOBE Institute, University of Copenhagen, 1353 Copenhagen, Denmark;
- 5Section for Evolutionary Genomics, The GLOBE Institute, University of Copenhagen, 1353 Copenhagen, Denmark;
- 6School of Environmental Sciences, Norwich Research Park, University of East Anglia, NR4 7TJ Norwich, United Kingdom;
- 7Biodiversity Research Institute (CSIC-Oviedo University-Principality of Asturias) Oviedo University, E-33600 Mieres, Spain;
- 8A.RE.NA, Asesores en Recursos Naturales, 27003 Lugo, Spain;
- 9University Museum, Norwegian University of Science and Technology, 7012 Trondheim, Norway;
- 10Centre for Ecological Genomics and Wildlife Conservation, Department of Zoology, University of Johannesburg, 2006 South Africa
Abstract
Understanding how large carnivores respond to increasingly human-dominated landscapes will determine their future adaptive potential. The Iberian wolf (Canis lupus signatus), a gray wolf subspecies endemic to the Iberian Peninsula (Portugal and Spain), has uniquely persisted in human-dominated landscapes, unlike many other wolf populations that faced widespread extinction across Europe during the twentieth century. In this study, we conducted a comprehensive genome-wide analysis of 145 historical and contemporary Iberian wolf samples to investigate whether hybridization with domestic dogs resulted in genetic introgression. We identified a dog-derived block on Chromosome 2 in Iberian wolves, displaying signatures consistent with introgression and high nucleotide similarity among introgressed individuals. Additionally, our estimates place the average timing of introgression between 6100 and 3000 years ago, with low sequence divergence to dogs from the Iberian Peninsula suggesting a single local origin for the hybridization event. Using forward genetic simulations, we show that the introgressed haplotype is most likely being maintained in Iberian wolves by selection. The introgressed dog variants are located within the MAST4 gene, which has been linked to neurological disorders, including cognitive and motor developmental delays, hinting at a potential role in cognitive behavior in Iberian wolves. This study uncovers a case of putative adaptive introgression from domestic dogs into wolves, offering new insights into wild canids’ adaptation to human-dominated landscapes.
Circa 10,000 years ago, humans started to change the planet, prompted by the advent of farming following the domestication of plants and animals (Frantz et al. 2020). Over time, large carnivores have been severely affected by these rapid environmental changes, mostly because of the increasing competition with humans (e.g., persecution mediated by livestock depredation), and many have gone (locally) extinct (Chapron et al. 2014). Notably, amid these challenges, some species managed to change their behavior and ecology to adapt to human activities in human-dominated landscapes (Benazzo et al. 2017). A remarkable example of such resilience is illustrated by some populations of the Eurasian gray wolf (Canis lupus) living in highly human-dominated landscapes (Sazatornil et al. 2016; Rio-Maior et al. 2019). Such landscapes are found in the Iberian Peninsula (i.e., Portugal and Spain), where the human population density ranges from 20 to 400 inhabitants per square kilometer in the wolf's distribution (Instituto Nacional de Estatística Portugal 2017) (vs. European average: 97 inhabitants per square kilometer) (Chapron et al. 2014).
The Iberian wolf subspecies (Canis lupus signatus) (Fig. 1C) diverged from other Eurasian wolves about 10,000 years ago and remains isolated (Silva et al. 2020). Despite the widespread extinction of wolf populations in Europe during the twentieth century (Mech and Boitani 2003), the Iberian wolf exhibits remarkable resilience regardless of intense human persecution (Sastre et al. 2011; Nores and López-Bao 2022). Notably, this subspecies not only persists in areas characterized by high human density but also exhibits a remarkable tolerance to high levels of human activity (Llaneza et al. 2012, 2016; Dennehy et al. 2021). Moreover, Iberian wolves show reduced levels of chronic stress compared with their Eastern European counterparts (Pereira et al. 2022). Genetic and spatial behavior analyses have unveiled a peculiar lack of long-distance dispersal and cryptic population structure in Iberian wolves, with negligible instances of dispersal beyond the range of the respective genetic cluster (Silva et al. 2018). This suite of features, particularly the lack of long-distance dispersal, hint at potential adaptations to increasingly human-dominated landscapes. In such environments, short-distance dispersal could confer an advantage, as individuals that disperse long distances tend to be less risk-averse, traveling through unknown and more human densely populated areas and resulting in decreased survival rates (Murray et al. 2010; Morales-González et al. 2022). However, the evolutionary mechanisms driving this behavior remain unknown.
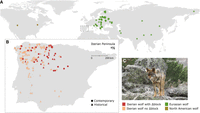
Wolf sample locations. (A) Geographical distribution of analyzed canid samples worldwide. Green and dark yellow dots denote samples from Eurasian and North American gray wolves, respectively. (B) Zoomed-in view of the sampling area within the Iberian Peninsula, outlined by the dashed line in A. Iberian wolves with and without the dog Δblock are depicted in red and pink, respectively. Circles and triangles represent contemporary and historical samples, respectively. For additional sample details, refer to Supplemental Tables S1, S3, and S4. (C) Image of an Iberian wolf (Canis lupus signatus); Photo credit: Raquel Godinho.
A consequence for wolves living in highly human-dominated landscapes is the occurrence of hybridization with domestic dogs (Vilà and Wayne 1999; Boitani 2003). Despite clear differences in behavior and morphology resulting from dog domestication, Eurasian gray wolves and dogs exhibit high genetic similarity (0.04–0.21% nucleotide differentiation) (Lindblad-Toh et al. 2005), and their hybrids can backcross to both wild and domestic individuals (Godinho et al. 2011). In the Iberian Peninsula, historical and contemporary events of wolf-dog hybridization have been documented (Godinho et al. 2011; Fan et al. 2016; Pacheco et al. 2017; Torres et al. 2017; Gómez-Sánchez et al. 2018; Lobo et al. 2023). Although hybridization has been mainly viewed as a conservation threat to wolves because the introgression of dog variants can disrupt their local adaptations (Rhymer and Simberloff 1996; Allendorf et al. 2001), more recent research has reported cases of adaptive introgression (Anderson et al. 2009; Schweizer et al. 2018; Pilot et al. 2021), suggesting it can also be a powerful force in the evolutionary response of wolves to new environmental conditions. Based on this, we hypothesize that ancient hybridization events and subsequent dog introgression could have influenced the evolutionary trajectory of the Iberian wolf, enabling its adaptation to human-dominated landscapes.
To test the “introgression fueled adaptation” hypothesis, we conducted a comprehensive genomic analysis of 48 historical (1912–2005) and 97 contemporary samples of Iberian wolves, alongside 67 worldwide wolves and 131 dogs (Fig. 1A,B; Supplemental Table S1). We used these data to (1) assess the evidence for dog introgression in the Iberian wolf genome, (2) determine whether introgressed variants display signatures of selection, and (3) explore the functional role of introgressed variants to elucidate their potential association with adaptations to human-dominated landscapes.
Results
Presence of a dog block in Chromosome 2 of Iberian wolves
To unveil signatures of dog introgression in the Iberian wolf genome, we employed a population genomic approach, analyzing 85,000 genome-wide single-nucleotide polymorphisms (SNPs; hereafter 85 K). These SNPs were genotyped using the Canine HD BeadChip microarray (Illumina) in contemporary samples of non–closely related individuals of the Iberian wolf (N = 95), the Eurasian wolf (N = 55), and dogs (N = 120; for details, see Methods) (Fig. 1A,B; see Supplemental Table S1). Using the criteria established by Lobo et al. (2023), we targeted wolf individuals measurably free of recent dog admixture to focus on older introgression events, setting a threshold of genome-wide dog ancestry below 10%. ADMIXTURE analysis clearly distinguished wolves from dogs at K = 2, with an average genome-wide proportion attributed to dogs below 1.6% in the wolf genome (Supplemental Fig. S1A). Similarly, all dogs had <1.5% of wolf ancestry in their genomes (Supplemental Fig. S1A), validating their use as a reference population. Principal component analysis (PCA) initially separated dogs and wolves and further distinguished Iberian wolves from other Eurasian wolf populations (Supplemental Fig. S1B), thus confirming the previously documented genetic distinctiveness of Iberian wolves (Pilot et al. 2014; Silva et al. 2020).
To identify signatures of dog ancestry across the Iberian wolf genome, we utilized local ancestry methods LAMP-ANC (Sankararaman et al. 2008; Pasaniuc et al. 2009) and ELAI (Guan 2014). In these analyses, Eurasian wolves and dogs were used as reference populations. Both local ancestry methods consistently assigned similar ancestries across the 38 autosomes, estimating that ∼1% of the Iberian wolf genome carries dog ancestry (Supplemental Figs. S2, S3). This 1% of dog ancestry results from small introgressed genomic regions, mostly stochastically distributed across individuals (Supplemental Figs. S2, S3).
Unlike neutral ancient introgression, if introgression conveys a selective advantage, population genetic theory predicts that introgressed genomic regions should be present at high frequency within the population (Aguillon et al. 2022). To identify such a pattern, we used the SNP-specific delta ancestry statistics (Δ) (Tang et al. 2007). We detected a “dog block” on Chromosome 2 of Iberian wolves as the top genome-wide outlier, corresponding to 12 standard deviations (SDs) from the mean Δancestry (Fig. 2A). This dog block was present in ∼30% of the Iberian wolf population based on genome-wide SNP data, irrespective of the number of mixture generations considered (ranging from 10 to 1000) (Supplemental Fig. S4). The block length is ∼2.6 Mb (hereafter Δblock) between positions 50.02 and 52.62 Mb (Fig. 2A; Supplemental Figs. S2, S3), encompassing 78 SNPs and 22 protein-coding genes (CanFam 3.1 Ensembl annotation) (Supplemental Table S2).
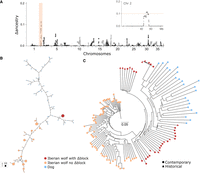
Evidence of a dog block in the Iberian wolf Chromosome 2. (A) Δancestry scores, indicating the excess of dog ancestry across the Iberian wolf genome, based on about 85,000 single-nucleotide polymorphisms from 95 contemporary samples. The Δblock position on Chromosome 2 is highlighted in orange. The inset offers a zoomed-in view of Chromosome 2, with the dashed line indicating the outlier detection cutoff (3 SD from the chromosome mean). (B) Median-joining network of Δblock haplotypes (78 SNPs) for contemporary Iberian wolf samples and village dogs from Iberia. Circle size is proportional to the frequency of each haplotype. (C) Neighbor-joining (NJ) tree for the Δblock using contemporary and historical samples (subset > 60 K SNPs; see Methods) of Iberian wolves and village dogs from Iberia. (B,C) Iberian wolves with and without the dog Δblock are colored in red and pink, respectively, and dogs are in blue. Circles and triangles denote contemporary and historical samples, respectively.
To further examine the haplotype structure and reconstruct phylogenetic relationships of Δblock within Iberian wolves and dogs, we used the 78 SNPs spanning this genomic region to construct both a neighbor-joining (NJ) tree and a median-joining network. The NJ tree revealed that all Iberian wolves with the Δblock (N = 21) were more closely related to dogs than to other wolves lacking the Δblock (Supplemental Fig. S6A). This relationship was further supported by a PCA (Supplemental Fig. S6B). Notably, Iberian wolves shared the same Δblock haplotype across the 78 SNPs (Fig. 2B), which is distinguished from a haplotype found in village dogs from Iberia by only eight 1-nt differences (Fig. 2B).
Prevalence of Δblock in historical Iberian wolf samples
For a recent temporal perspective on the prevalence of the dog Δblock in the Iberian wolf population, we analyzed a set of 48 historical Iberian wolf samples spanning periods from 1912 to 2005 (Fig. 1B; Supplemental Table S4; Pacheco et al. 2022). These samples, genotyped for 18,000 genome-wide SNPs overlapping with the 85 K SNP data set, underwent local ancestry analysis using LAMP-ANC.
The Δblock was identified in 15 historical Iberian wolves (Supplemental Fig. S7B,D) and confirmed as the top genome-wide outlier region with dog ancestry based on Δancestry statistics (Supplemental Fig. S7A,C). The Δblock was traced back to the oldest record in a wolf sampled in 1945. Additionally, a NJ tree recreated for Δblock, encompassing contemporary and historical Iberian wolf samples and village dogs from Iberia, revealed that all wolves with the dog Δblock clustered together and closer to dogs (Fig. 2C). This clustering suggests a common origin for the Δblock through the same introgression event.
Evidence that the dog block is an introgressed variant using whole-genome data
To enhance the resolution provided by the 85 K SNP data set (averaging one SNP every 25 kb), we complemented our population-based data set with whole-genome data. We sequenced the complete genome of 12 contemporary Iberian wolves (mean sequence coverage of 13.5×), each previously genotyped for the 85 K SNPs, and added publicly available whole-genome data from contemporary samples of two additional Iberian wolves, 10 Eurasian gray wolves, two North American gray wolves, 11 dogs, one Golden jackal, and an Andean fox, used as outgroup species (Fig. 1A,B; Supplemental Table S3). Employing the same local ancestry analysis in ELAI, we validated the presence of the dog Δblock in six Iberian wolf whole genomes: L474, L588, L590, L844, Wolf24, and Wolf39 (Fig. 3A; Supplemental Fig. S5). However, only in two individuals, L588 and L590, was the Δblock previously identified using the SNP data set, illustrating its limited genomic resolution. Wolf39 and L590 carried the longest dog block (Chr 2: 43.5–55.1 Mb), whereas it appeared fragmented in the other four wolves.
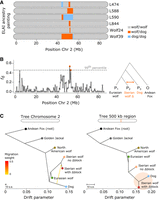
Signatures of excess allele sharing and phylogenetic discordance. (A) Representation of the Δblock on Chromosome 2 in six Iberian wolves using local ancestry analysis. Colors denote the attributed local ancestry: gray for homozygous wolf, orange for wolf/dog (heterozygous), and blue for homozygous dog. (B) Fraction of introgression (fd) across nonoverlapping 500 kb windows on Chromosome 2. The test followed the phylogenetic arrangement depicted on the right: Eurasian wolves as P1, Iberian wolves with the Δblock as P2, and dogs as P3, with the Andean fox as the outgroup. The dashed line indicates the threshold for outlier regions, with orange dots representing 500 kb windows surpassing the 99th percentile. (C) Population trees estimated on Treemix for Chromosome 2 and the 500 kb region ranked as the top window in the fd analysis, involving several canid species (Iberian, Eurasian, and North American wolves, dogs, Golden jackal, and Andean fox). The yellow arrow on the Chromosome 2 tree indicates migration (i.e., ancestry contribution) from dogs to Iberian wolves carrying the Δblock.
Having validated the presence of the dog Δblock in Iberian wolves, we delved into a more detailed analysis of allele sharing patterns across Chromosome 2. Employing a sliding window analysis of the fraction of introgression using the fd statistic (Martin et al. 2015), we uncovered a significant excess of allele sharing between Iberian wolves with the Δblock and dogs within a 500 kb genomic region consistent with the Δblock (Chr 2: 52.00–52.50 Mb, fd = 0.54) (Fig. 3B). In a scenario of random sorting of ancestral variation, this significant excess of shared variation would not be expected in relation to Eurasian wolves, thus suggesting postdivergence gene flow between Iberian wolves and dogs.
To further dissect the evolutionary history of this genomic region, we compared the population phylogenetic trees for the entire Chromosome 2 and for the 500 kb region using TreeMix (Pickrell and Pritchard 2012). The Chromosome 2 tree (Fig. 3C; Supplemental Fig. S8) is consistent with the genome-wide tree (Supplemental Fig. S8), recapitulating the species tree topology. Upon introducing migration into TreeMix (m = 1) for the Chromosome 2 tree, gene flow from dogs into Iberian wolves with the Δblock was detected (Fig. 3C). A pronounced discordance in the 500 kb tree relative to the expected species tree grouped Iberian wolves carrying the Δblock together with dogs (Fig. 3C).
Although the observed patterns of excess allele sharing and tree discordance align with the expectations of introgression, these can also emerge from alternative genomic processes, such as incomplete lineage sorting, population structure, or selection (Eriksson and Manica 2012; Smith and Kronforst 2013; Zheng and Janke 2018). However, only introgression should lead to exceptionally high levels of sequence identity between the donor and recipient species. Such high sequence similarity indicates more recent coalescence of variation in this part of the genome in Iberian wolves and dogs.
To further disentangle scenarios of incomplete lineage sorting and introgression, we compared levels of genetic differentiation (FST) and mean pairwise sequence divergence (dXY) at the genomic background between Iberian wolves with the Δblock and dogs with those estimated for the shared 500 kb region. Low FST and low dXY in the 500 kb region would support introgression over incomplete lineage sorting (Rosenzweig et al. 2016). Consistent with this scenario, FST levels across the 500 kb region were lower between Iberian wolves with the Δblock and dogs (0.05 vs. 0.27 at the genomic background level; Welch two-sample t-test: P = 1.49 × 10−9) (Supplemental Fig. S9) but not between other Eurasian wolves and dogs (Supplemental Fig. S9). Similarly, dXY levels across the 500 kb region were lower between Iberian wolves with the Δblock and dogs, particularly within a 100 kb window overlapping positions 52.4–52.5 Mb (0.0001 vs. 0.001 at the genomic background level; Welch two-sample t-test: P = 1.28 × 10−6) (Fig. 4C; Supplemental Fig. S10), and higher between other Eurasian wolves and dogs (Fig. 4B,C). Additionally, we also scanned Chromosome 2 haplotypes for regions of high sequence similarity between Iberian wolves, Eurasian wolves, and dogs using HybridCheck (Ward and van Oosterhout 2016) and found the top outlier regions consistent with the position of the 500 kb region (Supplemental Fig. S11).
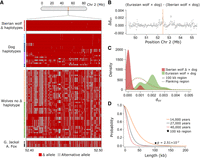
Haplotype sharing and sequence divergence between wolves and dogs. (A) Structure of the ∼100 kb haplotype, represented by 593 SNPs, found within the 500 kb window with the lowest dXY levels between Iberian wolves with the Δblock and dogs (Chr 2: 52.4–52.5 Mb). The top section shows the introgressed haplotype in Iberian wolves (red dots; L590 was homozygous across most positions, carrying two introgressed haplotypes); in the middle are the dog haplotypes (village dogs from Iberia in light blue dots, purebred dogs in dark blue, and village dogs from Asia and Africa in purple), followed by haplotypes found among wolves without the dog Δblock (Iberian in light orange dots, Eurasian in green, and North American in dark yellow). At the bottom are the Golden jackal and Andean fox haplotypes (gray and black dots, respectively). Each row represents a haplotype, and each position is colored according to whether it carries the introgressed allele (Δ in red) or the alternative allele (in gray). (B) Distribution of ΔdXY across all sites within the 6 Mb flaking region surrounding the introgressed block. This test identifies genomic regions with low sequence divergence specific to Iberian wolves and dogs and not in other Eurasian wolves (positive ΔdXY values). The orange bar indicates the position of the 100 kb region represented in A. (C) Density distribution of dXY between Iberian wolves with the Δblock and dogs (red) and other Eurasian wolves and dogs (green) in the 100 kb region (full line) and in the genomic background (dashed line). (D) Probability distribution of maintaining a haplotype of 0–200 kb length owing to ancestral shared variation, assuming a local recombination rate for the Δblock and three divergence time estimates between gray wolves and dogs (represented by distinct colors). The black star indicates the P-value associated with the introgressed haplotype (for 14,000 years ago).
We found that Iberian wolves with the Δblock and dogs shared a nearly identical 100 kb haplotype in the 100 kb window with low dXY (52.4–52.5 Mb). This 100 kb region was otherwise highly polymorphic and possessed 593 segregating sites. Iberian wolves with the Δblock and dogs from the Iberian Peninsula differed by an average of only three 1-nt differences within the 100 kb haplotype (Fig. 4A). In contrast, this 100 kb haplotype was highly divergent from those found in other Eurasian wolves, with an average of 186 1-nt differences across the 593 segregating sites (Fig. 4A). This supports that the dog Δblock originates from introgression caused by a single hybridization event and that the source was a dog from the Iberian Peninsula. The probability of finding only three 1-nt differences in a 100 kb haplotype with 593 segregating sites, where other Eurasian wolves possess on average 186 1-nt differences, is exceptionally low (binomial test: P = 3.90 × 10−91). Moreover, within the 100 kb region, we identified 18 SNP positions at which the ancestral allele was fixed in all gray wolves, whereas the derived allele was prevalent in dogs (75% frequency), was nearly fixed in village dogs from Iberia (97%), and was prevalent in Iberian wolves carrying the Δblock (60%) (Supplemental Table S5). These SNPs are positioned within a 5′-UTR intron of the MAST4 gene, which encodes a member of the microtubule-associated serine/threonine protein kinase (GeneCards, The Human Gene Database). A phylogenetic gene tree for MAST4 unambiguously grouped Iberian wolves with the Δblock closer to dogs (Supplemental Fig. S12).
Lastly, we estimated the probability of maintaining an identical 100 kb haplotype without recombination in dogs and gray wolves since the time of divergence as extremely small (ranging from P = 2.09 × 10−6 to 2.51 × 10−2 for 40,000 to 14,000 years ago) (for probability values using local or the chromosome-wide average recombination rates, respectively, see Fig. 4D; Supplemental Fig. S13; Skoglund et al. 2015; Fan et al. 2016; Frantz et al. 2016; Perri et al. 2021; Bergström et al. 2022). Cumulatively, these findings provide compelling evidence that this haplotype is of dog origin rather than an ancestral shared polymorphism between wolves and dogs.
Age of Δblock introgression in Iberian wolves
After finding support for introgression in Iberian wolves, we reconstructed the evolutionary history of the introgressed dog block, aiming to determine its onset date. We employed the STARTMRCA method (Smith et al. 2018), which allows leveraging both the decay of linkage disequilibrium (LD) and the number of accumulated mutations in the introgressed 100 kb haplotype and flanking regions. Average TMRCA estimates ranged between about 1360 (861–1857min-max) and about 670 (375–1072min-max) generations ago, assuming local or the chromosome-wide average recombination rates, respectively, and a mutation rate of 4.5 × 10−9 (Koch et al. 2019). The choice of a smaller mutation rate (4 × 10−9) (Skoglund et al. 2015) resulted in similar time estimates (Supplemental Fig. S14). Assuming a mean generation time of 4.5 years for wolves (Mech et al. 2016), this places the time since introgression between about 6100 and 3000 years ago.
Signatures of selection support adaptive introgression
To critically assess whether the observed pattern of admixture within the 100 kb region in Iberian wolves could be explained by neutral evolution, we conducted individual-based forward simulations using SLiM (Haller and Messer 2019). We explored multiple parameter combinations of number of migration events, number of migrants, and time since introgression (see Methods). In neutral simulations, the distinct combinations failed to reach the level of empirical admixture observed in the introgressed 100 kb region and in the genomic background (Supplemental Figs. S15–S17). Conversely, models that simulated adaptive introgression successfully recovered the observed empirical patterns of admixture in the introgressed 100 kb region (Supplemental Fig. S18). To investigate the genomic architecture and amount of selection required to retain an introgressed block, we simulated a range (one to six) of SNPs inside the simulated introgressed haplotype with a range of selection coefficients (s). Among alternative adaptive introgression models, those simulating strong selection acting over more than three adaptive SNPs (with a combined selection of 0.1) had a better fit to the observed level of admixture in this region (Supplemental Figs. S18, S19). Positive selection acting on multiple SNPs creates linkage that would have hindered the breakdown of the introgressed 100 kb haplotype. Moreover, we also evaluated distinct genetic dominance models—additive, dominant, or overdominant—while considering different timings for the introgression event. We found that the overdominance model (i.e., heterozygote advantage) provided the best fit to the empirical admixture and allelic frequencies observed for the SNPs within the 100 kb region (Supplemental Figs. S18, S19). However, for certain timings for the introgression (between 400 and 900 generations), we also observed successful replicates in the dominance model under lower selection coefficients (Supplemental Fig. S19). Combined, our simulation results strongly favor selection, either positive or balancing selection, maintaining the 100 kb introgressed haplotype in the Iberian wolf.
Discussion
Gene flow has been demonstrated to be widely pervasive in canids (Gopalakrishnan et al. 2018). This is exemplified by events of adaptive introgression in genes associated with coat color, immune response (Anderson et al. 2009; Schweizer et al. 2018), and hypoxia (Miao et al. 2017; Wang et al. 2020) between wolves and their domestic counterparts. Dog introgression has also been suggested as a potential powerful force in the evolutionary response of wolves facing new anthropogenic pressures (Newsome et al. 2017; Pilot et al. 2021), despite no strong empirical evidence. Our study validates the introgression of dog genetic variants in a wolf population persisting for millennia in densely human-populated areas (Llaneza et al. 2012, 2016; Dennehy et al. 2021). This finding suggests that introgression may be playing an important role in wolf adaptation to highly human-dominated landscapes.
Among Iberian wolves, the introgressed haplotype seems to coalesce into a single haplotype, strongly suggesting its origin from a single hybridization event. Such reduced nucleotide variability is unlikely to be explained by demographic events, such as the 1970s’ bottleneck experienced by the Iberian wolf (Sastre et al. 2011; Nores and López-Bao 2022; Clavero et al. 2023). Under this scenario, we would expect similar genome-wide signatures, which were not observed. Moreover, our study demonstrates that the introgressed haplotype was already present at the same frequency in the historical population, predating the bottleneck. SLiM simulations further demonstrate that, regardless of the amount or timing of gene flow simulated, neutral scenarios consistently failed to explain the differential levels of admixture observed in the introgressed haplotype. The presence of a single genomic region with dog ancestry consistently maintained at high frequency across the genome of Iberian wolves also suggests selection (Taylor and Larson 2019; Aguillon et al. 2022). Our simulations favored an overdominance model, suggesting heterozygote advantage, which raises the possibility of balancing selection maintaining the introgressed haplotype. Although balancing selection can facilitate introgression by conferring advantage to novel alleles (Fijarczyk et al. 2018, Schweizer et al. 2018), our data do not allow us to exclude the possibility of positive selection. Variables such as historical population size, the timing of selection onset, genetic drift, and past genetic structure within the Iberian wolf population could influence the rate of allele fixation under positive selection, potentially explaining the observed intermediate allelic frequencies.
Our estimates suggest an ancient introgression event occurring between 6100 and 3000 years ago, with the high haplotype similarity to dogs from the Iberian Peninsula indicating a local origin for the hybridization event. This timeframe coincides with significant human-driven landscape changes, including widespread deforestation and increased agricultural activity in the Iberian Peninsula (Tereso et al. 2016). The growing presence of dogs in human settlements during this period may have facilitated hybridization between wolves and dogs (Albizuri et al. 2021). Notably, previous studies have suggested that hybridization was already evident during the Chalcolithic in Iberia (Catagnano 2016). Abrupt climatic events documented for this period in the Iberian Peninsula (Bernal-Wormull et al. 2023) may have also impacted wolf population dynamics, potentially facilitating hybridization with dogs (Lobo et al. 2023).
One of the key challenges in investigating adaptive introgression is establishing a clear link between introgressed variants and specific functional or phenotypic traits in the recipient species (Jones et al. 2018; Suarez-Gonzalez et al. 2018; Grant and Grant 2019; Taylor and Larson 2019; Ferreira et al. 2023). The dog variants found on Chromosome 2 in Iberian wolves are located within a 5′-UTR intron of the MAST4 gene. Increasing empirical evidence reveals that MAST4 is associated with several neurologic disorders, including developmental cognitive and motor delays, as well as infantile spasms (Mala Cards: The Human Disease Database) (Strupp et al. 2020; Zhang et al. 2023). Additionally, MAST4 appears to be differentially expressed in the prefrontal cortex of atypical cases of frontotemporal lobar degeneration (Martins-de-Souza et al. 2012). Although this gene has been primarily studied in the context of neurobiology, emerging research has also linked it to bone development (Cui et al. 2022; Kim et al. 2022) and spermatogenesis (Lee et al. 2021), with knockout mice exhibiting reduced body size and increased infertility.
An experimental demonstration of the adaptive function of MAST4 in a protected wild large carnivore like the Iberian wolf is not feasible. However, although we did not directly assess the functional significance of the introgressed variants in the MAST4 gene, we hypothesize that these variants may be linked to immature cognitive development in Iberian wolves, mirroring the juvenile cognitive phenotype typically retained by domestic animals into adulthood (Wilkins et al. 2014). Although speculative, under this hypothesis, such immature cognitive development might partially contribute to the lack of long-distance dispersal observed in Iberian wolves, a behavior also common in domestic dogs and wolf pups (Jimenez et al. 2017; Morales-González et al. 2022). The frequency of introgression—∼30% using the SNP data, or 43% using the whole-genome data set—suggests that the dog-derived genetic contribution alone cannot fully account for the dispersal behavior of Iberian wolves and that other factors are likely also at play (Silva et al. 2018). We note, however, that the frequency may be underestimated owing to the limited genomic resolution of the SNP panel, as demonstrated by comparisons with whole-genome data. Future research, including functional genomic experiments in model vertebrate species (e.g., Bono et al. 2015; Li et al. 2024), will be essential to test our hypothesis and further explore the potential role of MAST4 in the cognitive behavior of Iberian wolves.
Methods
Population genomic approach: contemporary samples
Canine HD SNP BeadChip data
We generated genome-wide SNP data for a comprehensive sample set comprising 95 Iberian wolves, five Eurasian wolves, and 62 dogs (Supplemental Table S1). Iberian wolf samples, collected between 1996 and 2017, were obtained from muscle and blood samples (mainly from road kills), spanning the entire wolf distribution range in the Iberian Peninsula (Fig. 1B). The sampling strategy was designed to be representative of the entire Iberian wolf population. Dog samples, sourced from local shelters and collaborators, included muscle, blood, and buccal swabs. Eurasian wolf muscle samples were donated by collaborators, and no animals were sacrificed for this study. Total genomic DNA was extracted using the Qiagen DNeasy blood and tissue kit and quantified on the Qubit DNA quantification system (Thermo Fisher Scientific) using Qubit broad range assay reagents, following the manufacturer's protocol. DNA concentration across all samples was normalized to 50 ng/µL to be genotyped for approximately 170,000 genome-wide SNPs, using the Canine HD BeadChip microarray (Illumina). GenomeStudio software (Illumina) was employed for genotype calling following Illumina's guidelines. Our data set was expanded with two additional data sets genotyped using the same technique: 58 dogs, including 30 breeds, from the LUPA project (Lequarré et al. 2011; Vaysse et al. 2011) and 50 European wolves (Supplemental Table S1; Stronen et al. 2015). The three data sets were merged using PLINK v.1.9 (Purcell et al. 2007) after converting the SNP coordinates of the two additional data sets to the CanFam3.1 dog genome assembly using the liftOver tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver). SNP distribution and density along the genome were verified using the R/Bioconductor package karyoploteR (Gel and Serra 2017) in R (R Core Team 2017). The final data set, encompassing 95 Iberian wolves, 55 Eurasian wolves, and 120 dogs, excluded closely related individuals (identity-by-descent > 0.5). Only autosomal SNPs without multiple positions were retained, and filters for high call rates per sample (>0.95) and per SNP (>0.98) were applied. Loci with a minor allele frequency (MAF) <0.01 were removed, resulting in a final data set of about 85,000 SNPs. For global ancestry inference analyses (PCA and ADMIXTURE), we applied a light pruning on LD using r2 ≥ 0.8, with a sliding-window size of 50 SNPs; shifted; and recalculated every 10 SNPs (LD-pruned data set resulted in about 79,000 SNPs). All filtering processes were implemented in PLINK.
Global ancestry inference
Global ancestry proportions were initially explored through a PCA using PLINK. Subsequently, we used ADMIXTURE v.1.3 (Alexander et al. 2009) to estimate ancestry proportions (q) with a maximum likelihood model, focusing on K = 2 to distinguish between wolves and dogs. Separate runs were conducted for Iberian and Eurasian wolves. ADMIXTURE was run with the entire LD-pruned data set in 2000 iterations, implementing a 10-fold cross-validation procedure (Alexander and Lange 2011).
Local ancestry analysis
We employed local ancestry methods to infer the identity of individual chromosomal blocks within the Iberian wolf genome (Liu et al. 2013; Geza et al. 2019), testing two distinct methods: LAMP-ANC v.2.5 (Sankararaman et al. 2008; Pasaniuc et al. 2009) and ELAI (Guan 2014). LAMP-ANC, a non-LD-based method, estimates the most-likely ancestry per SNP within windows based on reference allele frequencies (Sankararaman et al. 2008). For this method, Iberian wolves (N = 95) were considered as the admixed population, and Eurasian wolves (N = 55) and dogs (N = 120) were set as reference populations. We assumed 10 generations since the beginning of admixture, considering a generation time of 4.5 years (Mech et al. 2016). LAMP-ANC is not designed for older admixture events, as additional generations lead to an overestimation of admixture proportions, especially when reference populations are closely related (Liu et al. 2013). We considered a genome-wide recombination rate of 9.7 × 10−9 (Wong et al. 2010; Campbell et al. 2016), a mixture proportion of 0.99:0.01 (based on global ancestry estimates), and an LD cutoff of r2 > 0.1, as LAMP-ANC assumes unlinked markers. Additionally, we ran ELAI, which employs a two-layer hidden Markov model (HMM) for local ancestry inference without requiring phased data or a prior window size definition (Guan 2014). ELAI can also infer an unsampled reference population based on allele frequencies of the admixed population (Seixas et al. 2018). Given the absence of a true Iberian wolf reference population, we ran ELAI considering three reference populations (Eurasian wolves, dogs, and an unsampled population) with the number of upper-layer clusters set to −C 3 and lower-layer to −c 15 (five times the number of −C, as recommended). ELAI was run using the filtered data set (about 85,000 SNPs), performing 20 expectation-maximization (EM) steps, and testing several mixture generations, starting with 10 generations and incrementing in intervals of 100 up to 1000.
SNP-specific Δstatistics and validation of introgression
Local ancestry methods, although very informative, lack statistical significance for each identified ancestry block. To address this, we computed SNP-specific Δstatistics (Tang et al. 2007) to find regions with a significantly elevated proportion of dog ancestry across individuals. The Δstatistics for parental population B at marker m is expressed as qBm – qB, where qBm denotes the mean ancestry at the marker m over all introgressed individuals, and qB is the mean genome-wide ancestry across all individuals. Therefore, positive Δancestry values indicate core hotspot regions for introgression from population B across the genome of all individuals. We computed Δancestry for all chromosomes using local dog ancestry estimates determined by ELAI. To minimize the risk of false positives, we focused exclusively on the top genome-wide regions as candidates for further analysis, rather than applying a genome-wide threshold of two or three SDs from the mean as in previous canid studies (vonHoldt et al. 2016; Pilot et al. 2021). Additionally, we only considered regions as outliers if they were also detected by LAMP-ANC. A region on Chromosome 2 emerged as the top genome-wide outlier, representing a 12 SD from the mean genome-wide Δancestry. We then identified outlier SNPs within Chromosome 2 by applying a threshold of three SD from the mean chromosome Δancestry, resulting in a block spanning 78 SNPs (Δblock).
To delve into the origin of the Δblock, we constructed an NJ tree using SNPs within this region, based on pairwise genetic distances between Iberian wolves and village dogs from Iberia (the most abundant dogs across the wolf range). The NJ tree distances were estimated with Tassel 5 (Bradbury et al. 2007) as 1 – identity by state (IBS), where IBS represents the probability that alleles drawn at random from two individuals at the same locus are the same. The NJ was built also with Tassel 5 and drawn with FigTree v.1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/). We also performed a PCA with PLINK, using the same set of 78 SNPs, to examine how individuals clustered. Additionally, a median-joining haplotype network for Δblock was constructed using POPART (Leigh and Bryant 2015) with reconstructed haplotypes of Iberian wolves and village dogs from Iberia generated in PHASE v.2.1 (Stephens and Donnelly 2003) with the LocusType option set to S for biallelic SNPs.
Population genomics approach: historical samples
Historical samples and in-solution capture enrichment
To explore the historical prevalence of the Δblock on Chromosome 2 in Iberian wolves, we analyzed a set of 48 historical samples collected across the former range distribution in the Iberian Peninsula, spanning from 1912 to 2005 (Fig. 1B; Supplemental Table S4). This historical data set, generated in parallel studies (Pacheco et al. 2022; Lobo et al. 2023), resulted from an in-solution target capture enrichment to obtain 100,000 genome-wide SNPs. The position of these SNPs was defined based on the coordinates available on the Canine HD BeadChip, ensuring its compatibility (full details about the methodology, such as bait design, capture experiment, sequencing, processing of raw reads, and genotyping, can be found in the original publication). Only genotypes with a read depth above four were retained, and concordance rates with the Canine HD BeadChip genotypes were estimated (>99%, based on two contemporary samples). Only samples with more than 20,000 SNPs and missing data <10% were kept, resulting in a final data set with about 23,000 SNPs. The historical and contemporary data sets were merged; filters for call rates per sample (>0.85) and per SNP (>0.90) were applied; and loci with MAF < 0.01 were removed, culminating in a combined data set of about 18,000 SNPs. All the previous steps were performed in PLINK.
Local ancestry analysis in historical samples
The introgression analysis in historical samples was performed using the local ancestry method LAMP-ANC, following the methodology outlined for the contemporary data set. Although LAMP-ANC is not as accurate as LD-based methods, we choose it because of its recommendation when the genome-wide SNP density is not too high (Sankararaman et al. 2008; Pasaniuc et al. 2009). We ran the analysis on the merged data set comprising historical and contemporary samples of Iberian wolves and dogs, encompassing about 18,000 SNPs. Reference populations comprised all dogs (N = 120) and contemporary Iberian wolves without any detected introgression signs on Chromosome 2 (N = 70). All the historical samples were considered as the admixed population. LAMP-ANC was run under the previously specified parameters. To mitigate potential biases from the limited number of loci in this data set (Chr 2 = 623 SNPs), we subsampled the historical data set, focusing solely on Iberian wolf samples carrying the Δblock on Chromosome 2 with at least 60,000 genome-wide SNPs (N = 13). This subset was merged with contemporary samples from Iberian wolves and dogs, and all the previously described filters were applied, resulting in a combined data set of about 48,000 SNPs. A second LAMP-ANC run was then conducted, employing these 13 Iberian wolves as the admixed population and maintaining all prior parameters. Outlier regions were identified for both LAMP-ANC runs using Δancestry. Subsequently, we constructed a NJ tree with all contemporary Iberian wolves, the subset of historical Iberian wolves (more than 60,000 SNPs), and village dogs from Iberia, using SNPs within the Δblock (69 SNPs). Genetic distances and the NJ tree were estimated with Tassel 5, and the tree was drawn using FigTree.
Whole-genome sequencing approach
Whole-genome sequences and SNP calling
To attain the requisite high resolution for detecting small genomic dog blocks and confirming the presence of the Δblock in Chromosome 2 of Iberian wolves, we generated whole-genomes for 12 of the contemporary Iberian wolves used in the SNP chip data set (Fig. 1B; Supplemental Table S3). DNA extraction was performed using a Thermo Scientific KingFisher instrument according to the manufacturer's guidelines, and resulting DNA fragments were fragmented in the Covaris LE220 plus focused ultrasonicator to achieve 350 bp fragment length. Fragmented DNA fragments were converted into BGISeq compatible double-stranded DNA sequencing libraries using the “single-tube” library building protocol BEST, as initially described by Carøe at al. (2018), and modified for BGISeq technology following the method of Mak et al. (2017). Subsequently, libraries were pooled and sequenced on a DNBSEQ-G400 instrument, using a PE 150 bp chemistry to an average depth of 13.5× per sample at BGI-Europe, Copenhagen. Sequence reads were processed and aligned using the BAM pipeline of PALEOMIX v.1.2.13.3 (Schubert et al. 2014). During the initial steps of the pipeline, low-quality and N bases were trimmed from the reads, adapter sequences were removed, and overlapping read pairs were collapsed using AdapterRemoval v.2.2.0 (Schubert et al. 2016) with default parameters. Reads were mapped to the dog reference genome CanFam3.1 (Lindblad-Toh et al. 2005) using BWA v.0.7.16a (Li and Durbin 2009) with the backtrack algorithm, considering a minimum base quality of zero to ensure that all the reads were retained in the process. Reads were subsequently filtered for PCR duplicates using Picard MarkDuplicates (https://broadinstitute.github.io/picard/). In the final step of the pipeline, local realignment around indels was performed with the IndelRealigner module of GATK v.3.8 (McKenna et al. 2010). Before SNP calling, we recalibrated the base quality scores (BQSR) using BaseRecalibrator/ApplyBQSR modules of the GATK v.4.1.8.1. Genotypes were then called for each sample using BCFtools v.1.10.2 (Li 2011) mpileup/call -m tools, with minimum Phred-scaled thresholds of 20 for base quality (BQ) and read mapping quality (MQ), and all the sites were emitted in the output. The genotypes were subsequently filtered using VCFtools (Danecek et al. 2011) to keep only biallelic and autosomal SNPs that have passed previous filters, had <20% of missing data across the 12 samples, and were supported by at least four reads.
To this data set, we added a set of 27 publicly available whole-genome sequences from several worldwide canids (14 gray wolves, including two Iberian wolves; 11 dogs; one Golden jackal; and one Andean fox) (Supplemental Table S3). Genotypes from the 27 canids were extracted from a VCF file containing 91 million variants and 722 canid genomes created by Plassais et al. (2019), available at NCBI's BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA448733. This VCF file was then filtered as described above and merged with the VCF file containing the genotypes of the 12 Iberian wolves, using the BCFtools merge tool. In the final data set, sites with <10% missing and with a MAF ≥1% were retained, resulting in 11.7 million SNPs (“variants only” VCF). Finally, we estimated missing and depth statistics across all individuals using VCFtools, ensuring that all individuals had <10% missing data and an average coverage above 17× across the 11.7 million SNPs.
Patterns of allele sharing and phylogenetic reconstruction
We first explored the genome-wide mixture proportion between wolves and dogs by conducting ADMIXTURE analysis for K = 2, replicating the approach used for the SNP chip data set. To validate the Δblock on Chromosome 2, we ran ELAI considering three reference populations (Eurasian wolves, N = 10; dogs, N = 11; and an unsampled population), while treating all Iberian wolves as admixed (N = 14), following the previously described parameters.
To quantify the fraction of introgression, we computed fd statistics (Martin et al. 2015) along Chromosome 2 (420,698 SNPs) in nonoverlapping 500 kb windows, each containing at least 100 SNPs, using the ABBABABAwindows.py script from the genomics_general package, available at GitHub (https://github.com/simonhmartin/genomics_general). This test was conducted based on the following phylogenetic configuration: P1, Eurasian wolves (N = 10); P2, Iberian wolves with the Δblock (N = 6, as defined by ELAI); P3, dogs (N = 11); and the Andean fox serving as outgroup. fd statistics will measure the excess shared variation between P3 and P2 that is not shared with P1, excluding the hypothesis of incomplete lineage sorting. Significantly introgressed regions were identified as those above the 99th percentile.
To unveil potential discordances in tree topology between introgressed regions and the expected species tree, we reconstructed population trees for Chromosome 2 and the top 500 kb outlier region from the window-based fd statistics. Population trees were constructed in TreeMix v.1.13 (Pickrell and Pritchard 2012), defining seven major groups: Iberian wolves with the Δblock (N = 6), Iberian wolves without the Δblock (N = 8), Eurasian wolves (N = 10), North American wolves (N = 2), dogs (N = 11), one Golden jackal, and an Andean fox set as the root. Allelic frequencies for each group were estimated with PLINK, and the output file was converted into the TreeMix input with plink2treemix.py script provided in TreeMix. Maximum likelihood trees were constructed by running TreeMix in blocks of 200 SNPs with 200 bootstrap replicates. For the Chromosome 2 tree, one migration event was incorporated into the analysis. TreeMix was run 100 times, and the trees were summarized with SumTrees of the DendroPy library (Sukumaran and Holder 2010) and visualized with FigTree. Individual-based NJ trees were also reconstructed using genome-wide SNPs (11.7 million) and SNPs for Chromosome 2 (420,698 SNPs). In both cases, SNPs were concatenated to generate pseudo FASTA alignments, and the genetic distances and the NJ trees were estimated using Tassel 5 and visualized on FigTree.
Genetic sequence divergence and haplotype reconstruction
To examine the genetic similarity between Iberian wolves carrying the Δblock, dogs, and other Eurasian wolves, we initially estimated FST levels between these groups along Chromosome 2 in nonoverlapping windows of 500 kb using VCFtools. Levels of differentiation between the Δblock and the genomic background (defined as the entire Chromosome 2 excluding the introgressed block) were compared using a Welch two-sample t-test in R. To identify the haplotype(s) of the Δblock, genotypes on Chromosome 2 were phased using Beagle v.5.1 (Browning and Browning 2007) with the aid of the dog recombination map retrieved from GitHub (https://github.com/cflerin/dog_recombination). HYBRIDCHECK v.1.0 (Ward and van Oosterhout 2016) was then employed to screen the haplotypes for blocks of high sequence similarity in 1 kb windows. Pseudo sequences of Chromosome 2 were created by concatenating phased SNPs for each individual. Pairwise comparisons were performed in sets of triplets (Iberian wolf with the Δblock–Eurasian wolf–Portuguese village dog) for each of the six individuals carrying the Δblock.
To distinguish between ancestral shared polymorphism and introgression, we estimate the mean pairwise sequence divergence (dXY) between haplotypes of Iberian wolves with the Δblock, Eurasian wolves, and dogs from the Iberian Peninsula. To ensure specificity, we used village dogs from Iberia as they likely represent the closest source for the donor of introgression. Considering that dXY analysis requires invariant sites, we considered that all the genotyped invariant sites that were not present in the combined VCF (“variants only”) were homozygous for the reference, using the ‐‐missing-to-ref option from BCFtools. Sites in a 6 Mb region (Chr 2: 49–55.5 Mb) surrounding the midpoint of the 500 kb outlier region from the window-based fd statistics were then extracted, resulting in 5.4 million sites. Flanking regions were treated as the genomic background. Haplotypes within this 6 Mb region were reconstructed using Beagle v.5.1, and dXY was calculated using the popgenWindows.py script from the genomics_general package in 10 kb nonoverlapping windows with at least 100 sites, using the -f flag set to haplo to handle haploid data. Welch's t-test was applied to compare sequence divergence between the genomic background and outlier regions. Density distributions were calculated using the R package sm (https://cran.r-project.org/web/packages/sm/index.html). Genotypes from outlier regions in dXY analysis were manually inspected using Tassel to identify signatures of allelic shared variation between dogs and Iberian wolves with the Δblock that were not observed with other wolves (e.g., major allele in dogs was only present in Iberian wolves with the Δblock).
Probability of maintaining the introgressed haplotype from shared ancestral variation
After identifying a common 100 kb haplotype in all Iberian wolves with the Δblock and dogs, we calculated the probability
of maintaining the introgressed haplotype owing to ancestral polymorphism. We followed the approach established by Huerta-Sánchez et al. (2014) and Miao et al. (2017), which follows a Gamma distribution:
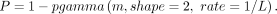
Here, L represents the expected length of a shared ancestral haplotype and is defined as L = 1/(r × t), where r is the recombination rate per generation per base pair, and t is the time since the divergence between wolves and dogs. We considered a divergence range of 40,000 to 14,000 years ago (Skoglund et al. 2015; Fan et al. 2016; Frantz et al. 2016; Perri et al. 2021; Bergström et al. 2022). To account for specific variability in recombination rates within the introgressed haplotype, we used the Chromosome 2 recombination map inferred for dogs by Campbell et al. (2016). Given the limited genomic resolution across the 100 kb haplotype, we estimated the recombination rate for the entire Δblock to be ∼0.37 cM/Mb. For comparison, we also performed the analysis using the sex-averaged recombination rate for Chromosome 2 reported by Campbell et al. (2016) at 0.84 cM/Mb. We report P-values for divergence estimates of 14,000, 27,000, and 40,000 years.
Individual-based forward simulation in SLiM
To assess the adaptive potential of the introgression, we implemented individual-based forward simulations in SLiM 3 (Haller and Messer 2019). The main goal was to investigate whether introgressed variants could have evolved under neutrality and to estimate the amount of selection required to recover the observed level of admixture in the empirical data. We simulated the entire Chromosome 2 of the dog reference genome CanFam3.1, along with its local estimates of recombination (Campbell et al. 2016), to capture realistic dynamics of the genomic background. Our model simulates two divergent lineages, representing dogs and wolves, that experience migration. We simulated the wolf lineage with an effective population size (Ne) of 3000 (Silva et al. 2020). Unique SNPs were introduced every 10 kb in the dog lineage across the entire chromosome to track their ancestry upon introgression into the wolf genome.
Initially, we simulated a neutral model across a range of scenarios for the timing and amount of introgression, considering parameters such as the number of migration events (single, one every generation, or one every 100 generations) and the number of migrants (one, 1%, 2%, or 4% of the wolf Ne). For each set of parameters, simulations ran for 1200 generations, with migration starting at generation 10, and 100 replicates were performed. Subsequently, we simulated different adaptive scenarios to investigate whether nonneutral models were a better fit to the empirical data. Given that neutral scenarios with higher and more frequent migration consistently led to a signal of greater chromosome-wide admixture than observed in the empirical data, we conducted the adaptive scenario using the parameters that reflected realistic dynamics in the neutral model (i.e., one migration event with a single migrant). In the adaptive simulations, we introduced positively selected alleles (referred to as “introgressed alleles”) inside the 100 kb introgressed haplotype in the dog genome. To test the impact of linkage and the genomic architecture of introgressed alleles, we simulated a range (one to six) of SNPs inside the introgressed haplotype. To investigate the magnitude of selection required for introgression to take place, we varied the selection (s) and the number of adaptive SNPs, calculating the combined selection by summing selection coefficients across adaptive SNPs as ∑s. Additionally, we investigated the mode of selection by varying the dominance coefficients (additive h = 0.5, dominant h = 1, or overdominance h = 2). Lastly, we assessed the true admixture proportion of ancestry-tracking SNPs and the frequency of introgressed SNPs inside the introgressed haplotype 100 kb, considering the estimated average time for introgression (see Results).
Estimating time of dog introgression
We estimated the time since introgression of the 100 kb haplotype in Iberian wolves using STARTMRCA (Smith et al. 2018), a Markov chain Monte Carlo (MCMC)–based method that leverages LD decay between the selected allele and nearby sites, along with new mutations in the introgressed haplotype. This method calculates the time to the most recent common ancestor (TMRCA), assuming the haplotype has been subjected to positive selection. It requires a panel of reference haplotypes without the selected allele with which the selected haplotype has recombined following introgression. For this, we used the haplotypes of Iberian wolves without the introgression to represent the reference haplotypes. We centered the analysis on Chr 2: 52,449,886 (central position in the introgressed 100 kb haplotype) and included 1 Mb of upstream and downstream sequence (Chr 2: 51,449,886–53,449,886), as recommended. This resulted in a genomic region comprising 1.8 million sites. We used the recombination map from Campbell et al. (2016) to infer the local recombination rate for the entire 2 Mb region, estimating it at ∼0.49 cM/Mb. TMRCA estimates were also calculated using the sex-averaged recombination rate for Chromosome 2, reported as 0.84 cM/Mb (Wong et al. 2010; Campbell et al. 2016). Additionally, we tested two widely used mutation rates for dogs and wolves, specifically 4 × 10−9 and 4.5 × 10−9 per base pair per generation (Skoglund et al. 2015; Koch et al. 2019). Ten independent MCMC chains were run, each with 50,000 iterations and a SD of 20 for the proposal distribution. The final 10,000 iterations from each chain were used to generate posterior TMRCA distributions. Estimates of TMRCA were then converted to time in years using a generation time of 4.5 years per generation (Mech et al. 2016). The results were visualized in violin plots using the R package vioplot v.0.3.5 (https://github.com/TomKellyGenetics/vioplot) in R.
Data access
The SNP genotypes of all contemporary samples generated in this study have been submitted to the Open Science Framework (OSF; https://osf.io/) under DOI 10.17605/OSF.IO/NSZ9K. All raw whole-genome sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1078274.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the European Union's Horizon 2020 Research and Innovation Programme under the Grant Agreement Number 857251. The authors also acknowledge the research support of the Portuguese Foundation for Science and Technology (FCT) under the projects PTDC/BIA-EVF/2460/2014, DivProtect/0012/2021, and UIDP/50027/2020. D.L., R.G., P.S., and C.P. were supported by FCT (PhD grant PD/BD/132403/2017 and research contract in project DivProtect/0012/2021 to D.L.; research contract 2022.07926.CEECIND to R.G.; research contract in project PTDC/BIA-EVL/31902/2017 to P.S.; and PhD grant PD/BD/135026/2017 to C.P.). C.V.O. was funded by the Earth and Life Systems Aliance (ELSA), Norwich Research Park. J.V.L.B. was supported by the Spanish Ministry of Economy, Industry and Competitiveness (RYC-2015-18932; CGL2017-87528-R AEI/FEDER EU), and by a GRUPIN research grant from the Regional Government of Asturias (AYUD/2021/51314). J.A. was supported by FEDER funds through the Operational Programme for Competitiveness Factors-COMPETE (POCI-01-0145-FEDER-029115) through FCT project PTDC/BIA-EVL/29115/2017. M.T.P.G. acknowledges ERC Consolidator Award 681396 “Extinction Genomics,” Danish National Research Foundation award DNRF143, and Norwegian Environment Agency project 18088069 for funding. G.H.A. is supported by the National Council of Science and Technology in Mexico (CONACYT) grant CVU 576734. We acknowledge the Portuguese Institute for Nature Conservation and Forestry (ICNF) for providing wolf samples in Portugal. Samples from wolves in Spain were collected by technical staff and wildlife agents of Consejería de Medio Ambiente del Principado de Asturias, Consellería de Medio Ambiente de la Junta de Galicia, Consejería de Medio Ambiente de la Junta de Castilla y León, Consejería de Agricultura, Medio Ambiente y Desarrollo Rural de la Junta de Castilla-La Mancha, Gobierno de Cantabria, and Parque Nacional Picos de Europa. Our thanks extend to all the collaborators and veterinarians whose assistance facilitated the collection of dog samples in Portugal and Spain. We are also indebted to the Museu Nacional de História Natural e da Ciência (Lisbon, Portugal), Museo Nacional de Ciencias Naturales (Madrid, Spain), and Estación Biológica de Doñana (Seville, Spain) for granting us access to the natural history collections of Iberian wolves. Our acknowledgments are extended to the owners of private collections who shared historical wolf samples. Furthermore, we express our gratitude to Sofia Mourão and Diogo Lima for laboratory and technical assistance, respectively. Finally, we are also indebted to the associate editor and two anonymous referees for all their helpful suggestions to improve the manuscript. This is scientific paper no. 33 from the Iberian Wolf Research Team (IWRT).
Author contributions: R.G., N.F., and D.L. designed the study. R.G. coordinated the project. R.G., D.L., and M.T.P.G. generated data. J.V.L.B. and L.L. coordinated the sample collection efforts in Spain. D.L., G.P., D.C., and C.P. conducted laboratory work. D.L. performed data analysis under the guidance of R.G., P.S., and C.V.O. H.E.M. performed SLiM simulations with the help of C.V.O. C.P. and G.H.A. provided technical support. J.A. and P.S. helped with bioinformatic scripting. D.L., C.V.O., and R.G. wrote the paper with input from all the other authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279093.124.
-
Freely available online through the Genome Research Open Access option.
- Received March 22, 2024.
- Accepted February 6, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








