
Accurate detection of tandem repeats from error-prone sequences with EquiRep
- 1Department of Computer Science and Engineering, The Pennsylvania State University, University Park, Pennsylvania 16802, USA;
- 2Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, Pennsylvania 16802, USA
-
↵3 These authors contributed equally to this work.
Abstract
A tandem repeat is a sequence of nucleotides that appear as multiple contiguous, near-identical copies arranged consecutively. Tandem repeats are widespread across natural genomes, play critical roles in genetic diversity and gene regulation, and are associated with various neurological and developmental disorders. They can also arise in sequencing reads generated by certain technologies, such as those used for sequencing circular molecules. A key challenge in analyzing tandem repeats is reconstructing the sequence of the underlying repeat unit. Although several methods exist, they often exhibit low accuracy when the repeat unit length increases or the number of copies is low. Furthermore, methods capable of handling highly mutated sequences remain scarce, highlighting a significant opportunity for improvement. Here, we introduce EquiRep, a tool for accurate detection of tandem repeats from erroneous sequences. EquiRep estimates the likelihood of positions originating from the same location in the unit through self-alignment, followed by a novel refinement approach. The resulting equivalence classes and consecutive position information are then used to build a weighted graph. A cycle in this graph with a maximum bottleneck weight covering most nucleotide positions is identified to reconstruct the repeat unit. We test EquiRep on two applications, identifying repeat units from satellite DNAs and reconstructing circular RNAs from rolling-circular long-read sequencing data, using both simulated and raw sequencing data sets. Our results show that EquiRep consistently outperforms or matches state-of-the-art methods, demonstrating robustness to sequencing errors and superior performance on long repeat units and low-frequency repeats. These capabilities underscore EquiRep's broad utility in tandem repeat analysis.
A tandem repeat is informally referred to as the appearance of multiple consecutive copies of the same sequence (termed as the repeat unit). Tandem repeats are commonly found in natural genomes, but they can also be introduced intentionally in certain sequencing protocols that produce reads composed of tandem repeats. Because of either mutations or sequencing errors, the observed sequences or reads are often not exact copies of the repeat unit but contain errors. Analyzing tandem repeats thus often requires reconstruction of the (unknown) unit from the erroneous, noisy sequences. Below, we first describe two biological applications involving tandem repeats. We then formally formulate the problem and present our algorithm.
The human genome consists of a vast array of repetitive elements, and many of them arise from a process called tandem duplication. In this process, a segment of the DNA is replicated multiple times, creating consecutive approximate repeat units. The length of these repeat units varies from a few base pairs (called short tandem repeats [STRs]) to 100 bp (called variable number tandem repeats [VNTRs]) and sometimes up to 1000 bp in satellite DNAs. Tandem repeats make up ∼8%–10% of the human genome and have been closely linked to several neurological and developmental disorders like Huntington's disease, Friedreich's ataxia, and fragile X syndrome (Siwach and Ganesh 2008; Usdin 2008; Hannan 2018). The repeat tracks associated with many of these diseases appear longer in certain affected individuals than is typically observed in the general population (Siwach and Ganesh 2008; Usdin 2008; Hannan 2018). For example, the GAA unit associated with Friedreich's ataxia appears five to 30 times normally, but 66 to more than 1000 times in affected individuals (Campuzano et al. 1996). More recently, longer repeat copies (25–30 bp) have been discovered to influence schizophrenia (Song et al. 2018) and Alzheimer's disease (De Roeck et al. 2018). Alpha satellite repeats of ∼171 bp (i.e., the so-called monomers) are found to be abundant in centromeric regions of many organisms and are essential for studying genome stability and evolutionary dynamics (Melters et al. 2013; Logsdon et al. 2024). To analyze tandem repeats, a critical step often involves the accurate reconstruction of the unit from either the assembled genome or unassembled (long) reads.
The rolling-circle amplification (RCA) is a recently refined sequencing technique that amplifies circularized template molecules, producing numerous tandem repeat copies of the original template. RCA can yield long tandem repeat units, with sequences often exceeding 150 bp and even reaching several kilobases in certain contexts. RCA followed by Pacific Biosciences (PacBio) or Oxford Nanopore Technologies (ONT) sequencing is a popular protocol adopted in many recent studies, especially for detection of full-length circular RNAs (Liu et al. 2021; Xin et al. 2021; Zhang et al. 2021). A crucial step in this process is the prediction of a consensus sequence derived from long reads, providing a highly accurate reconstruction of the original template (e.g., circular RNA). This step requires in silico intervention and typically employs widely used tandem repeat detection tools for consensus sequence prediction. It is important to emphasize that the reliability of circular RNA detection is therefore significantly influenced by the accuracy of the predicted consensus sequence during this intermediate step. Consequently, there is a pressing need for reliable tools capable of accurately predicting tandem repeat patterns of different kinds, accounting for the variability in unit length and copy number that may exist in different biological contexts. Addressing this gap is particularly essential for improving the accuracy and reliability of full-length circular RNA identification, especially considering that circular RNAs have emerged as promising biomarkers for numerous diseases (Rybak-Wolf et al. 2015; Wang et al. 2016; Kristensen et al. 2022).
Both above critical applications can be abstracted as this computational problem: Given a sequence R, decide if R contains tandem repeats (with mutations and errors) of a unit and, if yes, construct the sequence of the unit. Many methods have been developed, mainly driven by the development of sequencing technologies. Tools including mreps (Kolpakov et al. 2003), RepeatMasker (https://www.repeatmasker.org/), and INVERTER (Wirawan et al. 2010) are primarily designed to detect small repeat units from relatively low-error-rate data such as short-read sequencing data. They often do not perform well with higher repeat lengths and/or lower frequencies. Other tools like DeepRepeat (Fang et al. 2022), tandem-genotypes (Mitsuhashi et al. 2019), and ExpansionHunter (Dolzhenko et al. 2019) emphasize the quantification of tandem repeats more than unit reconstruction. Tandem Repeat Finder (TRF) (Benson 1999) is one of the most widely used tandem repeat detection tools. It is based on the idea of k-tuple matching and utilizes a probabilistic model followed by statistical analysis to make repeat predictions. It is also suitable for use in erroneous long reads given its ability to handle substitutions and indels. With the advent of third-generation sequencing and the resulting access to long-read data, new tools such as TideHunter (Gao et al. 2019) and mTR (Morishita et al. 2021) began to emerge. TideHunter is an efficient tandem repeat detection and consensus calling tool tailored for RCA-based long-read sequences. However, it faces challenges in accuracy when dealing with repeats of small length. Similarly, mTR struggles with repeats of low copy numbers, mostly owing to difficulty in finding a long cycle of short, infrequent k-mers. Despite the promising potential of long reads in revealing novel disease-associated tandem repeats and in reconstructing full-length circRNAs, tools capable of managing high error rates are rare. Those currently available also struggle to achieve satisfactory accuracy in challenging settings (such as too-short/long units and low copy numbers), as suggested by our experiments. Therefore, the task of accurately detecting tandem repeats from noisy sequences, particularly for longer units and low copy numbers, remains largely unresolved.
Here we present EquiRep, a new tool for reconstructing the tandem repeat unit from error-prone sequences. EquiRep stands out for its robustness against sequencing errors, as well as for its effectiveness in detecting repeats of low copy numbers. EquiRep employs a novel idea that identifies equivalent positions in the given sequence. This is achieved by self-local alignment followed by a critical refinement step that reduces the noises. The refined, equivalent positions are organized into equivalence classes. A graph is constructed in which nodes are equivalence classes and the identification of unit can be formulated as searching for a cycle in the graph with maximized bottleneck weight. We then evaluate the accuracy of EquiRep compared with leading methods across a variety of data sets over the two aforementioned applications, reconstructing repeat units from satellite DNA and circular RNAs from RCA data.
Results
We implemented the algorithm described in the Methods section as a new tandem repeat reconstruction tool named EquiRep. We compared EquiRep to four other repeat detectors: TRF, mTR, mreps, and TideHunter. For a given input sequence, each of these methods can generate multiple repeat patterns as the output, whereas EquiRep generates a single repeat pattern. If there were multiple predictions, we chose the unit corresponding to a criterion (e.g., maximum copy number) best for the method as the final predicted sequence. We evaluated these methods both on simulated and real data sets as follows.
Evaluation with simulated random sequences
The simulated random sequences are generated as follows: (1) generate a random string U constituting nucleotides (A,T,G,C) of length five, 10, 50, 100, 200, 500, and 1000 that serves as the ground-truth repeat unit; (2) concatenate multiple copies of the unit U to generate a longer sequence, with the frequency (number of copies) of the unit being three, five, 10, and 20; (3) introduce random errors—insertions, deletions, and substitutions at equal probabilities—at the rates of 10%, 15% and 20% into the concatenated string to simulate real-world sequencing errors and mutations; and (4) insert random strings, matching the length of the concatenated string (i.e., the repeat region), at both sides of the concatenated string.
For each of the settings (the combination of unit length, frequency of units, and error rate), we randomly and independently generated 50 sequences. We evaluated the methods’ predictions as follows. Let T be a ground-truth repeat unit, and let P be a prediction. We computed a rotation-aware edit distance between P and T. Specifically, because P may be a rotation of the T, we calculated the edit distance between T and all possible rotations of P and take the minimum value, defined as the rotation-aware edit distance. For each setting, we analyzed the 50 instances and report the following three metrics. First, we measured accuracy as the number of instances (out of 50) in which the method predicts the exact ground-truth unit (i.e., rotation-aware edit distance is zero). Second, we evaluated the proportion of close predictions, defined as cases in which the rotation-aware edit distance is <10% of the true unit length. Third, we reported the average of the normalized rotation-aware edit distance (distance divided by the unit length) across all 50 instances.
Figure 1, A–G, compares the accuracy on simulated data at a 10% error rate for various lengths and copy numbers. EquiRep consistently predicts a comparable or greater number of correct instances compared with other methods. The methods with a performance closest to that of EquiRep appear to be mTR and TRF; however, both struggle to maintain accuracy with large unit lengths. The accuracy of EquiRep is significantly higher than any of the other methods for unit lengths of 500 and 1000 bp, which demonstrates the ability of our tool to predict longer tandem repeats. Figure 2, A–G, compares the ratio of close predictions on simulated data at a 10% error rate. The ratio for EquiRep is high regardless of the copy number, and the trend tends to be consistent over the different unit lengths, unlike other methods. Figure 3, A–G, compares the averaged normalized rotation-aware edit distance. Observe that EquiRep consistently achieves the lowest distance, indicating that even when its predictions are incorrect, they remain the closest to the true sequence.
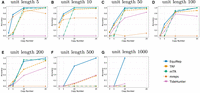
Comparison of accuracy on simulated data at a 10% error rate, for (A–G) unit lengths 5, 10, 50, 100, 200, 500, and 1000, respectively.
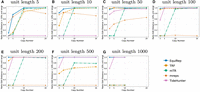
Comparison of proportion of close predictions (rotation-aware edits <10% of the unit length) on simulated data at a 10% error rate, for (A–G) unit lengths 5, 10, 50, 100, 200, 500, and 1000, respectively.
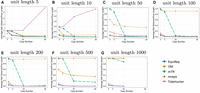
Comparison of average normalized rotation-aware edit distance on simulated data at a 10% error rate, for (A–G) unit lengths 5, 10, 50, 100, 200, 500, and 1000, respectively.
To better illustrate the distributions of the normalized rotation-aware edit distances between the predicted unit and the ground-truth, we show the fine-grained plots for all simulated settings on data with a 10% error rate, available in Supplemental Figures S5–S31.
A comparison with another approach, dot2dot (Genovese et al. 2019), on simulated data with 10% error rate, is available at Supplemental Figure S32, A–G, through Supplemental Figure S34, A–G. EquiRep outperforms dot2dot drastically on all settings.
Results for 15% and 20% error rates are available in Supplemental Figure S35, A–G, through Supplemental Figure S37, A–G, and in Supplemental Figure S38, A–G, through Supplemental Figure S40, A–G, respectively. For a higher error rate, TRF, mreps, and TideHunter see a sharp decline in accuracy as the unit length exceeds 10 bp. Conversely, mTR's ability to handle long, noisy reads allows it to achieve an accuracy close to that of EquiRep; however, its performance drops when the unit length reaches ≥500 bp. At such a long unit and with high sequencing errors, all methods struggle to accurately predict tandem repeats, particularly when the copy number is low. Overall, EquiRep outperforms other tools on the three metrics across different simulations.
Evaluation with data simulated with PBSIM2
To better mimic real long reads, we evaluated our method using data simulated by PBSIM2 (Ono et al. 2021). To simulate, we first generated sequences containing repeats positioned in the middle with random sequences flanking both ends. The repeat configurations were consistent with those described in the section “Evaluation with simulated random sequences,” including repeat units of lengths five, 10, 50, 100, 200, 500, and 1000, with each unit repeated three, five, 10, or 20 times. The following command (pbsim ‐‐depth 1 ‐‐hmm_model PC64.model ‐‐accuracy-mean 0.90) is subsequently used to simulate long reads using PBSIM2. Results were compared against the same set of alternative methods, detailed in Supplemental Figures S41, A–G, through Supplemental Figure S43, A–G. EquiRep consistently outperformed the competing methods in nearly all scenarios, highlighting its effectiveness on more realistic simulated reads.
Evaluation using simulated sequences with recurring k-mers in a unit
Genomic sequences are not purely random, often containing recurring substrings. We compared different methods on this scenario with simulations in which the repeat unit itself contains recurring structures. In this setting, predicting the correct repeat sequence is challenging as methods may encounter difficulties in distinguishing between such recurring k-mers in a single unit and identical k-mers across multiple units.
We used this approach to simulate the above sequences. First, for a given unit length l ∈ {50, 200, 500}, we generated a random k-mer of length k ∈ {5, 10, 20}, respectively. Second, we constructed the repeat unit by concatenating the random k-mer two or three times. After these concatenations, any remaining positions within the unit (i.e., l − 2k for two concatenations and l − 3k for three concatenations) will be filled with random nucleotides. Third, we concatenated multiple copies of the repeat unit to generate a longer sequence, with the frequency of units being three, five, 10, and 20. Fourth, we introduced random errors at rates of 10% and 20%. Fifth, at the end, we inserted random strings, matching the length of the concatenated string at both ends.
The same evaluation metrics for the previous simulations are also used here. Supplemental Figure S44, A–C, indicates accuracy (the ratio of fully correct instances) of EquiRep exceeding or equaling the other methods when the simulations have two copies of a k-mer within the unit at a 10% error rate. Supplemental Figure S45, A–C, shows that almost for all instances the edits predicted by our method are <10% of the unit length. Again, EquiRep achieves the lowest averaged distance as illustrated in Supplemental Figure S46, A–C. Supplemental Figures S47, A–C, through S49, A–C, demonstrate the results for data with a 20% error rate. There is a drastic decline in accuracy for all methods except mTR and EquiRep.
Nearly all repeat units generated by EquiRep have edits <10% of the unit length for copy numbers above 10, which highlights the reliability of our predictions, especially in challenging erroneous settings.
We also tested all methods on another set of data with three copies of repeating k-mers within the repeat unit, as shown in Supplemental Figure S50, A–C, through Supplemental Figure S52, A–C (for an error rate of 10%) and in Supplemental Figure S53, A–C, through Supplemental Figure S55, A–C (for an error rate of 20%). EquiRep is able to make better or similar predictions in all cases, indicating that its algorithm is the least affected by the presence of embedding k-mers within repeat units.
Evaluation using human satellite DNA data
We then tested all methods on reconstructing repeat units for satellite DNA in human Chromosome 5 (Paar et al. 2007). This known satellite DNA consists of 13 units (i.e., 13 monomers), each of which is ∼171 bp in size. To construct the input sequence for methods to predict, we concatenated the 13 monomers into a string denoted as (x). To create more testing instances, we introduced flanking regions on both sides of the concatenation denoted as (axa) and introduced errors of 1%, 5%, and 10% to (x) and (axa). To evaluate the predicted unit by different methods, we calculated the normalized rotation-aware edit distance between the predicted unit with each of the 13 known monomers and reported the averaged distance.
Table 1 shows the results. EquiRep consistently maintains a lower normalized distance, outperforming or matching all other tools. The values for EquiRep are similar to those of mTR when the input sequences have flanking regions at either end (axa), but our method is ∼87% better than mTR when just the repeat region is provided (x). Although TideHunter and TRF exhibit accuracy levels similar to ours, they fall short at higher error rates, for which EquiRep excels, with an 87% improvement.
Averaged normalized rotation-aware edit distance on human satellite DNA data
Evaluation using Caenorhabditis elegans centromere ONT data
We adopted a data set reported by Yoshimura et al. (2019), who studied the assembly of the Caenorhabditis elegans genome using Nanopore long-read data. We collected the raw long reads that are aligned to the centromere (listed in their Supplemental Fig. S4). Each of the long reads may contain more than one repeating region. Because our current method does not support detecting multiple repeating regions in a single input sequence, we manually extracted the rough region with repeats. Specifically, we first generated a dot plot for each long read, observed the repeating regions, and then manually cut out these regions and piped them to each of the methods. The ground-truth sequence of the unit is available, which is obtained by curating from PacBio HIFI data sets.
Table 2 presents the normalized rotation-aware edit distance between the predicted units and the ground truth. We report the average value across all cases. EquiRep achieves the second-best performance. For each method, we also report the number of cases in which the normalized rotation-aware edit distance is below 0.2, indicating high-quality predictions. EquiRep performs well in seven out of 13 cases, whereas the top-performing methods, mTR and TRF, achieve good predictions in eight cases.
Performance on raw ONT long reads from C. elegans centromere
Evaluation with RCA data
The set of real data is a RCA-based ONT sequencing protocol from isocirc (Xin et al. 2021) that has been used to detect a catalog of full-length circular RNAs from 12 human tissues. We considered a subset of 101 sequences from prostate tissue long-read ONT data (obtained from the NCBI Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/] under accession number GSE141693) for analysis. It is difficult to evaluate the repeats from the RCA-based long-read data owing to lack of reliable ground truth, so we evaluated these data in two different ways. First, we used a dot plot analysis. Dot plots have served as a common approach for visualizing and identifying the structural patterns of sequences such as repeats. We first aligned the input sequence to itself with LASTZ (Harris 2007) using specific parameters designed for generating dot plots. The alignment program generates a dot file that can be converted to an image file for visualization using a simple R (R Core Team 2021) script. The dot file can be used to estimate the repeat unit length (but not sequence of the unit). We treated this estimate as a benchmark for comparing the predictions of EquiRep and other tools. We reported the number of predictions that fall within 5%, 20%, 50%, and 80% error range of the true length. For the second approach, we first concatenated copies of the unit predicted to get a string A, which is longer than the input sequence. Then we got the “semiedit-distance,” which is the smallest edit distance between any substring of A and the input sequence. The idea behind this metric is that if the prediction is accurate, then the multiple concatenation of it should match the input sequence very well. We recorded the smallest edit distance and report the number of instances on which a method has a ratio (semiedit-distance)/(input sequence length) less than or equal to 0.1, 0.2, 0.3, 0.5, and 0.8.
Table 3 compares different methods in terms of the predicted repeat unit length, and Table 4 compares the normalized semiedit-distance. In both metrics, EquiRep demonstrates high accuracy, consistently outperforming mTR, TRF, and mreps. The results are also comparable to TideHunter, which is specifically optimized for RCA-based analysis. Given that the exact repeat sequences for this data set are not available, similar metric values in the table can be interpreted as comparable accuracy. It should be noted that although TideHunter excels on RCA data, its accuracy diminishes on shorter unit repeats, as indicated by the simulation results. This highlights that EquiRep is adaptable to a broad range of complex sequences and is versatile for various applications.
Performance on RCA data: number of predicted repeat lengths within error ranges of the true length and number of no repeats found (out of 101 instances)
Performance on RCA data: number of predicted repeat units with ratio of edit distance to input length less than various percentages (out of 101 instances)
In the above analysis of the RCA data sets, we observed that many repeat units exceed 1000 bp in length. This is consistent with the fact that many expressed circular RNAs are themselves >1000 bp. These observations also support the use of longer unit lengths (e.g., 500 bp and 1000 bp) in our simulated experiments (see section “Evaluation with simulated random sequences”).
Analysis of sensitivity of EquiRep to parameters
We conducted experiments to analyze the sensitivity of EquiRep to its three key parameters: (1) the score threshold (default: 25) used to identify significant paths from the initial matrix D; we tested the alternative values zero, 10, and 50; (2) the window size (default: 7) used for identifying local maxima in initial matrix D; we tested two other choices, five and nine; and (3) the number of iterations (default: 5) of iterative matrix refinement; we tested two other values, one and 10. To assess the effect of a choice of a parameter, we made it the only change to the default setting of EquiRep and then compared the variant with the default EquiRep. The same simulated data, used in the section “Evaluation with simulated random sequences,” with a 10% error rate was used here to obtain the results. We also used the same three metrics in the evaluation.
The results corresponding to the three parameters were given, respectively, in Supplemental Figure S56, A–G, through Supplemental Figure S58, A–G, Supplemental Figure S59, A–G, through Supplemental Figure S61, A–G; and Supplemental Figure S62, A–G, through Supplemental Figure S64, A–G. We can conclude that EquiRep is not sensitive to any of them, justifying its default choices.
Comparison of running time
Supplemental Table S1 presents the runtime of all methods on the simulated data from the section “Evaluation with simulated random sequences” with a 10% error rate. On average, mTR had the longest runtime, followed by EquiRep. TRF, mreps, and TideHunter were significantly faster. As noted in the Discussion, several modules in EquiRep are parallelizable, and we are optimistic about further improving its computational efficiency.
EquiRep is well-suited for processing a large number of error-prone long reads on multicore servers, as it operates on individual reads, allowing efficient batch processing that fully utilizes available cores. It is also likely that, in large-scale long-read data set, the majority of the long reads do not contain repeating regions. Fast filtering strategies, such as the seed-chaining procedure used in step 1 of EquiRep, can quickly discard such reads, leaving only a small subset that requires full processing by the complete EquiRep algorithm.
Discussion
In this paper, we present EquiRep, a robust and accurate tool for repeat detection. By leveraging a unique approach of grouping nucleotide positions into equivalence classes, EquiRep effectively builds a weighted graph to reconstruct repeat units with high accuracy. Our method addresses key challenges in detecting both short and long tandem repeats from highly erroneous sequences, areas in which existing tools often fall short. EquiRep was applied to two applications: reconstructing the repeat unit from satellite DNAs and reconstructing the circular RNAs from rolling-circular long reads. Through extensive testing using both simulated and real data sets, EquiRep outperforms or matches current state-of-the-art methods, demonstrating its robustness to sequencing errors and complex repeat patterns.
The task that EquiRep solves, reconstructing the repeat unit from erroneous sequence, is a general abstraction that can potentially be applied to other scenarios. One such application is to call circular consensus sequencing (CCS) reads from PacBio single-molecule real-time (SMRT) sequencing raw data, which produces multiple copies (with errors) of the circularized fragment. Several methods have been developed for calling CCS reads including the PacBio official consensus caller, DeepConsensus (Baid et al. 2023). We leave the comparison with these methods and the adaptation of EquiRep for CCS read generation for future work.
We demonstrated that EquiRep can be used to reconstruct the basic repeating unit of satellite DNA, known as the monomer. It is well known that satellite DNA is often organized into higher-order repeat (HOR) units, in which each HOR unit comprises multiple monomers, and these HOR units are themselves repeated in tandem. Currently, EquiRep does not capture this two-level structure of satellite DNA; it only reconstructs the repeat unit at the lower level, that is, the monomer. As part of future developments, we intend to extend EquiRep to identify and reconstruct HOR structures as well. This enhancement would enable the analysis of more complex, nested repeat architectures and make EquiRep particularly well-suited for characterizing satellite repeats in complete telomere-to-telomere (T2T) assemblies.
We are optimistic that the computational efficiency of EquiRep can be largely improved. Currently, the self-local alignment step presents a bottleneck in runtime. By improving this step, possibly through adapting more efficient alignment algorithms or parallel processing, we can substantially reduce its runtime. The second time-consuming step in EquiRep is matrix refinement. Matrix operations are inherently parallelizable, and the sparse property of the matrix can be leveraged to achieve acceleration. Although parallelization can improve performance, this approach benefits all tools when provided with additional resources. Therefore, to improve EquiRep's runtime from a design perspective, not just through scaling, we aim to streamline the pipeline itself. For instance, we are exploring faster local alignment strategies and are considering eliminating redundant steps, such as performing path-finding only once rather than twice as in the current design. We plan to explore these directions to make EquiRep more efficient and scalable for practical use.
We also aim for improving EquiRep's accuracy. The framework of EquiRep allows it to be improved in several ways. One approach is to enhance matrix refinement, which is crucial for producing accurate equivalence classes. The current method considers three mutually supportive pairs, but it can be extended to account for insertions and deletions. More precise modeling of insertions and deletions using equivalence classes, rather than single positions, is expected to improve node splitting, a key step in rescuing overcombines. Initial predictions of unit length might also help with guiding the search for repeat units within a specified range. Finally, improved heuristics for identifying cycles that combine both weights and optimal positional coverage would enable the weighted graph to represent complex repeat patterns more accurately. We plan to explore these strategies to enhance EquiRep's accuracy, which we expect will lead to improved performance on real data sets such as satellite repeats.
We realize that for short repeats (≤6 bp), there is often no clear notion of a true sequence owing to their imperfect nature. In such cases, in which detecting expansions and contractions rather than identifying a single consensus sequence might be more meaningful, EquiRep may have limited utility. For moderately long repeats (10–200 bp) found in telomeric or centromeric regions, as well as coding repeats like those in CEL or MUC1, a more defined repeat structure exists, and mutations within the repeat units can have important biological implications. Although EquiRep is applicable in such contexts, its current inability to automatically detect and resolve multiple repeat regions within a sequence introduces challenges for practical use. We will carefully take these factors into account as we continue to develop and refine the tool, with the goal of broadening its applicability and improving its usability. For very large repeats (≥500 bp) in RCA data, TideHunter demonstrates performance in both speed and accuracy. However, TideHunter is specifically optimized for RCA applications and does not perform as well in more general scenarios, particularly when dealing with shorter repeat lengths. In contrast, EquiRep is designed as a more versatile tool, aiming to provide robust performance across a broader range of repeat detection tasks and repeat size ranges.
There is a methodological similarity between EquiRep and some multiple sequence alignment approaches, such as Cactus (Paten et al. 2011a,b), as both use the concept of equivalent positions. This similarity arises naturally: In multiple sequence alignment, an ancestral sequence is assumed, and the observed nucleotides or residues that correspond to the same ancestral position are considered “equivalent.” EquiRep uses a similar intuition as the (unknown) number of copies are assumed to be mutated from the same repeat unit. The key difference lies in how these equivalent positions are constructed. Cactus derives equivalences from pairwise alignments, whereas EquiRep recognizes that the aligned positions obtained from the initial self-alignment are often inaccurate to serve as reliable equivalences. To address this, EquiRep introduces a novel, matrix-based iterative algorithm for more accurate reconstruction. Furthermore, EquiRep includes a heuristic that can split incorrect equivalence classes caused by overcombination. In contrast, Cactus produces smaller equivalence classes as the multiple alignment, without employing a similar correction mechanism. On top of these, we note that the two approaches are solving different tasks (multiple sequence alignment vs. reconstruction of the repeat unit) with different input data (multiple sequences vs. one sequence).
Methods
Given an error-prone (long) sequence/read R, EquiRep employs a four-step approach to determine the sequence of the true repeat unit U in it (if any).
-
Identifying substring S with repeating structure. From the input long read, this step determines the repeating region that potentially consists of multiple (mutated) repeats of a unit (see Supplemental Fig. S1).
-
Constructing classes of equivalent positions C. This step is the core part of the EquiRep framework. Equivalence classes are formed from equivalent positions using diagonal-free self-local alignment and a critical refinement step (see Supplemental Fig. S2). Details of the diagonal-free self-alignment are available in Supplemental Note S1.
-
Constructing candidate units from C. A weighted graph is created using equivalence classes as nodes and edges representing the connections between positions. A cycle with maximized bottleneck weight is identified to generate a candidate unit (see Supplemental Fig. S3). More candidates are generated using heuristics to handle false combinations and small unit sizes (see Supplemental Fig. S4).
-
Selecting the optimal unit. Among the multiple candidate units, the one that best satisfies a defined criterion is selected as the predicted repeat unit.
An extended version of the Methods with full descriptions of all the steps is provided as the Supplemental Methods.
Software availability
The EquiRep source code is freely available at GitHub (https://github.com/Shao-Group/EquiRep) and as Supplemental Material. The scripts, evaluation pipelines, and instructions that can be followed to reproduce the experimental results of this work are also available at GitHub (https://github.com/Shao-Group/EquiRep-test) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work is supported by the U.S. National Science Foundation (2145171 to M.S.) and by the U.S. National Institutes of Health (R01HG011065 to M.S.).
Author contributions: All authors designed and implemented the methods. Z.S. and T.Z. conducted the experiments. All authors discussed the results and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280750.125.
-
Freely available online through the Genome Research Open Access option.
- Received April 4, 2025.
- Accepted July 30, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








