
OMKar automates genome karyotyping using optical maps to identify constitutional abnormalities
- Siavash Raeisi Dehkordi1,2,5,
- Zhaoyang Jia1,5,
- Joey Estabrook2,
- Jen Hauenstein2,
- Neil Miller2,
- Naz Güleray-Lafci3,
- Jürgen Neesen3,
- Alex Hastie2,
- Alka Chaubey2,
- Andy Wing Chun Pang2,
- Paul Dremsek3 and
- Vineet Bafna1,4
- 1Department of Computer Science and Engineering, University of California San Diego, La Jolla, California 92093, USA;
- 2Bionano Genomics, Incorporated, San Diego, California 92121, USA;
- 3Institute of Medical Genetics, Center for Pathobiochemistry and Genetics, Medical University of Vienna, 1090 Vienna, Austria;
- 4Halicioğlu Data Science Institute, University of California San Diego, La Jolla, California 92093, USA
-
↵5 These authors contributed equally to this work.
Abstract
The whole-genome karyotype refers to the sequence of large chromosomal segments comprising an individual's genotype. Karyotype analysis, which includes identifying aneuploidies and structural rearrangements, is essential for understanding genetic risk factors, informing diagnosis and treatment, and guiding genetic counseling in constitutional disorders. The current karyotyping standard relies on microscopic chromosome examination, a complex and expertise-dependent process with megabase-scale resolution. Optical genome mapping (OGM) technology offers an efficient approach to detect large-scale genomic lesions. Here, we introduce OMKar, a computational method that generates virtual karyotypes from OGM data. OMKar integrates structural variants (SVs) and copy number (CN) variants into a breakpoint graph representation. It re-estimates CNs using integer linear programming to enforce CN balance and then identifies constrained Eulerian paths corresponding to full chromosome structures. OMKar is evaluated on 38 whole-genome simulations of constitutional disorders, achieving 88% precision and 95% recall for SV concordance and a 95% Jaccard score for CN concordance. We further apply OMKar to 154 clinical samples including 50 prenatal, 41 postnatal, and 63 parental genomes collected across 10 sites. It correctly reconstructs the karyotype in 144 cases, including 25 of 25 aneuploidies, 32 of 32 balanced translocations, and 72 of 82 unbalanced rearrangements. Identified disorders include cri-du-chat, Wolf–Hirschhorn, Prader–Willi, Down, and Turner syndromes. Notably, OMKar uncovers plausible genetic mechanisms in five previously unexplained cases. These results demonstrate the accuracy and utility of OMKar for OGM-based constitutional karyotyping.
Genomic structural variants involving the loss, amplification, or rearrangement of large genomic regions have been associated with many constitutional diseases (Stankiewicz and Lupski 2010). The Decipher database lists more than 2500 disorders, often caused by large structural changes in the genome, including trisomy, microdeletions and duplications, and other rearrangements (Firth et al. 2009). For molecular diagnosis, affected individuals undergo standard-of-care (SOC) testing from drawn blood, in which the extracted DNA is analyzed for genetic lesions. Genetic prenatal testing is also an important need, despite the recent advancements of noninvasive screening (NIPS) methods, typically utilizing maternal blood samples. Data from large studies suggest that although the negative predictive value (TN/(FN + TN)) was close to 100% for Down syndrome (trisomy 21) screening, the precision was in the 50%–81% range, and the numbers were similar for other disorders (Bianchi et al. 2014; Taylor-Phillips et al. 2016). Thus, a positive NIPS result is typically followed by a more invasive molecular diagnostic.
The current SOC for genetic diagnostic tests includes (1) karyotyping, (2) chromosomal microarray (CMA) (Shinawi and Cheung 2008; Miller et al. 2010), (3) FISH screening (Pergament et al. 2000; de Moraex-Malinverni et al. 2016), (4) panel sequencing, or (5) whole-exome sequencing. Karyotyping methods require considerable manual expertise and have a low resolution of 3–10 Mbp (Shaffer and Bejjani 2004). They can be combined with CMA or whole-exome sequencing to improve resolution for detecting copy number (CN) changes. These high-resolution methods (CMA, panel sequencing) do not easily detect CN neutral rearrangements. FISH requires knowledge of probes and is therefore limited in detecting novel variations. In contrast, ∼50% of all reciprocal translocations are de novo (Chang et al. 2013). Balanced rearrangements are found in 0.2% of individuals (up to 2.2% of the individuals with a previous history of miscarriage). Individuals with a balanced translocation may not directly present with a phenotype/syndrome, but during meiosis, a gamete could carry an unbalanced CN and result in fertility issues (Chantot-Bastaraud et al. 2008; Dai et al. 2022). However, balanced translocations would be very likely missed by exome sequencing/CMA.
Optical genome mapping (OGM) provides an exciting alternative to diagnostic technologies that lie between cytogenetics and exome sequencing in terms of resolution. OGMs are large enough to span repetitive and low-complexity regions, while still being able to capture smaller structural variations (SVs). Although OGM technology cannot call single-nucleotide substitutions or small insertions and deletions, it is well suited for calling aneuploidies, larger SVs, balanced and unbalanced rearrangements, and inversions and deletions (Balducci et al. 2022; Nilius-Eliliwi et al. 2023), especially with the development of advanced tools (Li et al. 2017; Raeisi Dehkordi et al. 2021). OGM has been used to successfully identify constitutional genomic lesions, despite some limitations (Dremsek et al. 2021; Mantere et al. 2021; Sahajpal et al. 2021; Dai et al. 2022). In principle, OGMs can be supplanted by long-read whole-genome sequencing (Goenka et al. 2022), but these methods are not yet readily available in a clinical setting. In fact, the demand for OGM-based diagnostics is increasing (Iqbal et al. 2023; Ghabrial et al. 2024; Levy et al. 2024a).
Automated karyotyping using OGM
The molecular karyotype of a donor can be described as a collection of genomic sequences, each sequence corresponding to one donor chromosome. Traditionally, the karyotype information was captured by cytogenetics, albeit at low resolution, and helped identify balanced and unbalanced rearrangements, aneuploidies, and other events that are directly relevant to constitutional disorders. In moving from cytogenetics to CMA and exome sequencing, much of that important information was lost. Current methods for SV calling typically do not capture the larger karyotype, making it harder to assign significance, for example, to a translocation event or to determine the locations of amplified genomic segments (Xiao et al. 2024).
Here, we present a method, OMKar, for automatically identifying karyotypes using OGM data. We tested our method using extensive simulations as well as on OGM data acquired from more than 100 prenatal and postnatal samples with constitutional disorders to gain an improved understanding of the power and limitations of the OGM technology for karyotyping.
Results
An overview of OMKar
A brief outline of OMKar (Fig. 1A–D) is presented here, with details in the Methods section. OMKar processes the output of the Bionano Solve pipeline (Bionano Genomics 2018), which includes SV, copy number variation (CNV), and contig alignment data. It generates a molecular karyotype (Table 1) in a custom text format (Supplemental Section S1) and also presents the karyotype as chromosomal clusters using International System for Human Cytogenomic Nomenclature (ISCN) language, with reference genome coordinates instead of cytogenetic bands. This approach bridges karyotyping and SV calling. Additionally, OMKar provides a graphical karyotype display.
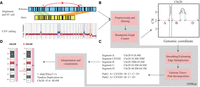
Overview of the OMKar method. (A) Input data. OMKar takes structural variant (SV) calls, copy number variation (CNV) calls, and sequence alignments as input. (B) Preprocessing and filtering. Low-confidence SVs and CNVs are removed. Chromosomes are segmented based on CNV boundaries and breakpoints, and a breakpoint graph is constructed in which vertices represent segment boundaries and edges represent segment continuity, reference adjacencies, or rearrangements. (C) Smoothing and path decomposition. Integer linear programming is used to estimate edge multiplicities while maintaining copy number consistency. Edge editing produces a Euclidean graph, and reconstructed paths are extracted using Eulerian tour and decomposition methods. (D) Interpretation and visualization. Structural variations are annotated using ISCN nomenclature; disrupted genes are identified; and results are compiled into an interactive HTML report with chromosome visualizations.
Terminology
Prior to our work, formal measurements of karyotyping accuracy were lacking. To address this, we developed two additional tools: KarSim, which generates random karyotypes in molecular karyotype and FASTA formats, and KarCheck, which compares two karyotypes by measuring their SV and CNV similarities. These tools help improve method comparisons and enable cross-technology evaluations.
The OMKar algorithm follows a multistep process: (1) preprocessing of input data, (2) construction of a breakpoint graph, (3) smoothing of edge multiplicities to generate a Eulerian graph, (4) computation of a Eulerian tour, and (5) chromosomal segregation and identification to derive the molecular karyotype. These steps are detailed in the Methods section.
OMKar runs efficiently on a desktop server
We tested the tool's performance on 154 clinical samples and 38 simulated data sets using a standard Linux machine (Intel Xeon CPU X5680 @ 3.33 GHz, 128 GB of RAM, and running Ubuntu 16.04.6 LTS). Although this represents a relatively powerful configuration, OMKar is designed to be efficient in computational resources, requiring only a single core and with a maximum RAM usage of 6 GB through all samples tested. OMKar was very efficient with a median runtime of 8.4 sec (range, 6.2–26.1 sec) (Supplemental Fig. S1). Including the time for image generation required for the HTML output, the median runtime increased to 21.3 sec (range, 15.0–48.5 sec). The runtime was correlated with the number of rearrangements (breakpoints).
OMKar reconstructs karyotypes in simulated data with high accuracy
A total of 552 SVs were simulated on 38 karyotypes at 100× coverage. The 38 karyotypes could be grouped into 803 chromosome clusters. Two hundred ninety-nine of the 803 clusters had at least one SV. Of the remaining 504 “nonevent” clusters, only six were reconstructed with an SV (false-positive), yielding a true-negative rate of 98.8%. We observed that the Bionano variant calling pipeline had a lower accuracy of 42.7% in capturing terminal SVs, which were simulated in the peri-telomeric regions (Supplemental Section S2; Supplemental Table S1). Therefore, in the following, we focused on clusters that contain nonterminal SVs, represented in 250 of 299 chromosomal clusters.
We first tested if OMKar could estimate the number of chromosomes correctly. Fourteen aneuploidies, a gain or loss of a chromosome, were simulated in nine of the 250 chromosome clusters with events, and OMKar correctly reconstructed 13 of them. OMKar reconstructed a normal number of chromosomes in 229 of the remaining 241 clusters (six FPs in the 208 clusters without terminal events, each with three or more balanced translocations; six FPs in the 33 clusters containing terminal events, owing to arm deletion).
For each cluster containing nonterminal SVs, we computed the Jaccard similarity (intersection over union) of the nonterminal SVs in the simulated and predicted clusters. The average Jaccard similarity across the 250 clusters was 84.8% (recall, 94.7%; precision, 87.5%), suggesting a high-quality karyotype reconstruction. To quantify performance according to the relative difficulty of the simulation, we estimated the complexity of each cluster as the number of breakpoint edges from nonterminal SVs. We denoted clusters with complexity score ≤6 as being low complexity and denoted them as high complexity otherwise. As expected, the performance on the low-complexity clusters (Jaccard, 89.9%; recall, 95.6%; precision, 92.2%) (Fig. 2A) was much better than on high-complexity clusters (Jaccard, 80.8%; recall, 89.9%, precision, 85.8%). The CNV comparison metric behaved similarly with high average Jaccard similarity across the 250 clusters at 96.0% and some degradation in performance from low to high complexity (Fig. 2B). The clinical cases described in the next section were of lower complexity (Fig. 2C). The distribution of the SV sizes suggests good representation of each SV type (Supplemental Fig. S2).
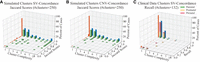
Validation statistics on simulated and clinical karyotypes. Each plot displays a 3D histogram of Jaccard score (or recall) of clusters in the sample group. The three axes separate clusters based on complexity, frequency of observations, and Jaccard score or recall. The frequencies of specific Jaccard scores are displayed in the orange projection, showing that the vast majority of samples have a high Jaccard score or recall. The frequencies of cluster complexity are shown in blue and reveal that the cluster complexity of simulated cases is generally higher than the clinical data. (A) Jaccard score of SV edges in simulations; (B) Jaccard score of CNV calls in simulations; (C) recall of SV calls on 132 clusters from prenatal, postnatal, and parental clinical samples.
In addition, we measured accuracy by directly investigating the SV edges (breakpoints) in the chromosome clusters (Table 2). A total of 839 SV edges were introduced in simulations, with 502 in low-complexity clusters. The overall accuracy was high: 96% for low-complexity and 89% for high-complexity clusters, in which the occurrence of closely spaced SV edges created challenges. The accuracy also varied depending on the SV type. For example, OMKar was successful in catching balanced translocations but had lower accuracy for tandem duplications and duplication inversions. These results support the conclusion that OMKar can accurately reconstruct the karyotypes, especially in samples with low rearrangement complexity.
Simulated nonterminal SV edge recall by event types
OMKar reconstructs karyotypes from prenatal, postnatal, and parental screenings with high accuracy
We applied OMKar to OGM data acquired from 154 samples (50 prenatal, 41 postnatal, and 63 parental) prepared at 10 different sites (Supplemental Table S2). Seven postnatal samples were biological replicates of three individuals, including samples that were mapped at different test sites. For each sample, a previous diagnosis of constitutional abnormality had been made using combinations of traditional cytogenetic methods of karyotyping, CMA, and FISH. These methods are not comprehensive, but they have high precision, so we first checked if OMKar could correctly reconstruct previously detected variations.
The union of calls from karyotyping, CMA, and FISH revealed 141 variations in 154 samples. OMKar was able to fully reconstruct 129 (91%) of the 141 variations, including 25/25 (100%) aneuploidies, 32/32 (100%) balanced reciprocal translocations, 38/39 (97.4%) deletions, 32/38 (84.2%) amplifications, and 1/3 (33.3%) unbalanced translocations. OMKar did not detect one inversion, one Robertsonian translocation, and one isodicentric chromosome. These 146 SVs formed 132 chromosome clusters, for which reconstructions of 121 (91.7%) were fully concordant. Some of the missed karyotypes were owing to SVs being masked (three clusters), a duplication size below OMKar threshold (two clusters), a Robertsonian translocation (one cluster), and an isodicentric chromosome (one cluster) (Fig. 2C; Supplemental Table S3).
Importantly, OMKar improved upon every other technology when considered in isolation (Supplemental Fig. S3). On the 65 samples for which karyotyping was performed, it detected only 56 (64%) of the 87 SVs. Similarly, CMA was applied on 76 samples and detected 94 (88%) of the 107 SVs; FISH was applied on 16 samples and detected 11 (58%) of the 19 SVs. Specifically, karyotyping mostly captured large rearrangements, whereas CMA and FISH mostly captured unbalanced events.
We tested OMKar consistency using biological replicates prepared and mapped at different test sites. Specifically, one postnatal sample was processed at six different test sites, and two postnatal samples were each processed at two test sites (Supplemental Table S2). In all cases, OMKar successfully reconstructed the correct karyotype.
Importantly, OMKar reconstructed additional SVs not caught by any of the other techniques. Specifically, after filtering out lower-quality SVs (Supplemental Section S3.1), OMKar detected 436 deletions, 506 amplifications, and 67 inversions, averaging 2.8 deletions, 3.3 amplifications, and 0.44 inversions as novel events per sample. These discoveries need to be experimentally validated. However, coverage support for novel SVs was similar to that of the simulations, in which OMKar achieved a precision of 88%.
In contrast with Bionano access, OMKar reconstructs the full karyotypes
OMKar takes as input the SV and CNV calls generated by Bionano Solve, which are also available through the Bionano Access software. In principle, a trained cytogeneticist could manually reconstruct the complete karyotype using Bionano Access information. However, in practice, particularly for samples with complex structural rearrangements, manual reconstruction is time-consuming and has potential for error. OMKar overcomes this limitation by fully automating this process, producing fast and consistent karyotype reconstructions.
To demonstrate the added value of OMKar, we compared its results with those presented by Bionano Access in three postnatal cases involving translocations. These included one canonical balanced reciprocal translocation and two cases with multiple, complex rearrangements.
Postnatal sample 1404 contains a balanced reciprocal translocation between Chromosomes 9p and 22q, along with a 240 kbp left-duplication inversion on Chromosome 22 located ∼1.3 Mbp upstream of the translocation breakpoint. Bionano Access correctly reported the presence of each individual event as discordant breakpoint edges (Supplemental Fig. S4A). In contrast, OMKar reconstructed the derivative chromosomes in their entirety, resolving the rearranged segment order (e.g., …74+ 75– 75+ 76+ 30− in derivative Chromosome 22) (Supplemental Fig. S4B), in which each number represents a distinct chromosomal segment and annotates the overall karyotype structure. It also generated an ISCN style representation (Supplemental Fig. S4C), and a chromosome ideogram (Supplemental Fig. S4D). An annotation of the balanced translocations and inverted duplication is provided for clarity (Supplemental Fig. S4E).
Postnatal sample 2281 (Supplemental Fig. S5) contains a set of complex interchromosomal rearrangements between the q-arm of Chromosome 2 and the p-arm of Chromosome 3. The Bionano Access output reports four distinct interchromosomal translocations, along with one deletion and one inversion. The four translocations share two approximate breakpoints but differ in orientation. Manual reconstruction of the karyotype by a cytogeneticist using Bionano Access information would be highly time-consuming. In contrast, OMKar generated a complete and coherent karyotype reconstruction without adding or omitting any calls (Supplemental Fig. S5B–D).
Postnatal sample 2282 (Supplemental Fig. S6) contains a set of complex inter- and intrachromosomal rearrangements between the p-arms of Chromosome 12 and 16. Bionano Access reports one interchromosomal translocation between the two chromosomes, followed by an intrachromosomal translocation, a deletion, and an inversion downstream from the initial translocation breakpoint on Chromosome 16. A CN gain is also observed on Chromosome 16 near the intrachromosomal translocation breakpoint. Notably, the CN gain does not span the entire region between the translocation breakpoint, suggesting a partial duplication or more intricate structural event. OMKar determined that it is not possible to reconstruct both homologous chromosome pairs without either excluding or introducing additional calls. OMKar automatically inferred a single additional translocation edge between segments 48 and 66 and generated a complete karyotype incorporating all reported SV and CNV calls (Supplemental Fig. S6B–D).
OMKar correctly reconstructs variations with partially missing calls
Ideally, unbalanced rearrangements are supported by both SV edges and by CNVs. However, the CNV call might be missed if the region is small, and the SV call might be missed if the breakpoints lay in regions of low-complexity that are masked by the Bionano pipeline. OMKar reconstructs karyotypes with rearrangements that are supported only by SVs or only by CNVs. Specifically, it infers missing SVs by adding additional edges to make the breakpoint graph Eulerian (Methods). OMKar outputs inferred variants as lower-confidence karyotype features.
We reanalyzed 40 deletions and 42 amplifications detected by OMKar that were cross-validated using complementary technologies. Of the deletions, 12 (30%) were reconstructed using only CNV calls, and four (10%) were reconstructed using only SV calls. Among the amplifications, 15 (36%) amplifications were reconstructed using only CNV calls.
In prenatal sample 205, an interchromosomal duplicated insertion was previously reported using a combination of karyotyping and CMA: ins(14;2) (q32;q36.1q31.2) (105.159M; 221.205M–178.043M). OGM reported a high-confidence interchromosomal SV call of Chr 14: 105.159M to Chr 2: 221.205M, with the correct orientation, but the other interchromosomal SV call was missing. It also reported a CN gain of the duplicated region of Chr 2. OMKar correctly inferred the missing SV call using support from the other SV and CNV calls and was able to automatically reconstruct this rearrangement.
OMKar identifies the genetic basis of previously diagnosed phenotypes
The OGM samples were generated based on different usage modalities (Supplemental Table S2). Prenatal testing in 50 samples was performed, either because of an abnormality detected by NIPS (44 samples) or because of elevated risk owing to family history or advanced maternal age. In contrast, 28 of 34 unique postnatal samples presented with a clinical phenotype. The 63 parental samples contained individuals who had experienced miscarriage, had a higher probability of translocation, or had a suspicion of infertility.
Among the pre- and postnatal samples, 20 had a previously diagnosed genotype-to-phenotype (G2P) mechanism, largely obtained through a manual analysis of CMA and karyotyping. OMKar automatically identified all 20 G2P mechanisms through a correlation between reconstructed karyotypes and an intersection with the DDG2P database (Methods). These were all aneuploidies, with phenotypes including triple-X, Jacobs, Turner, and Down syndromes. In contrast, for the (largely asymptomatic) parental samples, OMKar automatically and correctly reconstructed nine of 10 translocations (missed one Robertsonian) to explain eight infertility cases and one clinically remarkable child. The last case (ID:1999) highlighted the importance of parental karyotyping using OMKar. The sample was unremarkable in cytogenetic karyotyping but carried a balanced translocation between the p-arms of Chr 4 and Chr 7, t(4;7) (3,903,798;6,881,853). This balanced translocation resulted in the inheritance of an unbalanced translocation. The child carried a deletion of Chr 4: 0–3.9 Mbp, causal for the Wolf–Hirschhorn syndrome (deletion of Chr 4: 1.6–2.1 Mbp) (Wolf et al. 1965; Zollino et al. 2003).
Apart from the 10 cases, OMKar also identified a deletion, del(X) (31,614,556–31,831,572) in one parent (ID:19), which causes a monoallelic loss of the gene DMD, leading to Becker muscular dystrophy (BMD) (Ervasti et al. 1990). BMD can be inherited, and although the deletion was likely not causal for the observed infertility, it may have contributed in combination with other undiagnosed factors.
All of these confirmatory diagnoses either involved large CNVs that could be resolved by CMA or large translocation events that could be detected by karyotyping. We next investigated OMKar's capacity to detect smaller SVs.
OMKar reconstruction explains genetic basis of postnatal phenotypes
In addition to the larger variants discussed previously, after filtering (Supplemental Section S3.1), OMKar reported 28 balanced (copy-neutral) SVs and 144 unbalanced SVs in 21 postnatal samples with missing G2P explanations. Importantly, OMKar also provided novel G2P explanations for five of the 21 samples (Table 3).
OMKar G2P explanations for undiagnosed postnatal cases
Two samples showed unbalanced events with a loss of genes important for neurodevelopment. For sample 2081, it was a deletion call (10.4 Mbp). The karyotype for sample 2280 was more complex, with a deletion (5.1 Mbp) in the middle of a translocated segment (segment 4c) (Fig. 3A–C). Previously, only karyotyping had been performed for both cases, and neither deletion was detected.
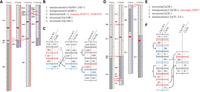
OMKar reconstructions in two postnatal samples: 2280 (panels A–C) and 2281 (panels D–F). The karyograms (panels A, D) and the ISCN-formatted description (panels B, E; slightly altered for exposition) were automatically generated by OMKar. The karyograms displayed show the “segment view” for easier referencing. Panels C and F describe the SV interpretation process after path decomposition, with black brackets indicating concordant blocks, red indicating deletion blocks, and blue indicating insertion blocks.
Among 13 balanced events reconstructed by OMKar in these five samples, boundaries in three samples (2276, 2281, and 2282) (see Table 3) interrupted a neurodevelopmental gene. Because precise coordinates of translocation were not reported via karyotyping or CMA, a genotypic basis was not previously diagnosed. Sample 2281 in particular illustrates the power of OMKar's reconstruction of a complex karyotype (Fig. 3D–F). The reconstruction revealed a transposition of a Chr 3 segment on to Chr 2, interrupting the MBD5 gene between segments 2C and 2D. The karyotype additionally included two inversions and a translocation, resulting in a highly rearranged chromosomal cluster with no change in CN.
Discussion
Karyotyping remains an essential tool in the diagnosis of constitutional genetic disorders, particularly those arising from large chromosomal rearrangements such as aneuploidies, translocations, and complex structural variants. Although traditional methods such as cytogenetic karyotyping, FISH, and microarray have long served as the standard for detecting these abnormalities, they are constrained by limited resolution, manual labor intensity, and an inability to detect balanced rearrangements and novel variations with high precision. In contrast, genomic technologies (whole-exome/genome sequencing) are very precise but do not easily provide chromosome-level characterizations. Long-read technologies are currently too expensive for clinical use. OGM thus represents a happy medium, offering a medium-resolution, robust alternative capable of detecting a broader range of SVs in clinical settings (Smith et al. 2022; Valkama et al. 2023). Recent clinical studies have shown OGM has high concordance (99.5%) with SOC methods over 1000 samples, with an increased detection rate of pathogenic or likely pathogenic variants (Iqbal et al. 2023; Broeckel et al. 2024). OGM is also recently incorporated into the ISCN (Hastings et al. 2024). For these reasons, we developed our tool starting with OGM data.
Our method, OMKar, bridges the gap between low-resolution techniques like cytogenetics and high-resolution sequencing methods by capturing large-scale rearrangements but also combining the information into an automated karyotype inference. It identifies key structural abnormalities such as balanced translocations, inversions, and duplications. The ability to automate karyotyping through OMKar not only reduces the manual workload but also enhances the speed and scalability of the analysis. This enables clinicians to analyze large data sets, improving diagnostic accuracy and potentially leading to faster treatment decisions.
In developing OMKar, we faced a significant challenge of resolving conflict between SV and CNV calls. Such conflicts were caused by either having one of the calls with high confidence while the other call was missed or masked or having SV and CNV call boundaries that were not identical. OMKar is designed to infer variations with partially missing or conflicting calls and resolve boundaries. Future developments in OGM technology and in algorithmic reconstruction should reduce these conflicts, resulting in higher-confidence calls.
OGM has a practical resolution limit for CNV detection, with high-confidence calls generally restricted to events ≥100–150 kb. In OMKar, we apply a conservative filtering threshold of 200 kb to exclude low-confidence CNV calls, thereby enhancing the specificity of the breakpoint graph and reducing computational complexity during reconstruction. Notably, smaller CNVs that overlap with SV calls are reintegrated into the analysis, and SVs with only partial CNV support are flagged as lower-confidence features. This approach preserves biologically relevant variation while maintaining the accuracy and tractability of the method and reflects the current resolution limitations inherent to OGM platforms.
OGM allows genome-wide detection of large SVs with high sensitivity, including balanced events like translocations and inversions that are often missed by sequencing. However, OGM cannot detect single-nucleotide variants (SNVs) or small indels, and it has reduced sensitivity for small CNVs (<100 kb), centromeric regions, and mosaicism. Despite these limitations, OGM fills a critical gap between cytogenetics and sequencing in clinical genomics.
OMKar utilizes the Bionano pipeline for SV calling, which in turn is based on searching the human reference genome. With the availability of multiple genomes, SV calling can be improved using assembly graphs or even de Bruijn graphs as the reference (Lin and Pop 2011; Alipanahi et al. 2016; Mukherjee et al. 2019, 2021; Leinonen and Salmela 2020). Future work will seek to incorporate these technologies to improve SV calling and karyotyping.
OMKar (and OGM), although highly effective for most structural variants, shows reduced sensitivity in detecting mosaic chromosomal abnormalities, events occurring in regions of low complexity, and segmental duplications that can lead to nonallelic recombination such as Robertsonian translocations. Mosaicism, characterized by variation in chromosomal numbers within different cell populations, also poses a challenge for OGM, which is primarily focused on large, stable genomic rearrangements. Further refinement of OGM technology and its tools will be needed to broaden its applicability in clinical settings.
OMKar showed a performance gap between terminal and nonterminal SV. Previous results from FISH screening suggest that a small number (5%–10%) of developmental disorders that lead to intellectual disability are owing to “cryptic telomeric rearrangements” (Moeschler and Shevell 2006). However, this may be an underestimate because subtelomeric rearrangements are often seen as de novo variants (Luo et al. 2011). Our future research will focus on algorithms for identifying telomeric abnormalities, including ring chromosomes (Mostovoy et al. 2024).
OGM technologies cannot currently detect variations within centromeres or the short arms of acrocentric chromosomes (Bionano Genomics 2018, 2024). In particular, Robertsonian translocations involving rearrangements of the short arms of acrocentric chromosomes are not currently detected by OGM. Recent studies have suggested the use of long-read technologies like Oxford Nanopore Technologies for detecting them (Mostovoy et al. 2024). Because the core of OMKar algorithm, which includes the building of Eulerian graphs followed by path extraction, is agnostic of a specific sequencing technology, OMKar should be easily adapted to other technologies. As more data sets describing these events are made available on different sequencing platforms, we plan to develop karyotyping tools for those platforms.
In conclusion, OMKar has demonstrated significant potential in automating and improving the accuracy of karyotyping using OGM data. It offers a robust, scalable, and high-resolution approach to detecting constitutional genetic abnormalities, although ongoing improvements are necessary to fully address its limitations. As the tool continues to evolve, it could become an increasingly important method for research and clinical diagnostics, complementing and potentially surpassing traditional methods in terms of accuracy and efficiency.
Methods
Data preprocessing
OMKar filters the SV and CNV calls to ensure data quality and relevance of the SV to karyotyping. OMKar utilizes the default Bionano pipeline thresholds for calling CNVs and SVs to ensure that only reliable variants are used in karyotype reconstruction. The Bionano pipeline maintains a database of regions that are observed with SV or CNV and masks genomic regions that are frequently seen in normal samples. OMKar filters out CNV calls in the masked regions if they do not have supporting SVs. Finally, OMKar filters CNVs <200 kbp, and those are reincorporated as local changes after karyotype reconstruction (see Supplemental Section S3.1).
Breakpoints from SV calls are further processed, sorting them by chromosomal and genomic coordinates and merging adjacent breakpoints within a 50 kbp window while also ensuring precise representation of SVs by splitting CNV segments when breakpoints occur within their boundaries. The result of this preprocessing is a partitioning of each chromosome into a minimal number of segments, so that (1) each segment has a nearly uniform CN, (2) all breakpoints link only the end-coordinates of segments, and (3) segments span all regions of reference chromosomes with a CN ≥1.
Breakpoint graph construction
We use the genome segment partitioning to generate a breakpoint graph, 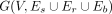 (Alekseyev and Pevzner 2009). Each vertex v ∈ V corresponds to the end-coordinate of a segment. The set of segment-edges is defined using Es = {(u, v) s.t. u and v are canonically the head and tail-node of the same segment}. The nonsegment edges include two sets: (1) the set of reference-edges, Er = {(v, u)}, where the head-node u of a segment is adjacent in reference coordinates to the tail-node v of another segment, and (2) breakpoint-edges (u, v) ∈ Eb joining vertex u to a nonadjacent vertex v. By definition, each vertex v is incident on exactly one segment-edge, at most one reference-edge, and possibly multiple breakpoint edges denoted by Eb[v].
(Alekseyev and Pevzner 2009). Each vertex v ∈ V corresponds to the end-coordinate of a segment. The set of segment-edges is defined using Es = {(u, v) s.t. u and v are canonically the head and tail-node of the same segment}. The nonsegment edges include two sets: (1) the set of reference-edges, Er = {(v, u)}, where the head-node u of a segment is adjacent in reference coordinates to the tail-node v of another segment, and (2) breakpoint-edges (u, v) ∈ Eb joining vertex u to a nonadjacent vertex v. By definition, each vertex v is incident on exactly one segment-edge, at most one reference-edge, and possibly multiple breakpoint edges denoted by Eb[v].
Smoothing edge multiplicities to generate a Eulerian graph
We use an integer linear programming (ILP) (Schrijver 1998) formulation to constrain the CN of each genomic segment. Let cv denote the CN assigned to the segment-edge incident on vertex v. The ILP assigns CNs rv ≥ 0 to the reference edge, se ≥ 0 to each edge in Eb[v], and auxiliary values xv while enforcing the following constraints: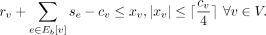 (1)
These two constraints ensure that the sum of CNs of outgoing edges from a segment is not greater than the segment's assigned
CN. Moreover, if a reference-edge incident on v is supported by a contig alignment that spans the adjacent segments, then rv ≥ 1. To make the graph Eulerian, we need to reduce the number of vertices with odd degrees. For this purpose, the binary
parameter ov is defined as follows:
(1)
These two constraints ensure that the sum of CNs of outgoing edges from a segment is not greater than the segment's assigned
CN. Moreover, if a reference-edge incident on v is supported by a contig alignment that spans the adjacent segments, then rv ≥ 1. To make the graph Eulerian, we need to reduce the number of vertices with odd degrees. For this purpose, the binary
parameter ov is defined as follows: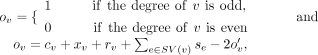 where
where 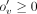 is an auxiliary variable added to the ILP. We minimize an objective function
is an auxiliary variable added to the ILP. We minimize an objective function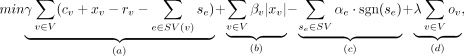 (2)
where section (a) penalizes for the discrepancy between observed plus slack CN of segment edges and the total CN of adjacent reference and
breakpoint edges; section (b) penalizes for using nonzero slack; section (c) provides a reward for using breakpoint edges at least once, and section (d) penalizes for having odd degree vertices. The actual implementation linearizes the nonlinear terms (Supplemental Section S3.2). The objective includes the parameters γ, αe, βv, λ, which were set empirically (Supplemental Section S3.3; Supplemental Table S4).
(2)
where section (a) penalizes for the discrepancy between observed plus slack CN of segment edges and the total CN of adjacent reference and
breakpoint edges; section (b) penalizes for using nonzero slack; section (c) provides a reward for using breakpoint edges at least once, and section (d) penalizes for having odd degree vertices. The actual implementation linearizes the nonlinear terms (Supplemental Section S3.2). The objective includes the parameters γ, αe, βv, λ, which were set empirically (Supplemental Section S3.3; Supplemental Table S4).
OMKar uses a fixed set of parameters designed to detect common structural variants, without data set–specific tuning. These parameters were applied consistently across 38 simulated genomes and 154 clinically diverse samples from 10 independent sites, demonstrating robustness and generalizability despite variability in sample type and data quality.
Computing Eulerian tours
Following the estimation of edge multiplicities, we utilize a breadth-first search (BFS) algorithm to identify all connected components within the graph, each component representing a chromosome cluster. For each connected component, denoted as C, our approach initiates with the identification of vertices that represent the telomeric regions of chromosomes. If C contains odd-degree nontelomeric vertices, we connect such pairs using dummy edges to transform C into a Eulerian structure. Algorithm 1 (Supplemental Section S3.4) is used to compute Eulerian tours originating from one of the telomeric vertices.
Chromosomal segregation and identification
In this formulation, each chromosome is a subpath with alternating segment and nonsegment edges. A connected component may incorporate multiple chromosomes. Therefore, in Algorithm 1, we compute Eulerian paths that force an alternation between segment and nonsegment edges. In case the only possible transition from segment edge (u, v) is to the segment edge (v, u), this represents a boundary between homologous chromosome pairs, and the subpaths are split at node v.
Eulerian decomposition is not unique, and multiple decompositions may exist. Let us assume that after path segregation, we
have a set of subpaths P = P1, P2, …. Now, consider two paths Pi and Pj that share the same segment s. In that case, a “crossover transition”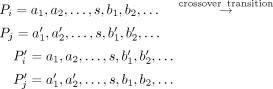 would generate another valid Eulerian path decomposition. We use this idea to heuristically refine the chromosomes based on
known biology. Specifically, OMKar counts the number of centromeres in each path. If there exists a pair of paths—one containing
two centromeres with at least one segment s in between and a second chromosome containing segment s but zero centromeres—OMKar performs a crossover transition on s to ensure that both paths now contain a single centromere. Finally, to standardize orientation, we flip the chromosomes so
that they are all oriented in the p-to-q direction. Each chromosome's orientation is identified by the centromeric segment
or, if it is acentric, by the majority orientation of all segments.
would generate another valid Eulerian path decomposition. We use this idea to heuristically refine the chromosomes based on
known biology. Specifically, OMKar counts the number of centromeres in each path. If there exists a pair of paths—one containing
two centromeres with at least one segment s in between and a second chromosome containing segment s but zero centromeres—OMKar performs a crossover transition on s to ensure that both paths now contain a single centromere. Finally, to standardize orientation, we flip the chromosomes so
that they are all oriented in the p-to-q direction. Each chromosome's orientation is identified by the centromeric segment
or, if it is acentric, by the majority orientation of all segments.
Event interpretation
Structural variants have somewhat conflicting definitions within the Genomics and Cytogenetics community. We developed an event interpretation module to describe SVs using the ISCN, described in Supplemental Table S5. OMKar automates the interpretation as follows (see Supplemental Section S3.5): It aligns SVs in reconstructed chromosomes with their wild-type (WT) counterparts, identified by centromeric or segmental makeup. It uses the longest common subsequence algorithm to create blocks and classify them as concordant, insertion, or deletion. Adjacent blocks with the same classification and contiguity are combined. Each insertion or deletion block is assigned an ISCN based on unique block-type signatures, in which indel size allowance determined via simulation (Supplemental Table S5; Supplemental Fig. S7). The system favors interpretations involving single, complex SVs over multiple simpler ones, providing a comprehensive explanation for the observed chromosomal deviations (Supplemental Section S3.5).
Report
Based on the interpreted SVs, disrupted genes that are present in the DDG2P database (Thormann et al. 2019) are reported. For balanced SVs, we looked at the boundaries within a resolution of 5 kbp for disrupted genes that might lead to a loss of function (resolution determined empirically with simulations) (Supplemental Section S4). For unbalanced SVs, we looked at the entire affected regions, for either gain or loss of gene product. Lastly, the allelic (monoallelic/biallelic) and mutational (loss/gain/altered gene product) requirements are used to filter for disrupted gene output and phenotype prediction.
An HTML report is compiled for ease of reading. It includes the decomposed paths (chromosomes), the corresponding visualization of the chromosome in both cytoband and segment views (Supplemental Section S3.6), the interpreted SVs under the ISCN language, and the disrupted developmental genes.
KarSim module for simulating karyotypes
The KarSim module (Supplemental Section S5.1; Supplemental Fig. S8) generates a molecular karyotype file, a FASTA file, and a history log of events for downstream use, including KarCheck comparisons, while also allowing for the simulation of different sequencing technologies.
Usage in simulation tests
Random karyotypes were generated to simulate a common genetic disorder from Decipher Database's CNV syndromes (Firth et al. 2009), followed by seven to 14 random de novo SVs from Supplemental Table S6. Certain genomic regions, including centromeres and telomeres, were masked during the analysis to ensure accurate structural variant placement. More details on the masked regions can be found in Supplemental Section S5.1. SVs were placed with breakpoints at least 50 kbp from masked regions, ensuring no segments <50 kbp were generated.
After generating the parameterized-random molecular karyotypes, simulated data were processed with OMSim (Miclotte et al. 2017) to generate OGM molecules with added noise. The standard Bionano Solve pipeline (v3.7) (Bionano Genomics 2018) was applied to compute CNVs, SVs, and contig alignments, which were then used as input for OMKar to reconstruct the final virtual karyotype. Full details on the simulation process and parameters can be found in Supplemental Section S5.3.
The KarCheck module for comparing karyotypes
Karyotypes from the simulation (Kt) are compared with reconstructed karyotypes (Kr) using the KarCheck module (Supplemental Section S6; Supplemental Fig. S8). Preprocessing is initially applied to Kt and Kr to divide chromosome groups into clusters. To achieve comparability, segments are further divided so both karyotypes share the same set of segments (Supplemental Section S6.1). For each chromosome cluster, three metrics are reported: (1) chromosome count concordance, (2) Jaccard similarity of SV edges, and (3) Jaccard similarity of CN.
SV similarity computation
SV similarity computation is performed by comparing nonsegment edges in the pair of chromosome clusters. The edges are matched (allowing for some tolerance) (for analysis, see Supplemental Section S4), and a Jaccard similarity score (intersection over union) is computed to measure similarity (Supplemental Section S6.2). OGM reads do not map with high confidence in the telomere, acrocentric p-arm, and acrocentric centromere regions. Therefore, these prefix/suffix regions were excluded in simulations and during SV similarity computations.
CN similarity comparison and metrics
CN similarity comparison is done by binning the whole genome (excluding prefix/suffix masked region) into spanning, nonoverlapping bins of 50 kbp, with a tolerance of ±100 bp (exact size chosen to maximize the size of the last bin on the chromosome). Each bin is used to store the average CN within that region, and a bin is termed “with CNV” if deviates more than 0.05 from diploid (chosen based on OGM's resolution). A Jaccard similarity is computed between the bins with CNV (Supplemental Section S6.3).
Ethics review statement
This study involved human participants and was approved by the ethics committees of the Medical University of Vienna (ethical code 2229/2019) and Keçiören Teaching and Research Hospital (ethical code 2012-KAEK-15/2083). The study adhered to the principles outlined in the Declaration of Helsinki. Informed consent was obtained from all participants prior to their inclusion.
Additionally, the study was conducted in accordance with the Declaration of Helsinki and received approval from the institutional review boards of the Western IRB–Copernicus Group (WCG) under study numbers 20203726 and 20212956. This approval included provisions for informed consent or waived authorization for the use of deidentified, banked samples for research purposes. All protected health information (PHI) was removed, and data were anonymized (coded and double-blinded) before accessioning for the study.
Software availability
The OMKar source code is publicly available under an open-source license at GitHub (https://github.com/siavashre/OMKar) and as Supplemental Code. The repository includes detailed documentation, example data sets, and scripts for reproducing key results presented in this study. To facilitate reproducibility and ease of deployment, we also provide a Docker image of OMKar that can be executed on Linux, macOS, or Windows or in cloud environments without manual dependency installation. A Conda version is also provided. The code for generating the figures is also available in the Supplemental Material.
Data access
The optical genome mapping (OGM) data sets generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB90248, and the European Genome-phenome Archive (EGA; https://ega-archive.org/) under accession number EGAS00001008245. All raw data used for karyotype reconstruction are included under these accession numbers. Metadata and analysis files are organized per sample, and detailed instructions for accessing and interpreting the data are provided in the repositories.
Competing interest statement
V.B. is a cofounder and a member of the scientific advisory board of Boundless Bio and Abterra and holds equity in both companies. A.H. was an employee of Bionano Genomics at the time of the work and owns a limited number of stock shares in the company. A.W.C.P. is currently an employee of Bionano Genomics. All other authors declare no competing interests.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grant R01GM114362. V.B. is a cofounder and scientific advisory board member of Boundless Bio (BBI) and Abterra, holding equity in both companies. BBI and Abterra were not involved in this research. We thank Gautam Kathir for developing the initial HTML report and Christopher Day for discussions on database-related aspects. We also acknowledge the authors of the two multicenter studies (Iqbal et al. 2023; Broeckel et al. 2024; Levy et al. 2024b) for providing 98 clinical OGM data sets used in the evaluation of OMKar. Some deidentified data used in this study originated from a study sponsored by Bionano Genomics, specifically from the “Validation of Optical Genome Mapping for the Identification of Constitutional Genomic Variants in a Postnatal Cohort” study (NCT05295277; https://clinicaltrials.gov/study/NCT05295277?term=Optical%20Genome%20Mapping&rank=6).
Author contributions: S.R.D. and Z.J. designed and implemented all software and analyses for this study, contributed to result interpretation, and cowrote the manuscript. J.E. assisted with software implementation. J.H. and N.M. provided data and contributed to the interpretation of cytogenetic results. N.G.-L. and J.N. provided clinical data. A.H. and A.C. provided data and contributed to the interpretation of cytogenetic results. A.W.C.P. provided data, contributed to cytogenetic interpretation, and codesigned the study scope. P.D. provided clinical data, contributed to result interpretation, and assisted in manuscript writing. V.B. codesigned the study scope, helped design the algorithms, contributed to result interpretation, and cowrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.280536.125.
-
Freely available online through the Genome Research Open Access option.
- Received February 13, 2025.
- Accepted August 15, 2025.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








