
KAS-ATAC reveals the genome-wide single-stranded accessible chromatin landscape of the human genome
- 1Cancer Biology Programs, School of Medicine, Stanford University, Stanford, California 94305, USA;
- 2Department of Genetics, School of Medicine, Stanford University, Stanford, California 94305, USA;
- 3Department of Applied Physics, Stanford University, Stanford, California 94305, USA;
- 4Center for Personal Dynamic Regulomes, Stanford University, Stanford, California 94305, USA;
- 5Chan Zuckerberg Biohub, San Francisco, California 94158, USA
-
↵6 These authors contributed equally to this work.
Abstract
Gene regulation in most eukaryotes involves two fundamental processes: alterations in genome packaging by nucleosomes, with active cis-regulatory elements (CREs) generally characterized by open-chromatin configuration, and transcriptional activation. Mapping these physical properties and biochemical activities, through profiling chromatin accessibility and active transcription, is a key tool for understanding the logic and mechanisms of transcription and its regulation. However, the relationship between these two states has not been accessible to simultaneous measurement. To this end, we developed KAS-ATAC, a combination of the kethoxal-assisted ssDNA sequencing (KAS-seq) and assay for transposase-accessible chromatin using sequencing (ATAC-seq) methods for mapping single-stranded DNA (and thus active transcription) and chromatin accessibility, respectively, enabling the genome-wide identification of DNA fragments that are simultaneously accessible and contain ssDNA. We use KAS-ATAC to evaluate levels of active transcription over different CRE classes, to estimate absolute levels of transcribed accessible DNA over CREs, to map nucleosomal configurations associated with RNA polymerase activities, and to assess transcription factor association with transcribed DNA through transcription factor binding site (TFBS) footprinting. We observe lower levels of transcription over distal enhancers compared with promoters and distinct nucleosomal configurations around transcription initiation sites associated with active transcription. We find that most TFs associate equally with transcribed and nontranscribed DNA, but a few factors specifically do not exhibit footprints over ssDNA-containing fragments. We anticipate KAS-ATAC to continue to derive useful insights into chromatin organization and transcriptional regulation in other contexts in the future.
Active cis-regulatory elements in most eukaryotes are usually characterized by low levels of nucleosome occupancy, a unique property first appreciated more than four decades ago (Wu 1980; Keene et al. 1981; McGhee et al. 1981). Therefore, active CREs are found in regions of increased chromatin accessibility. The genome-wide profiling of chromatin accessibility has been a key tool for identifying active regulator elements and assessing their dynamics across time and space. Methods for carrying out this mapping use the preferential cleavage or labeling of physically accessible DNA by various enzymes. Cleavage by DNase I was the first such approach to be widely adopted, initially coupled to microarray readouts (Dorschner et al. 2004; Sabo et al. 2006) and later combined with high-throughput sequencing in the form of DNase-seq (Crawford et al. 2006; Boyle et al. 2008; Thurman et al. 2012). An alternative approach has been to use methylation and either short- or long-read readouts to map chromatin accessibility both genome-wide and at a single-molecule level (Kelly et al. 2012; Krebs et al. 2017; Shipony et al. 2020). The most widely used assay for mapping open chromatin is ATAC-seq (Buenrostro et al. 2013), which is based on the preferential insertion of the hyperactive Tn5 transposase into accessible DNA.
Apart from identifying putative regulatory elements, ATAC-seq can also be used to map nucleosome positioning in the vicinity of CREs (Schep et al. 2015), and both DNase-seq and ATAC-seq can also footprint the interactions between individual transcription factors (TFs) and DNA, as TFs protect DNA from cleavage/insertion (Hesselberth et al. 2009; Pique-Regi et al. 2011; Neph et al. 2012a,b; Stergachis et al. 2014; Vierstra and Stamatoyannopoulos 2016; Vierstra et al. 2020).
Gene regulatory inputs in the form of TF occupancy and chromatin remodeler activity are integrated into changes in transcriptional activity at promoters. Therefore, mapping active transcription has also long been a key objective (contrasted with conventional RNA-seq profiling of steady-state polyadenylated transcripts [Mortazavi et al. 2008; Nagalakshmi et al. 2008; Sultan et al. 2008; Wilhelm et al. 2008], the levels of which reflect several additional layers of regulation of RNA stability).
To this end, several methods for mapping nascent transcription— based on adaptations of the nuclear run-on techniques or capturing RNA polymerase molecules associated with DNA, such as NET-seq (Churchman and Weissman. 2011), GRO-seq (Core et al. 2008, 2012), PRO-seq (Kwak et al. 2013), and variations (Tome et al. 2018)—have been developed. However, these methods often involve very elaborate experimental protocols, and thus, their deployment has been relatively limited. The recently developed KAS-seq assay (Wu et al. 2020), in contrast, provides a straightforward method for identifying RNA polymerase-associated DNA based on the highly specific covalent labeling of unpaired single-stranded guanine residues with N3-kethoxal (Fig. 1A); N3-kethoxal adducts can subsequently be subjected to a click reaction and the addition of biotin, which is then used to specifically pull-down labeled DNA fragments. Most ssDNA in the genome is found in the context of the transcriptional bubbles of elongating and paused RNA polymerases, with the rest arising from active replication and secondary structures such as G-quadruplexes (Bochman et al. 2012).
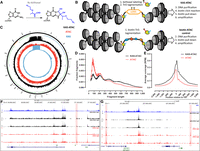
Overview of the KAS-ATAC assay. (A) N3-kethoxal specifically covalently labels unpaired guanine bases. (B) KAS-ATAC captures DNA fragments that are simultaneously single-stranded and accessible, by first labeling ssDNA with N3-kethoxal and then carrying out an ATAC-seq reaction. After purification of DNA and a click reaction that conjugates biotin onto kethoxal-labeled DNA, accessible ssDNA is specifically pulled down using streptavidin beads and amplified. In parallel, a control “biotin-ATAC” library can be prepared using biotinylated Tn5 that then undergoes the same pull-down procedure as KAS-ATAC DNA fragments (if the goal is to quantify the relative abundances of accessible ssDNA and accessible DNA). (C) KAS-seq, ATAC-seq, and KAS-ATAC mitochondrial genome profiles in human GM12878 cells. (D) Fragment length distribution in biotin-ATAC-seq and KAS-ATAC libraries (GM12878 cells). (E) Genome-wide TSS metaprofiles for biotin-ATAC-seq and KAS-ATAC libraries (GM12878 cells). (F,G) Representative genome browser snapshots showing ATAC-seq, KAS-seq, and KAS-ATAC profiles around nuclear loci in GM12878 cells.
Unlike RNA-based methods for mapping RNA polymerase activity and unlike previous methods for mapping ssDNA, such as KMnO4 footprinting (Hayatsu and Ukita 1967; Rubin and Schmid 1980; Mueller and Wold 1989; Sasse-Dwight and Gralla 1989; Kouzine et al. 2013, 2017), KAS-seq provides the unique opportunity to directly map the relationship between chromatin accessibility and polymerase activity (and potentially other ssDNA structures), that is, to what extent are various active CREs subject to active transcription, what nucleosomal configurations is this activity associated with, and what TFs may be enriched or depleted in terms of their physical associations with DNA on actively transcribed DNA fragments, as kethoxal labels the same DNA molecule that accessibility is measured on rather than a separate RNA one.
To address these questions, we developed KAS-ATAC, a method for jointly profiling ssDNA and chromatin accessibility (Fig. 1B). We applied KAS-ATAC together with appropriate controls to evaluate the relative degree of transcription in different classes of regulatory elements and chromatin states in the human genome, as well as to estimate the absolute abundance of transcribed and accessible/single-stranded structures. In addition, we map nucleosomal and subnucleosomal structures associated with active transcription and comprehensively measure TF footprints on polymerase-bound DNA molecules.
Results
KAS-ATAC maps identify genomic DNA fragments that are simultaneously accessible and contain single-stranded DNA
We developed KAS-ATAC by taking advantage of the complementary properties of the Tn5 transposase, which preferentially inserts into open chromatin, and the N3-kethoxal compound, which covalently attaches to unpaired G bases in ssDNA with very high specificity (Fig. 1A). N3-kethoxal adducts can then be subjected to click chemistry and biotin addition, and DNA fragments thus labeled can be stringently purified using streptavidin pull-down. To capture DNA fragments that are simultaneously accessible and containing ssDNA, we combined these two properties into KAS-ATAC as follows (Fig. 1B). First, cells are subjected to N3-kethoxal treatment to label ssDNA, after which the kethoxal is washed away. Next, cells are immediately processed through the standard ATAC-seq protocol, with Tn5 inserting into open chromatin regions. DNA is then isolated, and a click reaction is carried out, followed by streptavidin pull-down and library amplification. To control for sample loss and other potential biases associated with the pull-down procedure, we generated not just paired regular ATAC-seq libraries (from kethoxal-treated cells, but without a click reaction and biotin pull-down, in order to control for any effects of the kethoxal treatment) but also (for some key experiments) a “biotin-ATAC” control, in which no kethoxal labeling was carried out, but a transposase carrying biotinylated adapters was used, and then these fragments were pulled down following the same procedure that was applied for KAS-ATAC (Fig. 1B).
We carried out KAS-ATAC (together with parallel KAS-seq and ATAC-seq) in GM12878 lymphoblastoid cells and in HEK293 embryonic kidney cells as these have long been among the main cell lines of The ENCODE Project Consortium (The ENCODE Project Consortium 2012, The ENCODE Project Consortium et al. 2020) and as a wealth of other functional genomic data exists for them. Because we expected to recover less DNA than for regular ATAC libraries given that KAS-ATAC captures only a subset of the accessible genome and that the traditional ATAC-seq protocol only uses 50,000 cells as input, we pool at least two separate transposition reactions of 50,000 cells each for a given KAS-ATAC library.
As an initial assessment of the content of KAS-ATAC libraries, we examined KAS-ATAC, ATAC-seq, and KAS-seq profiles over the mitochondrial genome (Fig. 1C; Supplemental Fig. 1). This was for two reasons: first is the well-known high abundance of mitochondrial reads in ATAC libraries owing to the absence of nucleosomes in the mitochondrial nucleoid, leaving it highly susceptible to Tn5 insertion (Buenrostro et al. 2013; Marinov et al. 2014), and second is that the mitochondrial genome contains the most abundant ssDNA structure in cells, the D-loop in its noncoding region (Shadel and Clayton 1997). The D-loop is the major site of replication and transcription initiation in mitochondria (Cantatore and Attardi 1980; Montoya et al. 1982) and exists as a triple-stranded structure; that is, one of these three strands has numerous unpaired guanines and is accordingly highly enriched in KAS-seq libraries. We observe that although regular ATAC-seq libraries are slightly depleted over the D-loop, KAS-ATAC libraries are highly enriched over this region of Chr M, with a broadly similar, but also distinct in some places, profile compared with KAS-seq libraries. Thus, we conclude that KAS-ATAC indeed captures DNA fragments that are both accessible and single-stranded. Based on KAS-ATAC and ATAC-seq coverage over the D-loop, we estimate that we obtain at least 30× enrichment in KAS-ATAC over the transposition background. The overall fraction of reads originating from mitochondria was similar between all our matching KAS-ATAC and ATAC control libraries (Supplemental Fig. 2).
Next, we examined the broad properties of KAS-ATAC data sets in the nuclear genome. Typical ATAC-seq libraries exhibit a characteristic periodic fragment length distribution (Buenrostro et al. 2013), featuring a prominent subnucleosomal peak, corresponding to DNA fragments originating from within CREs; a second peak of size roughly that of the protection footprint of single nucleosomes in the immediate vicinity of CREs (as well as less frequent transposition events throughout the genome); a third dinucleosomal peak; and so on. We observe a similar picture in KAS-ATAC libraries (Fig. 1D); however, although in ATAC-seq libraries the subnucleosomal peak is often (though not always) the strongest, all KAS-ATAC libraries we have examined feature relatively more prominent nucleosomal peaks (Supplemental Fig. 3). This is to a certain extent counterintuitive and unexpected, given that the strongest KAS-seq peaks are those in the promoter regions of genes, corresponding to paused and initiating RNA polymerases; therefore, one may expect subnucleosomal fragments to dominate KAS-ATAC libraries. However, on the other hand, much of ssDNA globally originates from transcribed intragenic regions, where nucleosomes are mostly intact. Indeed, we compared fragment length distributions across ENCODE chromatin states (annotated by the ChromHMM algorithm) (Ernst and Kellis 2012), and we observed that the mononucleosomal peak is most prominent relative to the subnucleosomal one in the actively transcribed intragenic states (e.g., state 5 “Tx”) (Supplemental Figs. 5 and 4).
KAS-ATAC libraries also display a distinct signature around transcription start sites (TSSs), where a sharper peak is observed right at the TSS in metaprofile plots (Fig. 1E; Supplemental Fig. 6), likely corresponding to fragments associated with initiating/paused RNA polymerase.
The single-stranded accessible chromatin landscape is distinct from total open chromatin genomic maps
We then surveyed local single-stranded accessible profiles around the genome. Figure 1, F and G, shows representative KAS-seq, KAS-ATAC, and ATAC-seq snapshots in GM12878 cells. Both ATAC-seq and KAS-ATAC exhibit enrichment over promoters and distal regulatory elements in the genome; however, in a number of cases, KAS-ATAC peaks over promoter-distal CREs are considerably weaker than ATAC-seq signal over the same regions (e.g., highlighted areas in Fig. 1F,G).
We quantified global differences between ATAC and KAS-ATAC by carrying out differential accessibility analysis using DESeq2 (Love et al. 2014) on the set of common peaks called for both assays. In GM12878 cells, 1948 and 4641 regions were identified as preferentially enriched in ATAC-seq and KAS-ATAC, respectively, and we identified 2064 and 10,581 such peaks in HEK293 cells (Fig. 2A,B). We compared these differential regions against ChromHMM (Ernst and Kellis 2012) chromatin state annotations for these two cell lines and found that they are not uniformly and randomly distributed across chromatin states (Fig. 2C). KAS-ATAC libraries are relatively enriched over active TSSs (“TssA” state) and very strongly enriched over transcribed regions (“Tx” state), as well as over intragenic enhancer regions (“EnhG1/2”). They are depleted over active intergenic enhancers and over weak enhancers (“EnhG1” and “EnhWk”), and bivalent TSSs and enhancers. These observations generalized the anecdotal trend observed at individual loci; accessible intergenic enhancers appear to be less frequently engaged with polymerase bubbles than promoter regions. ATAC-seq and KAS-ATAC profiles around promoters and distal CREs also corroborate this conclusion (Fig. 2D); the two assays exhibit broadly similar profiles within promoters, but in distal CREs, it is often the case that KAS-ATAC signal is weaker.
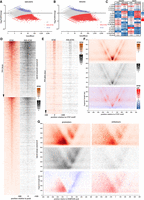
The single-stranded accessible chromatin landscape is distinct from total open chromatin genomic maps. (A) DESeq2 analysis of GM12878 ATAC and KAS-ATAC libraries. (B) DESeq2 analysis of HEK293 ATAC and KAS-ATAC libraries. (C) Enrichment of KAS-ATAC/ATAC differential peaks over ChromHMM chromatin states. Enrichment is calculated as the log2 ratio of the observed versus expected fractions of peaks overlapping each state (based on the overlap of the total set of peaks with ChromHMM states). (D) ATAC-seq and KAS-ATAC profiles around promoter-proximal and distal peaks (GM12878 cells). (E) ATAC-seq and KAS-ATAC profiles around occupied CTCF sites in GM12878 cells (excluding promoter-proximal CTCF peaks; CTCF ChIP-seq peak calls were obtained from ENCODE accession ENCFF827JRI). (F) ATAC-seq, KAS-ATAC, and KAS-ATAC/ATAC differential V-plot (Henikoff et al. 2011) around occupied CTCF sites. (G) ATAC-seq, KAS-ATAC, and KAS-ATAC/ATAC differential V-plot around RAMPAGE (Batut et al. 2013) peaks in promoter and in distal CRE regions (RAMPAGE peaks were obtained from ENCODE accession ID ENCSR000AEI).
Next, we turned our attention to the properties of sites occupied by the CTCF protein, which is the main insulator factor in mammalian cells (Bell et al. 1999). We excluded promoter-proximal CTCF sites from our analysis and focused on distal CTCF peaks only. KAS-ATAC signal is notably weaker at CTCF sites compared with ATAC-seq (Fig. 2E). CTCF also has a very strong effect on nucleosome positioning through its very robust and persistent occupancy of its own binding sites, often fixing the positions of more than a dozen nucleosomes around it (Fu et al. 2008); CTCF sites thus display a characteristic V-plot (Henikoff et al. 2011) signature in ATAC-seq data sets, with a strong subnucleosomal footprint corresponding to the binding of CTCF itself and larger fragments including neighboring nucleosomes surrounding it (Fig. 2F). This signature is also observed in KAS-ATAC data, although with a reduced magnitude. However, it has been previously reported that G-quadruplexes promote CTCF binding in mammalian cells (Wulfridge et al. 2023), and this could account for our observations of relatively weak KAS-ATAC signal in these data sets at CTCF sites. Another possibility is that this signal might be the product of background DNA “breathing” or DNA replication intermediates that CTCF associates with, as there is only modest enrichment of CTCF sites in regular KAS-seq libraries (Supplemental Fig. 9). Alternatively, although we observe very high enrichment of kethoxal-labeled ATAC fragments over the general accessible background, it is possible that we observe a residual accessibility signal, although KAS-ATAC levels around CTCF are inconsistent with (i.e., too high) our estimate for KAS-ATAC enrichment over ATAC (≥30×).
We also analyzed fragment structure around transcription initiation sites. To this end, we took advantage of existing ENCODE TSS annotations for total RNA obtained from the RAMPAGE (Batut et al. 2013) data sets, which we divided into promoter-proximal and promoter-distal groups, and then analyzed ATAC and KAS-ATAC fragment distributions (in a transcription directionality-aware manner) (Fig. 2G). ATAC-seq V-plots around promoters feature a prominent mass of subnucleosomal and some mononucleosomal fragments immediately upstream of the TSS (corresponding to occupancy by regulatory factors, and general TFs), another group of subnucleosomal fragments immediately around the TSS (likely corresponding to RNA polymerase initiation complexes), as well as +1 nucleosome and dinucleosomal-fragment masses downstream from the TSS. This signature is preserved in KAS-ATAC but with important differences; the upstream subnucleosomal fragments are depleted in KAS-ATAC, whereas the initiation complex ones are enriched, as is the +1 nucleosome and the downstream dinucleosome. Although transcription initiation in mammals has long been demonstrated to be bidirectional (Core et al. 2008; Seila et al. 2008), these results suggest possible preferential absolute abundance of polymerase bubbles in the primary direction of transcription (with a cautionary note that the upstream divergent TSS may not be as well positioned relative to the primary TSS and, thus, the V-plot signal could be somewhat diluted). In addition, engaged polymerase at the promoter and immediately downstream from it is associated with intact nucleosomes.
In contrast to promoters, transcribed distal CREs exhibit a similar picture in its broad structure in ATAC-seq data, but this signature is very weak in KAS-ATAC, and there is no preferential enrichment of KAS-ATAC fragments downstream from the transcription initiation site.
We also compared KAS, ATAC, and KAS-ATAC data sets to existing precision run on (PRO-seq) (Kwak et al. 2013) maps of active transcription at the RNA level. We observe similar correlation between KAS and KAS-ATAC and PRO-seq and better correlation between KAS/KAS-ATAC and PRO-seq over enhancer elements than over promoters (Supplemental Fig. 7). Finally, we compared KAS, ATAC, and KAS-ATAC in the context of the activity-by-contact (ABC) model (Fulco et al. 2019), relating biochemical activity at enhancer elements with promoter output; we observe similar predictive performance for the different modalities in terms of their ABC scores (Supplemental Fig. 8).
Estimating the absolute abundance of accessible ssDNA
KAS-ATAC also offers an opportunity to address the question of how often accessible DNA fragments contain ssDNA inside them in absolute terms. To answer this question, we used a paired biotin-ATAC data set so that we could control as closely as possible for the inevitable loss of material during sample handling and obtain comparable absolute-molecular-content ATAC and KAS-ATAC libraries. To maximize sample recovery, we also combined 12 normal ATAC samples (of 50,000 cells each, for a total of 600,000 cells input) into each of these libraries, which were generated in HEK293 cells. We sequenced them to saturation and estimated the total library complexity using the preseq algorithm (Fig. 3A; Daley and Smith 2013). As expected, the total molecular complexity of the KAS-ATAC libraries is lower by ∼50% than that of ATAC-seq ones.
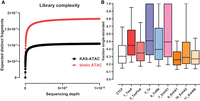
Estimating absolute levels of single-stranded accessible DNA. (A) Estimates for total library complexity in KAS-ATAC and biotin-ATAC HEK293 samples. (B) Distribution of the ratios between the number of KAS-ATAC molecules and biotin-ATAC molecules for ATAC-seq peaks found in different chromatin states and for CTCF occupancy sites.
We used the total molecular complexity of the KAS-ATAC and biotin-ATAC samples to calculate the total number of fragments (that was derived from about 600,000 cells) for each ATAC-seq peak found in a given ChromHMMM state, as well as for CTCF peaks, and then estimated the ratio between the two to evaluate how often accessible chromatin regions are also associated with ssDNA (Fig. 3B; Supplemental Fig. 10). The median estimate we obtain for active TSSs is that ∼45% of ATAC fragments contain ssDNA. The highest such estimates are for peaks in the actively transcribed (“Tx”) and intragenic enhancer (“EnhG1”) states, at ∼50%, but they often approach 100%. In contrast, for active intergenic enhancers, the median fraction of ssDNA fragments is in the 20%–25% range (“EnhA1/A2”). For CTCF sites, the median estimate is ∼30%. These results should be taken with some caution because of the possibility of capturing some baseline DNA “breathing” during kethoxal labeling, but overall, they corroborate the observations of lower levels of transcription at intergenic enhancers and of the presence of ssDNA structures at CTCF sites, while also quantifying their abundance in cells.
TF footprints in KAS-ATAC
Finally, we asked whether TFs are associated with DNA in a similar way regardless of whether transcriptions (or other processes generating ssDNA) are occurring in the immediate vicinity. To this end, we calculated aggregate TF footprints over TF motifs within ChIP-seq peaks for each TF in GM12878 and HEK293 cells, including all such TFs for which ChIP-seq data are available from the The ENCODE Project Consortium et al. (2020) and for which motifs are annotated in the Cis-BP database (Weirauch et al. 2014). We applied bias-correction utilizing bias models trained using the ChromBPNet framework (for details, see Methods), as Tn5 insertion bias is a well-documented confounding issue in ATAC-seq data sets (Sung et al. 2016; Baek et al. 2017; Li et al. 2019; Bentsen et al. 2020; Zhang et al. 2021).
For most TFs, we find a similar depth of footprints in both the ATAC and KAS-ATAC data sets (Fig. 4A–D; Supplemental Figs. 11–15). For example, CTCF footprints in HEK293 cells are largely the same in both the total ATAC library and within ssDNA-containing fragments (Fig. 4A), with concordant footprint depths and fine-scale features. Analogous conclusions apply to factors such as the bZIP family factor ATF3 (Fig. 4B), the nuclear receptor NR2F1 (Fig. 4C), the YY1 zing finger TF (Fig. 4D), and most others (Supplemental Figs. 12–15). Thus, most TFs appear to physically associate with DNA fragments in which active transcription is ongoing in the immediate vicinity.
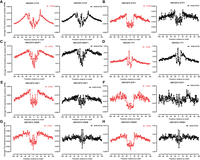
Transcription factor (TF) footprints in KAS-ATAC and ATAC data sets. Shown are the aggregate average Tn5 insertion profiles (for details, see Methods) around the motifs for the indicated factors that are located within ENCODE ChIP-seq peaks for the TF in the given cell line: (A) HEK293 CTCF, (B) GM12878 ATF3, (C) GM12878 NR2F1, (D) HEK293 YY1,(E) GM12878 USF1, (F) GM12878 E4F1, (G) GM12878 CREM, and (H) GM12878 CREB1.
However, we also find several TFs for which strong footprints are only observed in ATAC-seq libraries and not in KAS-ATAC. Examples include the bHLH TF USF1 (Fig. 4E), the zinc finger E4F1 factor (Fig. 4F), and the bZIP family TFs CREM and CREB1 (Fig. 4G,H). These are factors that appear to preferentially associate with DNA fragments that are not actively undergoing transcription, although the precise mechanisms underlying this phenomenon are still unclear for each individual case.
Discussion
In this work, we describe the KAS-ATAC assay, which we developed to identify the subset of physically accessible DNA fragments in the genome that also contain embedded ssDNA structures, the bulk of which is the result of the opening of the DNA double-strand associated with engaged RNA polymerase molecules. We use KAS-ATAC to answer several questions regarding the relationship between active transcription/ssDNA formation and fine-scale chromatin architecture.
We show that the genomic regions that are jointly accessible and containing ssDNA are preferentially located in promoters and inside gene bodies. In comparison, distal enhancers, although often comparably accessible to promoters, are considerably less frequently transcribed; in contrast, intragenic enhancers are enriched in the KAS-ATAC population of fragments, consistent with them being located within actively transcribed regions of the genome. These observations are in line with results obtained from active transcription mapping at the RNA level (Core et al. 2014; Tome et al. 2018). The promoter and enhancer classes of elements are also characterized by subtle differences in their subnucleosomal and nucleosomal organization as it relates to the act of transcription, with the single-stranded accessible footprint signature of enhancers being much less asymmetric than that of promoters with respect to the primary direction of transcription. We also observe a substantial presence of CTCF footprints in the KAS-ATAC data sets even though, as an insulator protein, CTCF is not thought to be associated with transcription-related ssDNA structures. Previous studies have suggested that G-quadruplexes, which do show up as robust ssDNA structures in KAS-seq data sets, may promote CTCF occupancy (Wulfridge et al. 2023), but it is also possible that we observe postbiotin pull-down residual CTCF-associated ATAC-seq fragments, although the magnitude of the observed KAS-ATAC CTCF footprints is inconsistent with the very high degree of enrichment of KAS-ATAC fragments over the background ATAC population that we estimate. This issue remains an open question to be resolved in the future.
We also evaluate the absolute fraction of accessible DNA that is engaged in transcription for different classes of regulatory elements, estimating that up to half of fragments within promoters contain ssDNA, whereas in distal enhancers, this fraction is less than one-fourth of fragments. These values should be taken with a cautionary note about the possibility of DNA breathing contributing some of these percentages, namely, they are upper bounds, but nevertheless, they are the first direct genome-wide estimates for these parameters. We hope that true single-molecule quantification will become available in the future and conclusively resolve these questions.
Finally, we demonstrate that TFs physically associate with actively transcribed DNA, and most of them do so with comparable frequency to non-ssDNA-containing DNA. This is not fully unexpected because in many cases the fragments that are enriched for by the KAS-ATAC assays are sufficiently large to accommodate both an elongating polymerase and TFs occupying their cognate sites nearby. However, a number of TFs are, in fact, depleted in KAS-ATAC libraries and are preferentially associated with non-ssDNA DNA fragments. We currently have no obvious mechanism that could account for such a preference; our study is, to the best of our knowledge, the first time it has been observed, and it will be an important question to more specifically investigate in the future.
We expect KAS-ATAC to be a useful tool for further future exploration of the detailed relationship between transcription/ssDNA structures and the physical organization of chromatin in many other contexts, as will the combination of kethoxal labeling and other molecular readouts of the chromatin structure.
Methods
Cell culture
GM12878 and HEK293T cells were grown using the standard protocols for each cell line: RPMI 1640, 2 mM L-glutamine, 15% fetal bovine serum and 1% penicillin–streptomycin for GM12878 cells and DMEM + sodium pyruvate, 6 mM L-glutamine, 10% fetal bovine serum and 1% penicillin–streptomycin for HEK293T cells.
KAS-seq
KAS-seq experiments were executed as previously described (Marinov et al. 2023a).
For HEK293 cells, media was removed and room-temperature 1× PBS was used to wash them, and then, they were dissociated with trypsin, which was quenched with media. Cells were pelleted at room temperature and then resuspended in 500 μL of media supplemented with 5 mM N3-kethoxal (final concentration). GM12878 cells were pelleted down by centrifugation at room temperature and also resuspended in 500 μL of media supplemented with 5 mM N3-kethoxal (final concentration). Cells were incubated for 10 min at 37°C with shaking at 500 rpm in a ThermoMixer and then pelleted by centrifuging at 500g for 5 min at 4°C. Genomic DNA was extracted using the Monarch gDNA purification kit (NEB T3010S) following the standard protocol but with elution using 175 μL 25 mM K3BO3 (pH 7.0).
The click reaction was carried out by combining transposed DNA, 5 μL 20 mM DBCO-PEG4-biotin (DMSO solution, Sigma-Aldrich 760749), 20 mM K3BO3 (pH 7.0), and 20 μL 10× PBS in a final volume of 200 μL.
Biotinylated DNA was purified using AMPure XP beads and sheared on a Covaris E220 instrument down to ∼150–200 bp size.
For streptavidin pull-down of biotin-labeled DNA, 10 μL of 10 mg/mL Dynabeads MyOne streptavidin T1 beads (Invitrogen 65602) was separated on a magnetic stand and then washed with 300 μL of 1× Tween washing buffer (TWB; 5 mM Tris-HCl at pH 7.5, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween 20). The beads were resuspended in 300 μL of 2× binding buffer (10 mM Tris-HCl at pH 7.5, 1 mM EDTA, 2 M NaCl); the sonicated DNA was added (diluted to a final volume of 300 μL if necessary); and the beads were incubated for ≥15 min at room temperature on a rotator. After separation on a magnetic stand, the beads were washed with 300 μL of 1× TWB and heated at 55°C in a ThermoMixer with shaking for 2 min. After removal of the supernatant on a magnetic stand, the TWB wash and 55°C incubation were repeated.
Final libraries were prepared on beads using the NEBNext Ultra II DNA library prep kit (NEB E7645) as follows. End repair was carried out by resuspending beads in 50 μL 1× EB buffer and adding 3 μL NEB ultra end repair enzyme and 7 μL NEB ultra end repair enzyme, followed by incubation for 30 min at 20°C (in a ThermoMixer, with shaking at 1000 rpm) and then for 30 min at 65°C.
Adapters were ligated to DNA fragments by adding 30 μL blunt ligation mix, 1 μL ligation enhancer, and 2.5 μL NEB adapter, incubating for 20 min at 20°C; adding 3 μL USER enzyme; and incubating for 15 min at 37°C (in a ThermoMixer, with shaking at 1000 rpm).
Beads were then separated on a magnetic stand and washed with 300 μL TWB for 2 min at 55°C, 1000 rpm in a ThermoMixer. After separation on a magnetic stand, beads were washed in 100 μL 0.1× TE buffer, resuspended in 15 μL 0.1× TE buffer, and heated for 10 min at 98°C.
For PCR, 5 μL of each of the i5 and i7 NEB Next sequencing adapters were added together with 25 μL 2× NEB ultra PCR mater mix. PCR was carried out with 30 sec of 98°C incubation; 12 cycles of 10 sec at 98°C, 30 sec at 65°C, and 1 min at 72°C; followed by incubation for 5 min at 72°C.
Beads were separated on a magnetic stand, and the supernatant was cleaned up using 1.8× AMPure XP beads.
Libraries were sequenced in a paired-end format on an Illumina NextSeq instrument using NextSeq 500/550 high-output kits (2 × 38 cycles).
Tn5 production
Tn5 transposase was produced as previously described (Picelli et al. 2014), with the following modifications of the adapter oligos for biotin-ATAC experiments:
-
Tn5MErev, /5Phos/CTGTCTCTTATACACATCT;
-
Tn5ME-A, /5Biotin/TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG; and
-
Tn5ME-B, /5Biotin/GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG.
ATAC-seq experiments
ATAC-seq experiments were carried out following the Omni-ATAC protocol as previously described (Corces et al. 2017; Marinov et al. 2023b). Briefly, about 50,000 cells (with or without prior kethoxal treatment) were centrifuged at 500g, resuspended in 500 μL 1× PBS, and centrifuged again. Cells were then resuspended in 50 μL ATAC-RSB-lysis buffer (10 mM Tris-HCl at pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630, 0.1% Tween-20, 0.01% digitonin) and incubated on ice for 3 min. Subsequently 1 mL ATAC-RSB-wash buffer (10 mM Tris-HCl at pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, 0.01% digitonin) were added; the tubes were inverted several times; and nuclei were centrifuged at 500g for 5 min at 4°C.
Transposition was carried out by resuspending nuclei in a mix of 25 μL 2× TD buffer (20 mM Tris-HCl at pH 7.6, 10 mM MgCl2, 20% dimethyl formamide), 2.5 μL Tn5 transposase (custom produced), and 22.5 μL nuclease-free H2O and incubating for 30 min at 37°C in a ThermoMixer at 1000 RPM.
Transposed DNA was isolated using the MinElute PCR purification kit (Qiagen 28004/28006) and PCR-amplified as previously described (Corces et al. 2017; Marinov et al. 2023b): 3 min at 72°C; 30 sec at 98°C; 10 cycles of 10 sec at 98°C, 30 sec at 63°C, and 30 sec at 72°C. Libraries were purified using the MinElute kit and then sequenced on an Illumina NextSeq 550 as 2 × 38mers.
KAS-ATAC experiments
For KAS-ATAC experiments, cells were processed and subjected to N3-kethoxal treatment as described above for KAS-seq.
Subsequently, they were washed by centrifugation and removal of media, resuspension in 500 μL 1× PBS, and centrifugation. The ATAC-seq step was then carried out by resuspending cells in 50 μL ATAC-RSB-lysis buffer, incubating on ice for 3 min, adding 1 mL ATAC-RSB-wash buffer, centrifuging at 500g for 5 min at 4°C, and transposing in a total volume of 50 μL. DNA purification was carried out using the MinElute PCR purification kit.
Multiple parallel ATAC reactions were carried out, including about 50,000 cells each, and then were pooled together for the click reaction and biotin pull-down.
Click reaction was carried out as described above, by combining 175 μL transposed DNA, 5 μL 20 mM DBCO-PEG4-biotin (DMSO solution; Sigma-Aldrich 760749), 20 mM K3BO3 (pH 7.0), and 20 μL 10× PBS in a final volume of 200 μL. Biotinylated DNA was purified using the MinElute PCR purification kit.
For streptavidin pull-down of biotin-labeled DNA, 10 μL of 10 mg/mL Dynabeads MyOne streptavidin T1 beads (Invitrogen 65602) were separated on a magnetic stand and then washed with 300 μL of 1× Tween washing buffer (TWB; 5 mM Tris-HCl at pH 7.5, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween 20). The beads were resuspended in 300 μL of 2× binding buffer (10 mM Tris-HCl at pH 7.5, 1 mM EDTA, 2 M NaCl); the biotinylated DNA was added (diluted to a final volume of 300 μL if necessary); and the beads were incubated for ≥15 min at room temperature on a rotator. After separation on a magnetic stand, the beads were washed with 300 μL of 1× TWB and heated at 55°C in a ThermoMixer with shaking for 2 min. After removal of the supernatant on a magnetic stand, the TWB wash and 55°C incubation were repeated.
Libraries were prepared on beads using the same settings as described above (3 min 72°C; 30 sec at 98°C; 10 cycles of 10 sec at 98°C, 30 sec at 63°C, 30 sec at 72°C). Beads were separated on a magnetic stand, and the supernatant was cleaned up using the MinElute PCR purification kit.
Biotin-ATAC experiments
ATAC-seq experiments were carried out as described above but using a custom-produced biotinylated Tn5 transposase to tagment chromatin.
Purified transposed DNA was then subjected to streptavidin pull-down and PCR amplification on beads as described above.
ATAC-seq, KAS-seq, and KAS-ATAC data processing
Computational processing was carried out as previously described (Marinov and Shipony 2021; Marinov et al. 2023a).
Demultiplexed FASTQ files were mapped to the GRCh38 (hg38) assembly of the human genome as 2 × 36mers using Bowtie (Langmead et al. 2009) with the settings -v 2 -k 2 -m 1 ‐‐best ‐‐strata. Mitochondrial mapping reads were filtered out, and duplicate reads were removed using picard-tools (version 1.99).
Reads were also mapped separately to the mitochondrial genome using the settings -v 2 -a ‐‐best ‐‐strata, both to estimate the extent of mitochondrial contamination and to generate mitochondrial genome coverage tracks.
Browser track generation, fragment length estimation, TSS enrichment calculations, and other analyses were carried out using custom-written Python scripts (see Data access). The RefSeq annotation was used for evaluation of enrichment around TSSs.
Peak calling was carried out using MACS2 (Feng et al. 2012), with the following settings: -g hs -f BAMPE ‐‐to-large ‐‐keep-dup all ‐‐nomodel.
Absolute library complexity was estimated using the lc_extrap function in the preseq package (Daley and Smith 2013).
For differential accessibility analysis, a set of unified peaks was compiled across all libraries and conditions being compared, and read counts were calculated for each peak. Differential analysis was the carried out using DESeq2 (Love et al. 2014).
Final mapping statistics for all data sets used in this study are available in Supplemental Table 1.
TF motif analyses
As a first step, motif locations for all human TF motifs in the CIS-BP database were identified genome-wide using FIMO (Grant et al. 2011). These maps were then intersected with ChIP-seq peaks from The ENCODE Project Consortium (Supplemental Table 2).
Aggregate footprints were generated for each TF by calculating the average number of Tn5 insertions (shifted by +4/−4 bp) normalized to read per million (RPM). Bias correction was carried out using a genome-wide bias prediction track for a ChromBPNet model (https://github.com/kundajelab/chrombpnet) trained on the very deeply sequenced main ENCODE ATAC-seq data set for GM12878 cells by subtracting the rescaled predicted bias profile from the observed ATAC/KAS-ATAC insertion profile.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE264534. Except where explicitly indicated otherwise, data were processed using custom-written Python scripts available at GitHub (https://github.com/georgimarinov/GeorgiScripts) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Anusri Pampari, Zohar Shipony, and Anna Shcherbina for technical assistance and members of the Greenleaf and Kundaje laboratories for helpful discussions and suggestions. This work was supported by National Institutes of Health grants (P50HG007735, RO1 HG008140, U19AI057266, UM1HG009442, and 1UM1HG009436 to W.J.G.), the Rita Allen Foundation (to W.J.G.), the Baxter Foundation Faculty Scholar grant, and the Human Frontiers Science Program grant RGY006S (to W.J.G.). W.J.G. also acknowledges support by grants 2017-174468 and 2018-182817 from the Chan Zuckerberg Initiative. S.H.K. was supported by Medical Scientist Training Program (MSTP) training grant T32GM007365 and the Paul and Daisy Soros Fellowship. Fellowship support was also provided by the Stanford School of Medicine Dean's Fellowship (G.K.M.).
Author contributions: G.K.M. and S.H.K. conceived the project. S.H.K. and G.K.M. carried out experiments. G.K.M. analyzed data. G.K.M. and S.H.K. wrote the manuscript with input from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279621.124.
- Received May 24, 2024.
- Accepted November 19, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








