
Parameter-efficient fine-tuning on large protein language models improves signal peptide prediction
Abstract
Signal peptides (SPs) play a crucial role in protein translocation in cells. The development of large protein language models (PLMs) and prompt-based learning provide a new opportunity for SP prediction, especially for the categories with limited annotated data. We present a parameter-efficient fine-tuning (PEFT) framework for SP prediction, PEFT-SP, to effectively utilize pretrained PLMs. We integrated low-rank adaptation (LoRA) into ESM-2 models to better leverage the protein sequence evolutionary knowledge of PLMs. Experiments show that PEFT-SP using LoRA enhances state-of-the-art results, leading to a maximum Matthews correlation coefficient (MCC) gain of 87.3% for SPs with small training samples and an overall MCC gain of 6.1%. Furthermore, we also employed two other PEFT methods, prompt tuning and adapter tuning, in ESM-2 for SP prediction. More elaborate experiments show that PEFT-SP using adapter tuning can also improve the state-of-the-art results by up to 28.1% MCC gain for SPs with small training samples and an overall MCC gain of 3.8%. LoRA requires fewer computing resources and less memory than the adapter tuning during the training stage, making it possible to adapt larger and more powerful protein models for SP prediction.
Signal peptides (SPs) are short amino acid sequences typically located in the N-terminals of nascent polypeptides and are universally present in many proteins of a wide range of prokaryotic and eukaryotic organisms (Owji et al. 2018). Most SPs direct proteins to enter the secretory (Sec) pathway for translocation across the prokaryotic plasma membrane or the eukaryotic endoplasmic reticulum membrane. SPs containing a twin-arginine motif (R-R) target proteins to the twin-arginine translocation (Tat) pathway (Palmer and Berks 2012). The primary difference between the Sec and Tat pathways is that the Sec pathway transports proteins in unfolded conformation, whereas the Tat pathway translocates fully folded proteins (Palmer and Stansfeld 2020).
Upon successful translocation of the protein across the membrane, the SP is precisely cleaved at a specific cleavage site (CS) by signal peptidase (SPase). Subsequently, the mature protein is released on the trans side of the membrane (Freudl 2018). The SPases are categorized into three groups: SPase I, II, and III (sometimes referred to as SPase IV) (Dalbey et al. 2012). SPase I cleaves general Sec SPs, whereas SPase II and SPase III cleave SPs from lipoproteins and prepilin proteins, respectively. SPase I (Sec/SPI), SPase II (Sec/SPII), or SPase III (Sec/SPIII) can handle the processing of Sec substrates, whereas Tat substrates are exclusively processed by SPase I (Tat/SPI) or SPase II (Tat/SPII).
The SPase CS is recognized by SPase. Most SPs have a common tripartite structure, comprising a positively charged n-region, a central hydrophobic h-region spanning approximately five to 15 residues, and a c-region housing the CS for SPase I. Lipoprotein SPs, cleaved by SPase II, are recognized through the presence of a lipobox in the c-region. Prepilin SPs, subject to processing by SPase III, exclusively consist of a vital translocation-mediating region, as opposed to the conventional tripartite structure (Owji et al. 2018). The amino acid composition and length of the SP regions exhibit diversity, which allows them to adapt to the specific requirements of various proteins within distinct cellular contexts. Although these SP regions are recognizable, the absence of clearly defined consensus motifs presents a significant challenge to SP prediction.
With the advances in machine learning and deep learning technologies, numerous applications for SP prediction have been developed and widely used in bioinformatics research. SignalP versions 1–4 (Nielsen et al. 1997; Nielsen and Krogh 1998; Bendtsen et al. 2004; Petersen et al. 2011) are machine learning–based methods designed to predict Sec-translocated SPs cleaved by SPase I (Sec/SPI) and the corresponding CS locations. SPEPlip (Fariselli et al. 2003) employs a neural network approach combined with PROSITE patterns (Hulo et al. 2004), allowing for the identification of SPs cleaved by Spase I (Sec/SPI) and lipoprotein SPs cleaved by Spase II (Sec/SPII). DeepSig (Savojardo et al. 2018) utilizes convolutional neural networks (CNNs) and grammar-restrained conditional random fields (CRFs) to predict Sec-translocated SPs cleaved by Spase I and their CS. SignalP 5.0 (Almagro Armenteros et al. 2019) incorporates CNN and long short-term memory networks to predict Sec substrates cleaved by Spase I (Sec/SPI) or Spase II (Sec/SPII), as well as Tat substrates cleaved by Spase I (Tat/SPI). In contrast to its predecessors, SignalP 6.0 (Teufel et al. 2022) stands out as a remarkable tool capable of predicting all five types of SPs (Sec/SPI, Sec/SPII, Tat/SPI, Tat/SPII, Sec/SPIII) through ProtTrans (Elnaggar et al. 2022), a robust protein language model (PLM) pretrained on the UniRef100 data set (Suzek et al. 2007, 2015) with a mask language model objective. Nevertheless, its performance in predicting SP with limited training samples leaves room for improvement.
Large PLMs, such as ProTrans and ESM-1 (Rives et al. 2021), have become foundational tools for various biological modeling tasks related to proteins. Recently, ESM-2 increased the number of parameters in the transformer model, which has led to substantial advancements in downstream protein prediction tasks (Lin et al. 2023). The most common approach using pretrained PLMs for downstream tasks involves fine-tuning these models by updating all the parameters to leverage the information from the pretrained model effectively. Although fine-tuning a model has proven to be a competitive strategy, the extensive fine-tuning process becomes impractical for PLMs owing to significant computational requirements or a lack of large training samples. To tackle this challenge, a new strategy of prompt-based learning has emerged, focused on parameter-efficient fine-tuning (PEFT) for large language models (LLMs), such as adapter tuning (Houlsby et al. 2019), prompt tuning (Lester et al. 2021), and low-rank adaptation (LoRA) (Hu et al. 2021). These techniques introduced parameters within the pretrained model, keeping all remaining parameters frozen during the training phase to mitigate the effects of catastrophic forgetting (Kirkpatrick et al. 2017). The gradients of these frozen parameters are neither computed nor stored during back-propagation, substantially reducing computational and memory costs, as well as the need for large training samples. Moreover, these approaches have demonstrated competitive performance compared with fine-tuning for various natural language processing tasks (He et al. 2021; Li and Liang 2021; Liu et al. 2022; Chen et al. 2023; Dettmers et al. 2024) and protein structure–related tasks (Wang et al. 2022, 2023).
In this paper, we present a novel SP prediction framework, PEFT-SP, designed to harness the capabilities of PLM for SP and CS prediction. PEFT-SP consists of the ESM-2 model, a linear CRF model, and PEFT modules, including adapter tuning, prompt tuning, and LoRA. The ESM-2 model serves as the backbone for encoding amino acid sequences and is kept frozen during the training phase. The CRF probabilistic model takes the representations generated by ESM-2 as input and predicts all five types of SPs and their corresponding CS. The PEFT method fine-tunes ESM-2 to suit the SP prediction task better. Our framework is an end-to-end solution, focused exclusively on optimizing parameters within CRF and PEFT modules. To demonstrate the effectiveness of our framework, we conducted a comprehensive performance comparison against existing SP predictors, including a state-of-the-art tool, SignalP 6.0. Our results indicate that PEFT-SP using LoRA with ESM2-3B surpasses both the state-of-the-art tool and fine-tuned ESM-2 models across all five SPs. Notably, PEFT-SP using LoRA significantly improves SP performance with limited training data. Additionally, we thoroughly investigated the performance of PEFP-SP using different PEFT methods with the ESM-2 model family for SP prediction.
Our framework PEFT-SP outperforms the current state-of-the-art model, SignalP 6.0, in two types of SPs with limited training samples, and it achieves comparable or superior performance in three other SP types with larger training data sets. We comprehensively evaluate ESM-2 fine-tuned and PEFT-SP using different combinations of the PEFT methods (including prompt tuning, adapter tuning, and LoRA) with the ESM-2 model family in the context of SP prediction. Enhancing the efficient utilization of PLMs is crucial in improving SP prediction performance, particularly given the continuous expansion in the scale of LLMs in recent years. Although PEFT achieves success in natural language understanding, to the best of our knowledge, this is the first study whose authors explore the effectiveness of PEFT on PLMs for SP prediction. Furthermore, to facilitate user-friendly SP prediction, we developed a web server leveraging our proposed PEFT-SP framework. The interface allows users to easily conduct SP prediction in batches.
Results
Comparisons with state-of-the-art methods
Because the well-trained models of SignalP 6.0 for nested cross-validation are not publicly available, we retrained it using the same data sets and default hyperparameters reported in the original paper. We employed PEFT-SP using LoRA for each model from the ESM-2 model family and trained them independently. We evaluated the MCC1 and MCC2 scores (see Methods) for each SP type within each organism group across test sets. Additionally, we calculated the mean MCC1 and MCC2 scores across all SP types and organisms.
PEFT-SP using LoRA with a ESM2-3B backbone achieved the best performance (as shown in Fig. 1A,B). It consistently outperformed SignalP 6.0 in the SP types (Sec/SPIII and Tat/SPII) with limited training samples, except for Tat/SPII in Gram-positive bacteria. It achieved a maximum MCC1 gain of 79.8% and MCC2 gain of 87.3% in Sec/SPIII for Archaea. It attained a mean MCC1 improvement of 5.6% and a mean MCC2 improvement of 6.1%. For SP types (Sec/SPI, Sec/SPII, and Tat/SPII) with sufficient training data, PEFT-SP using LoRA with ESM2-3B demonstrated superiority to SignalP 6.0, with MCC1 gains ranging between 1.8% and 7.2% and MCC2 gains ranging between 0.6% and 18.5%. It performed slightly worse than SignalP 6.0, with MCC1 differences ranging between 0.3% and 3.0% and MCC2 differences ranging between 0.4% and 11.5%, in Sec/SPI and Sec/SPII for Archaea and in Tat/SPII for both Gram-negative and Gram-positive bacteria. The Wilcoxon signed-rank test comparing all MCC2 values generated from SignalP 6.0 and PEFT-SP using LoRA with ESM2-3B yielded a P-value of 0.039, indicating a statistically significant improvement. The area under the ROC curve and area under the precision-recall curve analysis on SignalP 6.0 and PEFT-SP using LoRA with ESM2-3B further revealed the superior predictive capabilities of our method on most SP types (Supplemental Figs. S1, S2). We also visualized the confusion matrices for each organism group in Supplemental Figure S3. These matrices illustrate that PEFT-SP, using LoRA with the ESM2-3B backbone, exhibits strong performance in SP-type prediction with fewer classification errors compared with SignalP 6.0.
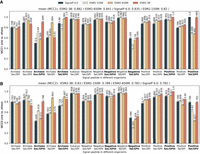
PEFT-SP using LoRA and SignalP 6.0 performance in terms of MCC score for each SP type across different organisms. The bold text in the x-axis represents the SP type with small training samples. The MCC1 and MCC2 scores are shown above the bars. The sorted mean values for MCC1 and MCC2 are listed at the top. (A) MCC1 scores performance on a negative class composed of soluble and transmembrane proteins. (B) MCC2 scores performance on a negative class comprising soluble and transmembrane proteins and other SP types.
We also computed precision and recall for CS prediction in PEFT-SP using LoRA and SignalP 6.0 (as shown in Fig. 2). Regarding precision, the PEFT-SP using LoRA with a ESM2-3B backbone outperformed SignalP 6.0 in the Tat/SPII SP type, which is particularly notable given the limited training data for this type.

Results of PEFT-SP using LoRA and SignalP 6.0 in precision and recall for CS prediction across different organisms. The precision and recall were calculated within a tolerance window size of 0. The bold text in the x-axis represents the SP type with small training samples.
Comparisons with other baseline models
Considering the excellent performance of the PEFT-SP using LoRA with the ESM2-3B (Lin et al. 2023) backbone, we compared it against all other baseline models. The performances for all baseline models were initially reported in SignalP 6.0 (Teufel et al. 2022). We included these performances in the benchmark. The benchmark also included the performance of SignalP 6.0, both when trained by our team and as reported in the original paper. The original baseline models were obtained from their publicly available web services, and all performance measurements were conducted on the same test sets generated through nested cross-validation. It is worth noting that, except for SignalP 6.0, the baseline models were trained on SP types with large training samples, and consequently, their performance regarding Sec/SPIII and Tat/SPII SP types has not been reported. Table 1 demonstrates that PEFT-SP using LoRA with ESM2-3B outperformed all baseline models (Gomi et al. 2004; Käll et al. 2004, 2007; Bendtsen et al. 2005; Chou and Shen 2007; Bagos et al. 2008, 2009, 2010; Rahman et al. 2008; Reynolds et al. 2008; Zhang and Shen 2017; Savojardo et al. 2018; Almagro Armenteros et al. 2019; Teufel et al. 2022) in Sec/SPI for Eukarya, Sec/SPI and Sec/SPII for Gram-negative organisms, and all SP types for Gram-positive bacteria. Benchmark results for the recall of CS prediction in Sec/SPI, Sec/SPII, and Tat/SPI in four tolerance windows can be found in Supplemental Tables S1–S4.
Benchmark results for SP prediction in Sec/SPI, Sec/SPII, and Tat/SPI
Comparisons with fine-tuning and other PEFT methods
We compared PEFT-SP using different PEFT methods with ESM2-3B, as well as SignalP 6.0 and the fine-tuned ESM2-3B model. We trained all models independently with the same data sets generated from nest cross-validation. The performance of each model was measured using MCC2 by cross-validation.
Table 2 shows that the fine-tuning approach outperformed SignalP 6. This suggests that the ESM2-3B model holds promise as a potential candidate for other PEFT methods. The PEFT-SP using LoRA performed better than PEFT-SP using prompt tuning and adapter tuning regarding the mean MCC2. Moreover, the PEFT-SP using LoRA has fewer trainable parameters than fine-tuning and other PEFT methods during the training stage, reducing the computing resource and memory storage. The number and percentage of trainable parameters for PEFT-SP are listed in Supplemental Table S5.
Benchmark results of MCC2 for SignalP 6.0, Fine-tuning ESM2-3B, and PEFT-SP models using different PEFT methods with the ESM2-3B backbone
To comprehensively analyze the effectiveness of PEFT-SP using various PEFT methods with the ESM-2 model family, we benchmarked their results based on MCC1 and MCC2 for SP prediction (as presented in Supplemental Tables S6, S7, respectively), and precision and recall for CS prediction (as presented in Supplemental Tables S8, S9, respectively). According to the benchmark results of MCC1 and MCC2, PEFT-SP using LoRA with ESM2-3B still performed the best compared with other combinations. PEFT-SP using adapter tuning with ESM-650M performed better than SignalP 6.0. It achieved a maximum MCC2 (MCC1) gain of 28.1% (50.0%) in the SP types with limited training samples and a mean MCC2 (MCC1) gain of 3.8% (2.7%) across all SP types. For Archaea Sec/SPII and Negative Sec/SPIII, LoRA tuning underperforms the fine-tuning method. These discrepancies could be attributed to the distribution of sequence representations. The t-distributed stochastic neighbor embedding (t-SNE) (Van Der Maaten and Hinton 2008) plot (as shown in Supplemental Fig. S4) shows Sec/SPI and Sec/SPII cluster together, indicating a lack of discriminative patterns between the two groups. Although LoRA tuning preserves the backbone model better than fine-tuning to reduce the effect of catastrophic forgetting, it also more heavily relies on the frozen backbone model to capture the underlying features relevant to the task. Hence, LoRA may not always outperform fine-tuning.
Visualization and interpretation of attention weights
To understand why the PEFT-SP using LoRA with ESM2-3B outperformed others, we visualized the attention weights in the last layer of the transformer in the ESM2-3B model to investigate the motifs of SP types. We used the sequence logos to visualize the attention weights, inspired by MULocDeep (Jiang et al. 2021). For a comprehensive comparison, we created a gold standard and predicted label sequence logos for each SP type. We compared the relationship between these logos at each sequence position using the Spearman's rank correlation. The SP type with the highest correlation is shown in Figure 3, A–C, and corresponding logos for other SP types are presented in Supplemental Figures S5–S8.
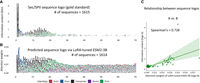
The sequence logo of Sec/SPII generated with (A) known Sec/SPII sequences (represented by information content) and (B) predicted Sec/SPII sequence patterns via LoRA-tuned ESM2-3B (represented by attention weights). (C) Comparison between the gold-standard sequence logo and the predicted sequence pattern using Spearman's rank correlation between information content and attention weights. Each dot represents one position on the sequence.
Based on the sequence logos (Fig. 3A–C), the gold-standard sequence logo (predicted label sequence logo) shows regions with high information content (attention weights) in the N terminus and low information content (attention weights) when it is far away from the N terminus, indicating that LoRA tuning enables the frozen ESM2-3B model to capture relevant signals for SP prediction. We compared the relationship between the information content of the gold-standard logo and the attention weights of LoRA-tuned ESM2-3B for the first 70 positions at the N terminus by Spearman's rank correlation. The Spearman's rank correlation between these two logos is 0.728, indicating that the attention weights can reflect the information content and be used to predict motif. The sequence patterns between the two logos also look similar; for example, the dominant amino acid is L followed by A and V at the peak positions of the logos. This finding is consistent with the general features of Sec and Tat SP types mentioned in previous studies (Freudl 2018; Teufel et al. 2022).
Ablation test on PEFT-SP using LoRA with ESM-2 model family
The PEFT-SP using LoRA with ESM2-3B demonstrated superior performance compared with the ESM2-150M and ESM-650M models overall, yet it fell short of outperforming ESM2-150M and ESM2-650M in specific SP types. Because the number of LoRA modules in the ESM-2 model family is the most critical factor influencing the number of tunable parameters that contribute to the performance of models, we delved deeper into its impact by training PEFT-SP with varying numbers of LoRA modules and assessing MCC2 across cross-validation. Both LoRA hyperparameters, rank and alpha, were set to eight.
As detailed in Supplemental Table S10, models with more layers of LoRA modules tended to exhibit better performance than those with fewer layers. This observation implies that the increased number of tunable parameters contributes significantly to enhancing the overall MCC2 performance. Although models with fewer LoRA modules may outperform the SP types with sufficient data, they exhibit much poorer performance in the SP types with limited data. This tendency suggests that fewer LoRA modules may bias the model toward certain SP types with sufficient data. Additionally, when the number of LoRA modules is close to the number of transformers in the ESM-2 model family, the model may exhibit poorer performance in the SP types with limited data.
Web server for PEFT-SP
To make PEFT-SP accessible, we developed a user-friendly web server building on MULocDeep, which is publicly available at https://www.mu-loc.org/peftsp/. Users can submit jobs by uploading their sequences in the FASTA format through the website. All the jobs are queued for computing allocation. Our web server applies the guest privacy policy, enabling users to retrieve their results without logging in. We have also created a comprehensive user guide providing instructions on the functions and usage of the PEFT-SP website.
Discussion
This work presents PEFT-SP as a novel SP prediction framework. It takes protein sequence as input without organism identifier. PEFT-SP using LoRA with ESM2-3B demonstrated its capability to effectively handle SP types with limited training data sets and deliver performance comparable to or better than the baseline models across all SP types. The effectiveness of PEFT-SP using LoRA can be primarily attributed to the following factors: (1) Larger scaling of models can improve future performance of SP prediction; (2) PEFT-SP leverages the property of the ESM2-3B backbone model, which captures the evolutionary aspects of protein sequences, making good representation of sequences; and (3) PEFT-SP employs LoRA, a lightweight fine-tuning method, to adapt PLM to SP prediction while preserving the high quality of the PLM.
We explored fine-tuning and different PEFT methods with the ESM2 family model for SP prediction. The fine-tuned models from the ESM2 model family performed better than the SignalP 6.0, suggesting ESM2 models may be better suited for PEFT methods than other PLMs. That said, it is important to choose an appropriate PEFT method for a given task to achieve optimal performance. For example, although prompt tuning showed superior performance compared with the state-of-the-art tools in numerous tasks, we did not observe the same level of performance in SP prediction, possibly owing to the ESM2-3B model being too small. PEFT-SP using adapter tuning outperforms SignalP 6.0 but introduces a massive number of training parameters compared with PEFT-SP using LoRA. LoRA tuning relies heavily on the PLM, making it sensitive to the quality of the representations generated by ESM2-3B. The sequences in different SP types may have very similar distributions, which could limit the performance of PEFT-SP if the pretrained ESM2-3B fails to distinguish between these SP types effectively.
To our knowledge, this is the first study to explore PLM's effectiveness when using the PEFT approach for SP prediction tasks. There are several directions for future work: (1) PEFT-SP, combined with PEFT methods, such as integrating PEFT-SP using LoRA and adapter tuning, potentially yielding complementary improvement; (2) modification of PEFT-SP to enhance interpretability, thereby unveiling the underlying motifs associated with SP; and (3) exploration of the use of structure-aware PLM models as backbones, incorporating protein structure information to enhance SP prediction further. We believe this study opens exciting new possibilities for applying PLMs not only in the SP prediction task but also in other protein analysis tasks, such as targeting peptide prediction and protein cellular localization.
Methods
Pretrained large PLMs
In recent years, several PLMs have emerged. For example, ProtTrans, ESM-1, and ESM-2 models have been trained on sequences from the UniRef (Suzek et al. 2007, 2015) protein sequence database using a masked language modeling objective. These models are specifically designed for protein feature extraction and can function as a foundation for fine-tuning in SP prediction tasks. The state-of-the-art model SignalP 6.0 utilized ProtBert, derived from ProtTrans, as its backbone model. Unlike ProtBert, which has only a single version available, the ESM-2 model family has various larger models. Previous studies have established a relationship between model scale and learning of protein structure, suggesting that further scaling up of the models may continue to improve the performance (Rao et al. 2020; Meier et al. 2021; Rives et al. 2021). The ESM-2 model family encompasses varying model sizes ranging from 8 million parameters to a substantial 15 billion parameters. The ESM-2 model family, including ESM2-150M, ESM2-650M, and ESM2-3B, outperforms other PLMs from ProtTrans and the ESM-1 model family in protein sequence–related tasks (Lin et al. 2023). According to the t-SNE analysis with the Calinski–Harabasz index, the pretrained ESM-2 model family generated better clustering of sequence representations than the pretrained ProtTrans, suggesting the ESM-2 model family may be promising backbone models (Supplemental Fig. S9). Furthermore, we individually replaced the backbone in SignalP 6.0 with three backbones from the ESM-2 model family and fully fine-tuned them on the SP data sets. The ESM-2 model family also outperformed ProtBert on the SP prediction (Supplemental Fig. S10). Hence, based on the solid performance in these protein sequence–related tasks, we employed the ESM-2 model family as the pretrained backbone in our subsequent experiments. It is worth noting that although ESM-15B is available and exhibits excellent predictive capabilities, we had to exclude it from our study because of computational resource limitations.
Unlike existing SP prediction models that require appending an organism identifier to the protein sequence, PEFT-SP with a
ESM-2 backbone (as shown in Fig. 4A) takes only the protein sequence S as input, encoding it into token embeddings. The token embeddings of the sequence are then fed into a stack of multiple transformer
layers, designed to learn contextual relationships between amino acids. Each transformer layer consists of a self-attention
mechanism and position-wise feed-forward networks (FFNs) surrounded by separate residual connections. In the self-attention
mechanism, the attention function processes the input feature X and transforms it into three different vectors of dimension d: query (Q), key (K), and value (V). This transformation uses three weight matrices: Wq, Wk, and Wv. Subsequently, the scaled dot-product attention calculates the scaled attention score by performing a dot product between
the Q and the K values associated with the amino acid token. It then converts this score into a probability distribution using the Softmax
function: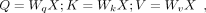 (1)
(1)
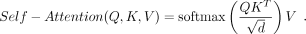 (2)
(2)
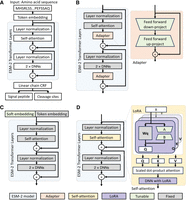
The architectures for the ESM-2 model and PEFT-SP using different PEFT modules. The light green modules are tunable during training, whereas the gray modules are fixed. (A) The ESM-2 backbone model uses amino acid sequences for SP and CS prediction. (B) PEFT-SP using adapter tuning contains a bottleneck architecture. (C) PEFT-SP using prompt tuning appends soft embedding into token embedding. (D) PEFT-SP using LoRA adds trainable rank decomposition matrices into the self-attention layer.
The output of the self-attention mechanism is a representation achieved through the weighted summation of values. The FFN
module is constructed from two linear transformations, each activated by the rectified linear unit function (Agarap 2018), yielding a sequence of hidden states. We removed the special tokens (CLS and SEP) introduced from the backbone, and retained
a sequence of hidden states h with the same length as the input sequence S: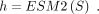 (3)
(3)
PEFT methods for ESM-2
PEFT is a technique to improve the performance of LLM on various downstream tasks. It achieves efficiency by introducing tunable parameters while freezing the original parameters in the backbone model. Thus, the model can be tailored to new tasks with reduced computational overheads and fewer labeled training samples. The PEFT methods reduce computing resources during the training stage by updating the introduced parameters only. Unlike the original configuration of adapter tuning and LoRA, which incorporate related modules into all transformer layers, we specifically inserted them into the bottommost L transformer layers within the ESM-2 model. The idea comes from the LLaMA-Adapter (Zhang et al. 2023), designed to enhance the fine-tuning of representations with higher-level semantics.
Adapter tuning
The adapter tuning (Houlsby et al. 2019) incorporates adapter modules with a bottleneck architecture within the transformer layer of the ESM-2 model. These adapter modules are introduced as distinct components, positioned after the projection phase following self-attention and after the two feed-forward layers. Each adapter module comprises a residual connection and a bottleneck architecture, which compress the input data into a bottleneck layer with reduced dimensionality and subsequently reconstruct the data to match the original input size. The fusion of the ESM-2 model with adapter modules is illustrated in Figure 4B.
Prompt tuning
The prompt tuning (Lester et al. 2021) method involves the addition of trainable embeddings, as “soft prompts,” into the sequence embeddings, which serve as inputs to the ESM-2 model. Considering the high sensitivity of prompt tuning to prompt initialization, prompts are initially set using embeddings of randomly selected amino acids. All parameters within the ESM-2 model remain fixed throughout the training process, whereas the soft prompts are continuously updated using gradients. Including soft prompts in the input sequence introduces extra hidden states generated by the ESM-2 model. To ensure that the length of the hidden states matches the sequence length, we omitted the hidden states associated with the soft prompts. An overview of the ESM-2 model with prompt tuning is provided in Figure 4C.
Low-rank adaptation
LoRA (Hu et al. 2021) is built on the idea that trainable weights have a low “intrinsic rank.” This characteristic enables the weights to learn
effectively, even when randomly projected into a smaller subspace. We performed lightweight fine-tuning of ESM-2 (Lin et al. 2023) by introducing trainable rank decomposition matrices into the transformer architecture, implementing LoRA (as shown in Fig. 4D). Specifically, this reparameterization is applied to the projection matrices of the query, key, value, and FFN modules
within the transformer. The trained weight matrix, denoted as W0 ∈ Rd×k, is coupled with a low-rank decomposition matrix ΔW = BA, where B ∈ Rd×r, A ∈ Rr×k, and r ≪ min(d, k). Both W0 and ΔW are simultaneously employed on the same input, and the output vectors they generate are combined elementwise. Hence, the
functions for transforming X into a query (Q), key (K), and value (V) within the transformer, as shown in Equation 1, undergo the following modifications: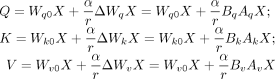 (4)
where α is a scaling constant. We initialized A with random Gaussian initialization and B to zero, resulting in ΔW = BA to zero and preserving the original knowledge in the ESM-2 model at the beginning of training. During the training state,
the pretrained weight matrix W0 is kept frozen, and the low-rank decomposition matrix ΔW is trainable by updating A and B with a gradient. The hyperparameters α and r are determined through the hyperparameter tuning process.
(4)
where α is a scaling constant. We initialized A with random Gaussian initialization and B to zero, resulting in ΔW = BA to zero and preserving the original knowledge in the ESM-2 model at the beginning of training. During the training state,
the pretrained weight matrix W0 is kept frozen, and the low-rank decomposition matrix ΔW is trainable by updating A and B with a gradient. The hyperparameters α and r are determined through the hyperparameter tuning process.
Linear chain CRF
The linear chain CRF (Lafferty et al. 2001) is widely used in sequence labeling tasks, which can capture relationships between the labels in a sequence and the observed
data. The linear chain CRF takes the sequence of hidden states h from the ESM-2 model and assigns regions of y for each sequence position based on the dependencies between neighboring states. The linear chain CRF can be written as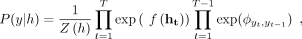 (5)
where Z(h) is a normalizing constant, f( · ) denotes a linear transformation that prepares the emissions for the CRF, and ϕ represents the transition matrix governing the probability of yt−1 following yt. To accurately predict the defined regions of the SP class, which include the n-region, h-region, c-region, and twin-arginine
motif, the ϕ transition matrix is constrained in the same way as SignalP 6.0 (Teufel et al. 2022).
(5)
where Z(h) is a normalizing constant, f( · ) denotes a linear transformation that prepares the emissions for the CRF, and ϕ represents the transition matrix governing the probability of yt−1 following yt. To accurately predict the defined regions of the SP class, which include the n-region, h-region, c-region, and twin-arginine
motif, the ϕ transition matrix is constrained in the same way as SignalP 6.0 (Teufel et al. 2022).
The Viterbi decoding process computes the most probable state sequence, encompassing the predicted SP class regions. For CS
prediction, the linear chain CRF predicts the regions of SP. The CS is identified based on the last state of the region for
the SP class within the most likely state sequence. The forward–backward algorithm calculates the marginal probabilities for
each sequence position. To predict the type of SP, the probability of a specific SP type is calculated by summing the marginal
probabilities associated with all states belonging to a particular type and then dividing by the sequence length: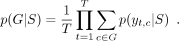 (6)
(6)
Loss function and regularization term
We constructed the loss function and regularization term following the approach of SignalP 6.0. We treated the SP region prediction task as a multilabel classification problem at the training stage. Specifically, positions around the boundaries of the regions were labeled as multilabel. For instance, a position near both the n-region and h-region was designated as both the n-region and h-region, reflecting the absence of a strict definition for region borders.
The loss function is derived from the negative log-likelihood of the linear chain CRF. At a specific position, the set of
ground-truth labels is denoted as Mt. The formulation of the loss function is as follows: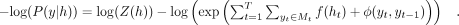 (7)
(7)
The regularization aims to promote diverse amino acid compositions within the three SP regions (n-region, h-region, c-region). We summed the marginal probabilities of all CRF states c belonging to region r for all positions with amino acid a, yielding a vector of scorer for each region r. Diversity scores are then calculated using cosine similarity for the vectors of scores between the n-region and h-region, as well as between the h-region and c-region, for each sequence. The mean of these diversity scores across all sequences is incorporated into the loss function with a scaling factor.
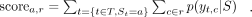 (8)
(8)
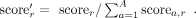 (9)
(9)
Model evaluation
We applied the same metrics used in baseline models, specifically the MCC, commonly employed in other SP prediction methods for a fair comparison. Considering that most existing methods involve binary classification for distinguishing SP from non-SP, we calculated MCC metrics (MCC1) using a data set in which negative samples comprised transmembrane and soluble proteins. In addition, we computed MCC metrics (MCC2) on a data set in which a specific SP type was designated as the positive sample, with all other SP types and non-SP as the negative samples.
Because the CS prediction in our method relies on the last state of the region for the SP class, the output of the CS prediction is the position of the CS in the protein sequence rather than the probabilities of the CS for each position in the sequence. Precision and recall are used to assess the performance of CS prediction within a tolerance window of up to three. Precision is the ratio of correct CSs to the total number of predicted CSs, whereas recall is the ratio of correctly predicted CSs to the total number of true CSs. For both metrics, a CS prediction was deemed accurate only if the predicted SP was also correct. For example, if the model predicts a CS within a Sec/SPI sequence but assigns Sec/SPII as the SP label, the sample is not considered in precision and recall calculation. These calculations are designed to measure the CS prediction under the correct SP prediction.
Data set
We utilized the benchmark data set obtained from the SignalP 6.0 web server (https://services.healthtech.dtu.dk/services/SignalP-6.0/), which includes a diverse set of protein sequences. The data set consists of 2582 Sec/SPI, 1615 Sec/SPII, 72 Sec/SPIII, 365 Tat/SPI, 33 Tat/SPII, 16,421 intracellular sequences, and 2615 transmembrane sequences. The SP types with limited training samples are Sec/SPIII and Tat/SPII. Each protein sequence in the data set is accompanied by information about its SP type and region labels at each position, with the final label associated with the SP type indicating the CS. The data set was initially acquired from four organism groups: Archaea, Eukarya, Gram-positive, and Gram-negative bacteria. The data set was partitioned into three subsets to ensure fairness and robustness, with similar sequences grouped within each partition but no significant sequence similarity between proteins of different partitions. Furthermore, each partition was meticulously balanced across the four organism groups. Our experimental design involved a threefold nested cross-validation process encompassing a twofold inner loop and a threefold outer loop. This configuration resulted in six distinct test sets. The distribution of the number of samples and the distribution of CS for SP types within each organism group are depicted in Supplemental Figures S11 and S12, respectively.
Experiment setting
We employed a pretrained PLM from the ESM-2 (Lin et al. 2023) model family as the backbone for our model, comprising ESM2-150M, ESM2-650M, and ESM2-3B. Throughout the training process, the backbone model remained frozen. In contrast, the linear chain CRF and all parameters introduced by our PEFT methods, including LoRA (Hu et al. 2021), prompt tuning (Lester et al. 2021), and adapter tuning (Houlsby et al. 2019), were trainable. The model was trained end-to-end with the Adamax (Kingma and Ba 2014) optimizer. For model selection, we computed a combined MCC2 score for SP prediction and an MCC score for CS prediction. The best model was chosen from the validation sets based on the mean of these evaluation values. To optimize hyperparameters, we utilized Gaussian optimization provided by Optuna (Akiba et al. 2019). The specific hyperparameters and their corresponding search ranges are detailed in Supplemental Table S11. All runs were trained on an Nvidia A100 GPU with a batch size of 20.
We first compared our PEFT-SP using LoRA with the state-of-the-art model SignalP 6.0. Subsequently, we evaluated PEFT-SP using LoRA against all baseline models trained on the SP data set with sufficient training samples. Finally, we extended our comparisons to fine-tuning and PEFT-SP using different combinations of PEFT methods (including prompt tuning, adapter tuning, and LoRA) with the ESM-2 model family. Our goal was to attain better performance in both SP and CS prediction compared with other existing methods.
Sequence logo
We generated gold-standard and predicted label sequence logos for each SP type. The gold-standard sequence logos were created using Shannon's entropy on amino acid frequencies, calculated with the ggseqlogo package (Wagih 2017). The predicted label sequence logos were generated using attention weights from the LoRA-tuned ESM2-3B model, calculated as the frequency of amino acids weighted by position attention weights. We visualized the sequence logos using ggseqlogo. The relationship between the two sequence logos was assessed using the Spearman's ranked correlation between the information content in the gold-standard sequence logos and the attention weights in the predicted label sequence logos.
Software availability
The SP data set was downloaded from the web service of SignalP 6.0. The source code of PEFT-SP and trained models are publicly available at GitHub (https://github.com/shuaizengMU/PEFT-SP) and as Supplemental Code. The web server is available at https://www.mu-loc.org/peftsp/.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Yuexu Jiang and Fei He for their useful discussions. This work was funded by the National Institutes of Health (R35-GM126985) and the National Science Foundation (DBI-2145226).
Author contributions: S.Z., D.W., and D.X. conceived and designed the study. S.Z. developed the methodology and performed the experiments. L.J. developed the web server. S.Z. wrote the original draft. D.L. and D.X. reviewed and edited the manuscript. All authors approved the final version.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279132.124.
- Received February 15, 2024.
- Accepted July 15, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








