
Abstract
The main way of analyzing genetic sequences is by finding sequence regions that are related to each other. There are many methods to do that, usually based on this idea: Find an alignment of two sequence regions, which would be unlikely to exist between unrelated sequences. Unfortunately, it is hard to tell if an alignment is likely to exist by chance. Also, the precise alignment of related regions is uncertain. One alignment does not hold all evidence that they are related. We should consider alternative alignments too. This is rarely done, because we lack a simple and fast method that fits easily into practical sequence-search software. Described here is the simplest-conceivable change to standard sequence alignment, which sums probabilities of alternative alignments and makes it easier to tell if a similarity is likely to occur by chance. This approach is better than standard alignment at finding distant relationships, at least in a few tests. It can be used in practical sequence-search software, with minimal increase in implementation difficulty or run time. It generalizes to different kinds of alignment, for example, DNA-versus-protein with frameshifts. Thus, it can widely contribute to finding subtle relationships between sequences.
Many methods have been developed for finding related parts of nucleotide or protein sequences. They usually follow a standard approach: define positive and negative scores for letter matches, mismatches, and gaps and then seek high-scoring alignments. Why is this bizarre, intricate approach a good way to find related sequences? Its result depends utterly on the score parameters, so how should we choose them?
The answer to both questions is that the approach is equivalent to probability-based alignment (Dayhoff et al. 1978; Durbin et al. 1998). This uses probabilities of matches, mismatches, and gaps, for example, their average rates in the kinds of related sequences we wish to find, and seeks high-probability alignments. An alignment's score is a log probability ratio (Frith 2020):
A more direct way to judge whether two sequence regions are related is to calculate their probability without fixing an alignment. In other words, use a score like this:
A similar sum-over-alignments method has been used with great success in the HMMER software (Eddy 2008). So, it is curious that many sequence-search tools developed more recently than HMMER do not use this approach. No one has described a sum-over-alignments method that fits easily into typical sequence-search software, with minimal increase in implementation difficulty or computational cost. HMMER is too complicated and specialized; for example, it has an enormous “implicit probabilistic model,” and sequence-length–dependent parameters (Eddy 2008). HMMER is designed to search a smallish “query” sequence against a large “database,” so it cannot find related parts of two large sequences, for example, two genomes.
It is important to know whether a similarity score (Equations 1, 2) is likely to occur by chance between unrelated sequences. The simplest definition is as follows: the probability of an equal or higher score between two sequences of random independent and identically distributed (i.i.d.) letters. Even this simplest definition, however, is hard to calculate. There is a solution for gapless alignment, in the limit where the sequences are long (Karlin and Altschul 1990). Gapped alignment relies on conjecture and simulation. BLAST can calculate it only for a few sets of score parameters, not including realistic DNA scores that distinguish transition (a:g, c:t) from transversion mismatches (Altschul et al. 1997). Some widely used aligners do not calculate it at all (Harris 2007). The ALP library can calculate it for arbitrary parameters, by complex simulations (Sheetlin et al. 2016). Notably, HMMER calculates it in an easier way based on other conjectures. The scope of those conjectures is unclear; for example, they seem to hold only after tweaking HMMER to use a “uniform entry/exit distribution” (Eddy 2008).
Here is described a minimal modification of standard alignment, which sums probabilities of alternative alignments (Equation 2). It can replace the alignment component of practical sequence-search software, with minimal increase in implementation difficulty or run time. This also makes it easier to calculate probabilities of similarity scores between random sequences, based on a clear conjecture.
Limitations of previous methods
Many previous studies have summed probabilities of alternative alignments. This section surveys some representatives to understand why this approach is rarely used to find related parts of sequences.
One line of research considered substitutions, insertions, and deletions occurring over time, so that match/mismatch/gap probabilities depend on the length of time (Bishop and Thompson 1986; Thorne et al. 1991; Thorne and Churchill 1995; Hein et al. 2000; Knudsen and Miyamoto 2003; Rivas and Eddy 2015). These methods achieve several aims. Given two related sequences, they can infer substitution/insertion/deletion rates per unit time, find the most likely alignment, and report the probability that any pair of letters descends from a common ancestor. Furthermore, Hein et al. (2000) used a log likelihood ratio to compare two hypotheses, that two whole sequences are related or not:
Another line of research (including the present study) simplifies things by ignoring time dependence and directly using probabilities of matches, mismatches, and gaps. Allison et al. (1992) estimated such probabilities from a pair of related sequences, allowing complex gap-length distributions. They also used a log likelihood ratio to compare hypotheses that two whole sequences are related or not.
In a pioneering study, Bucher and Hofmann (1996) used a log likelihood ratio to compare two hypotheses, that two sequences have a related segment or not:
HMMER uses a log likelihood ratio similar to Equation 4 (Eddy 2008). HMMER is more general, however. It finds related parts between a sequence and a sequence family: The family has position-specific letter and gap probabilities. The null hypothesis (no related parts) is compared to either a “unihit” hypothesis (one related part) or a “multihit” hypothesis (one or more related parts). Each hypothesis has probabilities for the length of the sequence. The hypotheses are roughly equal in this regard. HMMER achieves that by adjusting each hypothesis to fit the length of each sequence that is provided for analysis (Eddy 2008). Again, scores of related regions decrease with increasing sequence length. HMMER's DNA version, nhmmer, applies this analysis to sequence windows around potential matches in long sequences (Wheeler and Eddy 2013).
Lunter et al. (2008) compared DNA sequences accurately, using probability methods similar to the present study. In their scenario, they are given a pair of sequences that are assumed to have one related region, so they did not use any score to find related regions.
FEAST is practical heuristic software for finding related sequence parts, and it sums probabilities of alternative alignments (Hudek and Brown 2011). It does not determine whether similarities would be unlikely to exist between random sequences. It is slower than standard alignment, and its implementation difficulty was not made clear.
Several of these previous methods use a likelihood ratio, which is the optimal way to judge between two hypotheses with those likelihoods (Neyman and Pearson 1933). It is, however, not clear that comparing two hypotheses is the best way to find related sequence parts. For example, if we compare the human and mouse X Chromosomes, it is not very useful just to know whether or not they have zero related parts. There is a tradeoff between judging whether sequences are related, and how they are related. A single alignment specifies a relationship in detail but has the lowest power to detect subtle relationships. Summing over all alignments has the highest power but the least detail. We seek a compromise between these extremes.
The final previous idea that we shall examine is “hybrid alignment” (Yu and Hwa 2001). When hybrid alignment compares a sequence of length m to a sequence of length n, it considers m × n hypotheses for how they are related. Each hypothesis concerns the length-i prefix of one sequence and length-j prefix of the other. The hypothesis is that the sequences have a related region that ends exactly at the ends of these prefixes. The probability associated with this hypothesis is the sum of probabilities of all alignments that start anywhere and end at the ends of the prefixes. This sum of alignment probabilities is converted to a score (Equation 2). It is easy to calculate the probability of an equal or higher score between random i.i.d. sequences, provided that the alignment parameters satisfy a “conservation condition.” This calculation was not proven correct but was supported by theoretical arguments and tests on random sequences.
The present study builds on hybrid alignment. Hybrid alignment has been neglected, perhaps because its description was mathematically intricate, and it was presented as a “semiprobabilistic,” “hybrid” method. Actually, it has an entirely probabilistic interpretation: The point is that it adds probabilities of some alignments, rather than all or just one alignment. The formulas for calculating hybrid alignment scores correspond to a particular probability model (see Supplemental Fig. S2 in the work of Frith 2020). It is not necessary to use this exact model. Here, we shall use a model that makes the equations and algorithms as simple and fast as possible. Instead of alignments ending at the end of the length-i prefix of one sequence and length-j prefix of the other, we shall sum over alignments that pass through this (i, j) point. This treats uncertainty about starts and ends of related regions symmetrically. It is a good fit for practical heuristic aligners, which extend alignment forward and backward from a candidate starting point.
Methods
Review of standard alignment
Practical methods for finding related parts of sequences often have two steps. To cope with big data, the first step uses fast heuristics to find potentially related regions, and then, the second step defines the regions precisely by alignment. The heuristics are various: short exact matches, spaced matches, reduced-alphabet matches, groups of nearby matches, etc. (Altschul et al. 1997; Ma et al. 2002; Noé and Kucherov 2004; Roytberg et al. 2009). The second step is often done by extending alignments to the left and right of a small “core” alignment (Fig. 1B). This finds the highest-scoring alignment extension within a limited range, as shown in gray in Figure 1B. The range is adjusted as the algorithm proceeds, attempting to follow the highest score. The best way to do this is unclear: Several ways have been suggested (Altschul et al. 1997; Zhang et al. 1998; Suzuki and Kasahara 2017; Guidi et al. 2021; Liu and Steinegger 2023).
Some ways of extending an alignment in both directions from a small “core” alignment (black). In each direction, an optimum alignment extension is sought from among all possible ones within the gray area. (A) Simple toy method: The gray areas are blocks of predefined size n × n. (B) Realistic example: The gray areas are adjusted as the calculation proceeds, attempting to follow good alignments. (C) Unlimited extension from zero-size core.
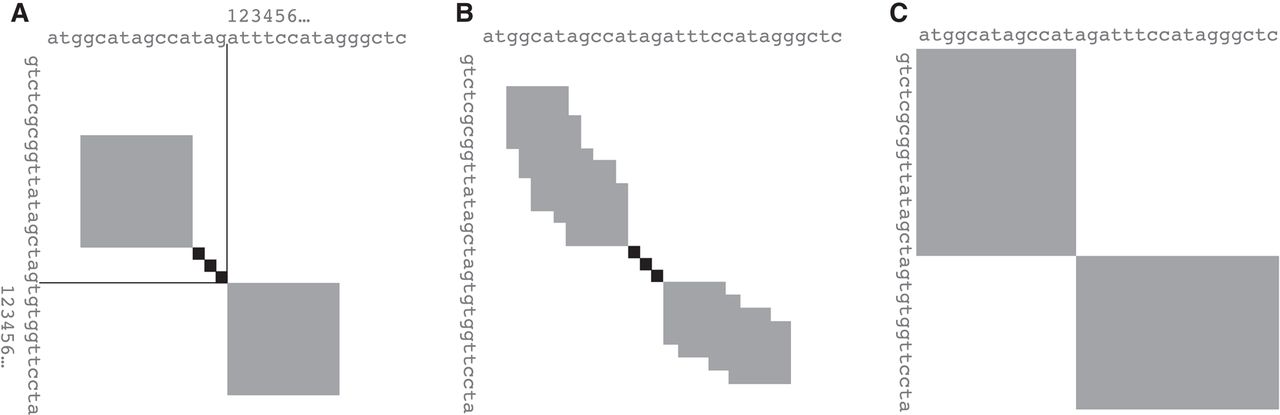
To avoid irrelevant complexity, here is the extension algorithm in a block of predefined size n × n (Fig. 1A). This algorithm extends forward (toward the lower-right in Fig. 1A): A similar algorithm extends backward. The two sequences are R (e.g., “reference”) and Q (e.g., “query”), with positions numbered as in Figure 1A. The score for aligning letter x (a, c, g, or t for DNA) in R to letter y in Q is Sxy. Let us use different scores for starting versus extending gaps, because gaps are somewhat rare (low start probability) but often long (high extension probability). So the score for a length-k deletion is aD + bD × (k − 1); for a length-k insertion, aI + bI × (k − 1).
Algorithm 1 finds the highest possible score for an alignment ending at Ri and Qj, with Ri aligned to Qj (Xij) or with Ri aligned to a gap (Yij) or with Qj aligned to a gap (Zij). Thus, w is the highest possible score for a global (end-to-end) alignment of R1, …, Ri and Q1, …, Qj. v tracks the best alignment score seen so far and is the final output. Several variants of this algorithm are possible. This variant has simple boundary conditions, while minimizing add, max, and array access operations (Cameron et al. 2004). The alignment score after left and right extension is
New extension algorithm
This section shows the new algorithm (Algorithm 2), and the next section explains how it sums probabilities. There are two changes from Algorithm 1. The first, which makes no difference to the result, is to work with exponentiated values: probability ratios instead of log probability ratios. This is so we can easily add them. This change means that −∞ ⇒ 0, 0 ⇒ 1, and addition ⇒ multiplication. The other change is that maximization ⇒ addition. This calculates the sum of probability ratios for all alignment extensions in the gray area (Equation 2) instead of the maximum probability ratio. The score with a left and right extension is
The core score is the same as in Equation 5. The core is typically a short, high-similarity, gapless alignment. Alternative alignments of the core region are not considered.
Note that the number of computational operations is unchanged: The only changes are replacing addition with multiplication, and maximization with addition. So, the computational complexity is unchanged. The practical run time depends on implementer expertise (e.g., using SIMD), but plausibly, it need not increase too much.
Unfortunately, the values (probability ratios) in Algorithm 2 may become too large for the computer to handle. This can be fixed by occasionally rescaling them (Supplemental Methods). Because the rescaling is only done occasionally, it does not increase the run time noticeably.
Parameters from probabilities
To use Algorithms 1 or 2, we must choose values for the parameters (e.g., , ). They are related to probabilities of matches, mismatches, and gaps. We need a precise definition of these probabilities. We shall use the definition in Figure 2. The main alignment probabilities are shown in the middle: αD is the deletion start probability, βD the deletion extension probability, αI the insertion start probability, and βI the insertion extension probability. Apart from starting an insertion or deletion, the other possibilities are to have a pair of aligned letters (probability, γ), or end the alignment (probability, 1 − γ − αD − αI). A pair of aligned letters has probability πxy of being letter type x in sequence R and y in sequence Q. There are also probabilities for letters left and right of the alignment: ωD and ωI (for sequence R and Q, respectively). An unaligned letter is of type z with probability ϕz in R, and ψz in Q.
A probability model for two sequences with a related region. The model produces different alignments with different probabilities. Starting at “begin,” the arrows are followed according to their probabilities (e.g., ωD vs. 1 − ωD). Each pass through a D state produces an unaligned letter x in one sequence (R), with the probabilities ϕx. Each pass through an I state produces an unaligned letter y in the other sequence (Q), with the probabilities ψy. Each pass through M generates two aligned letters, x in R and y in Q, with the probabilities πxy.
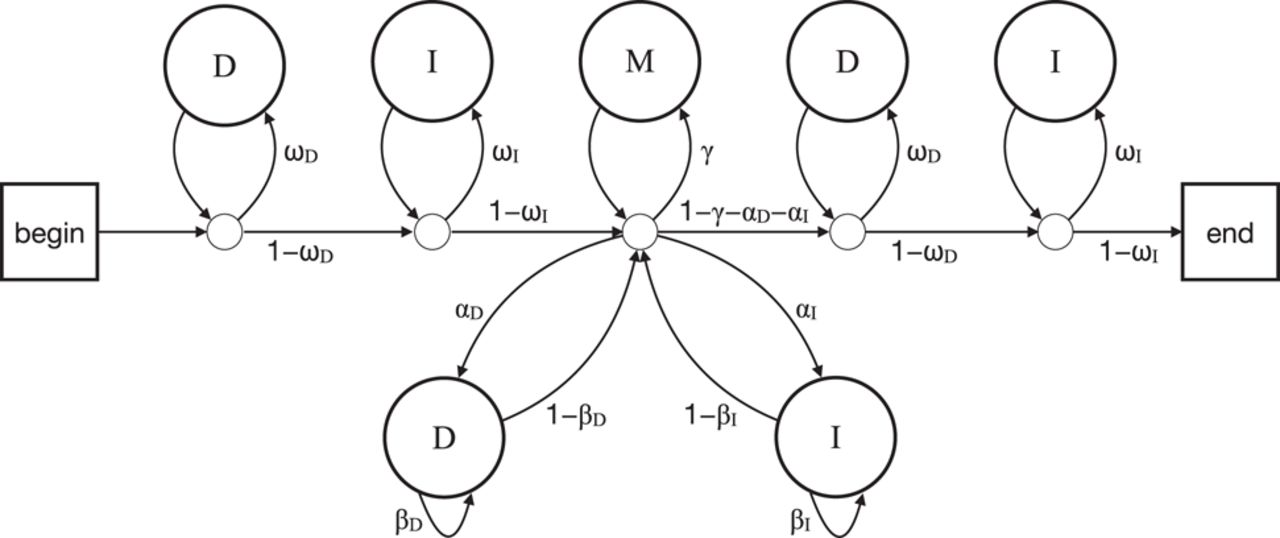
A “null alignment” is produced by a path that never traverses the αD, αI, or γ arrows. This corresponds to a length-0 alignment between the sequences. The score of a null alignment is traditionally zero, which is achieved by Equation 1. This means that an alignment score is determined solely by the aligned regions and not by flanking sequences or their lengths.
Having defined these probabilities, we can state the parameters for Algorithm 2, as shown previously (Frith 2020):
Figure 2 is not the only way to define the probabilities (Frith 2020). There are other ways that cause no change to Algorithm 1 but cause slight changes to Algorithm 2. The definition in Figure 2 seems to produce the simplest equations and minimizes the number of computational operations in Algorithm 2. This is a minor improvement over hybrid alignment, which defined probabilities with an unnatural asymmetry between insertions and deletions (see Supplemental Fig. S2 in the work of Frith 2020).
A nonheuristic similarity score
What would we do if we did not need fast heuristics? As mentioned in the introduction, we seek a compromise between using just one alignment and summing over all alignments. We shall use a compromise with two useful features: It is approximated by the heuristic approach (Fig. 1B), and we can tell if a similarity is unlikely to exist between random sequences.
When we compare a length-m sequence (R1, R2, …, Rm) to a length-n sequence (Q1, Q2, …, Qn), we shall consider (m + 1) × (n + 1) hypotheses for how they are related. Imagine cutting R into a prefix of length i and suffix of length m − i, and Q into a prefix of length j and suffix of length (n − j). These cuts define a point, illustrated in Figure 1C by the touching corners of the gray rectangles. The hypothesis is that the sequences are related by any alignment that passes through this point (including alignments that start or end there). The score of this hypothesis is given by Equation 2, with the sum taken over these alignments.
We can calculate this score for each (i, j). We first calculate , the sum of probability ratios of all alignments that end at the ends of the prefixes:
We then calculate , the sum of probability ratios of all alignments that start at the starts of the suffixes:
A minor point is that this score definition excludes alignments that pass through (i, j) while traversing the βD or βI arrow (Fig. 2). This makes it closer to typical heuristic methods that extend alignments from a high-quality gapless “core” (Fig. 1).
Conservation condition
We wish to know the probabilities of similarity scores between random sequences. Because our scores, , are similar to those of hybrid alignment, we shall assume (and test) that we can use the same approach. This approach requires that the alignment parameters satisfy a “conservation condition.”
The conservation condition is related to alignment length bias. The probabilities in Figure 2 may be biased toward short or long alignments between two given sequences. For example, if αD, αI, and γ are all low (almost zero) but ωD and ωI are high (almost one), shorter alignments will have higher probability. In the converse situation, longer alignments are favored. It was shown previously (Frith 2020) that the probabilities are unbiased when
The conservation condition depends on the precise definition of gap probabilities (Fig. 2), as explained previously (Frith 2020). The conservation condition for hybrid alignment was different (see equation 28 in the work of Yu and Hwa 2001), because they defined gap probabilities differently (see Supplemental Fig. S2 in the work of Frith 2020). The conservation condition generalizes an equation in the proven solution for probabilities of gapless alignment scores (see equation 4 in the work of Karlin and Altschul 1990).
Similarity scores occurring by chance
We can calculate the probability of a similarity score s between two random i.i.d. sequences: one with length m and letter probabilities ϕx, the other with length n and probabilities ψy. Chance similarities occur at a constant rate between any part of one sequence and any part of the other. The P-value is the probability of a score ≥s occurring, and the E-value is the expected number of distinct similarities with score ≥s. In the limit where m and n are large, the number of distinct similarities follows a Poisson distribution, which means that P = 1 − e−E. The prediction is that
These formulas have one unknown parameter, K. We can estimate K by generating some pairs of random i.i.d. sequences, calculating for each pair, and fitting K (Supplemental Methods).
Implementation details
This approach to finding related sequence parts was added to the LAST software. This implementation is just a proof of principle: The heuristics are not necessarily the best. LAST can either use Algorithm 1 with E-values from the ALP library or use Algorithm 2. In both cases, it gets a representative alignment by running Algorithm 1 and tracing back a way to get the maximum v. LAST runs these algorithms not in an n × n block (Fig. 1A) but in a range that is adjusted as the algorithm proceeds (Fig. 1B; Supplemental Methods).
Sometimes, LAST finds overlapping alignments from two different “cores” (Fig. 1B). To remove such redundancy, if two representative alignments share an endpoint (same coordinates in both sequences), LAST keeps just one, with highest similarity score.
LAST estimates K from random sequences. It is not clear how long these sequences need to be. Based on some tests (Supplemental Figs. S1, S2), LAST uses 50 pairs of length-500 sequences by default.
The match/mismatch/gap probabilities (πxy, αD, etc.) were determined using last-train, which finds related regions of given sequences and infers these probabilities (Hamada et al. 2017). last-train was modified to make the probabilities satisfy Equation 12. It sets ωD and ωI by assuming that they are equal and finding the unique value that satisfies Equation 12 (Supplemental Methods). last-train is not necessarily the best way to get the probabilities. As an alternative, a last-train option was added, which makes it work as usual except that the letter probabilities (π, ϕ, and ψ) are fixed to those of a BLOSUM or PAM matrix.
Results
Similarity scores occurring by chance
Let us test whether we can calculate probabilities of similarity scores between random sequences. Three sequence-search scenarios are considered, with typical DNA, at-rich DNA, and proteins.
The first scenario is finding ancient (shared with reptiles) repeats in the human genome. This can be done by comparing the genome to repeat consensus sequences from Dfam (Storer et al. 2021). Some are relics of transposable elements; others are source unknown. The match/mismatch/gap probabilities were found by comparing the genome to the consensus sequences with last-train. Then, pairs of random i.i.d. sequences were generated, with letter probabilities ϕx and ψy equal to those found by last-train. For each pair of sequences, the highest score smax was found by the nonheuristic algorithm. The observed scores are consistent with their probabilities predicted by Equation 15 (Fig. 3, left).
Nonheuristic similarity score (smax) between 10,000 pairs of random i.i.d. sequences (length, 10,000) for three sets of alignment parameters. The frequency has a 5% chance of being outside the dashed lines (2.5% each).
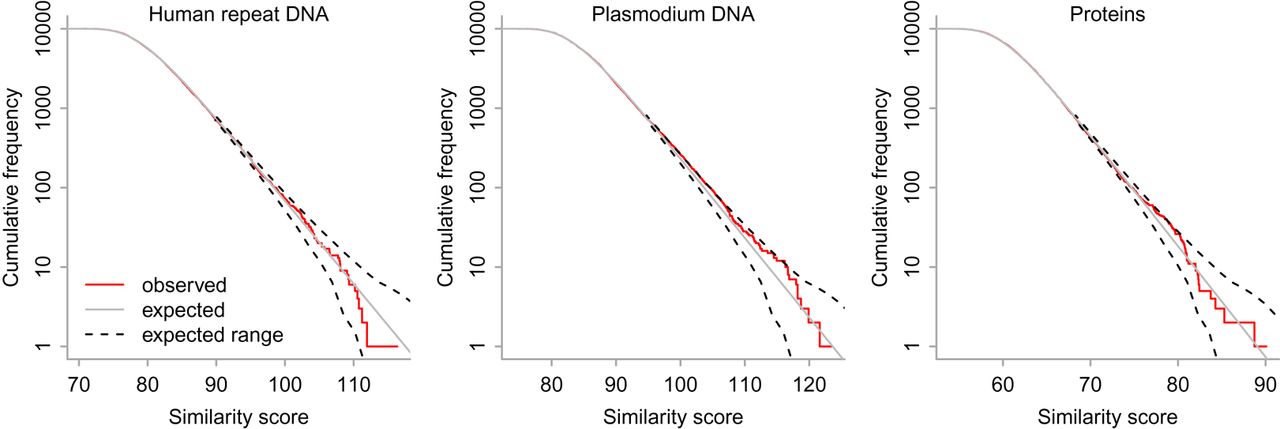
The next scenario was to find related parts of the Plasmodium falciparum and Plasmodium yoelii genomes, which are both ∼80% a + t. Alignment parameters were found by last-train, and then similarity scores were found between random at-rich sequences (Fig. 3, middle). Again, the observed scores are consistent with Equation 15.
The third scenario was to find related parts of proteins from Aquifex aeolicus (a heat-loving bacterium) and Pyrolobus fumarii (a heat-loving archaeon). Alignment parameters were found by last-train, and then, similarity scores were found between random sequences (Fig. 3, right). Yet again, the observed scores are consistent with Equation 15. This remains true if we use BLOSUM62 letter probabilities (Supplemental Fig. S3A).
Related versus biased sequences
A high similarity score could occur because the sequence regions have a common ancestor and/or similar composition bias (Fig. 4A). We can investigate this by comparing two biological sequences after reversing (but not complementing) one of them (but see Glidden-Handgis and Wheeler 2024). Then, there are no segments related by evolution, but there may be similarities owing to composition bias (Fig. 4A).
Alignments between parts of DNA sequences. (A) Reversed human Chromosome Y (upper) and UCON1 (lower). This is the highest-scoring alignment in the top-left panel of Figure 5. (B) Human Chromosome 6 (upper) and LFSINE_Vert (lower). (C) Human Chromosome 21 (upper) and human U2 (lower).

Thus, the reversed human genome was searched against the repeat consensus sequences, the reversed P. yoelii genome against P. falciparum, and reversed P. fumarii proteins against A. aeolicus proteins. In each case, Algorithm 2 found more high similarity scores than expected between random sequences (Fig. 5, upper row), owing to biased regions (Fig. 4A; Supplemental Fig. S4).
Heuristic similarity scores between reversed and nonreversed sequences, with or without biased-sequence masking.
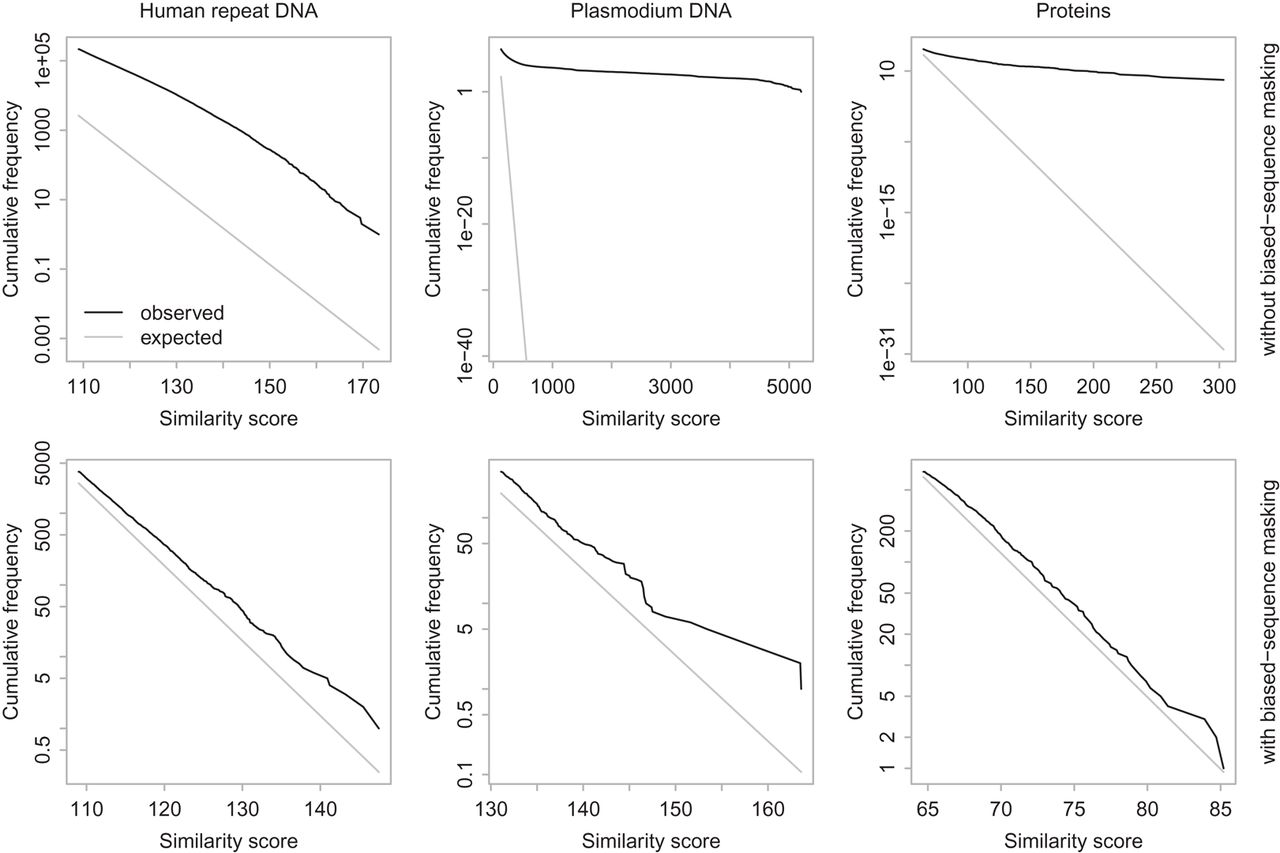
For standard alignment, these similarities can be avoided by detecting biased regions with tantan (Frith 2011a) and “masking” them. An effective masking scheme is to change when x or y is in a biased region (Frith 2011b). For Algorithm 2, this masking scheme was tried: ; another untested idea is . With this masking scheme, the similarity scores between reversed and nonreversed sequences are close to what is expected between random sequences (Fig. 5, lower row).
The new method can be more sensitive
Related sequence parts were sought by standard alignment (Algorithm 1) and by the new method (Algorithm 2), with biased-sequence masking. The two methods often find identical alignments, but with different scores and E-values. The new method tended to assign lower E-values, for alignments of the human genome to repeat consensus sequences (Fig. 6A) and for P. fumarii proteins to A. aeolicus proteins (Fig. 6B; Supplemental Fig. S3B). For example, both methods found the same alignment between human Chromosome 6 and the LFSINE_Vert consensus (Fig. 4B): The Algorithm 1 E-value is 22, and the Algorithm 2 E-value is 0.000031. The Algorithm 2 score is 185.3, much more than the highest score with reversed sequences (147.5) (Fig. 5, lower left). This means the new method can find related sequence parts more confidently.
E-values for identical alignments found by Algorithm 1 (horizontal axis) and Algorithm 2 (vertical axis). Each point is one alignment. The diagonal gray lines indicate equal E-values. These E-values were calculated with m = total length of all reference sequences (e.g., all A. aeolicus proteins) and n = total length of all query sequences (e.g., all P. fumarii proteins). For DNA, one query strand was searched against both reference strands, so m was multiplied by two. (A) Human repeat DNA; (B) proteins; (C) U2 DNA fragments; (D) U2 DNA fragments with fitted match/mismatch/gap probabilities.
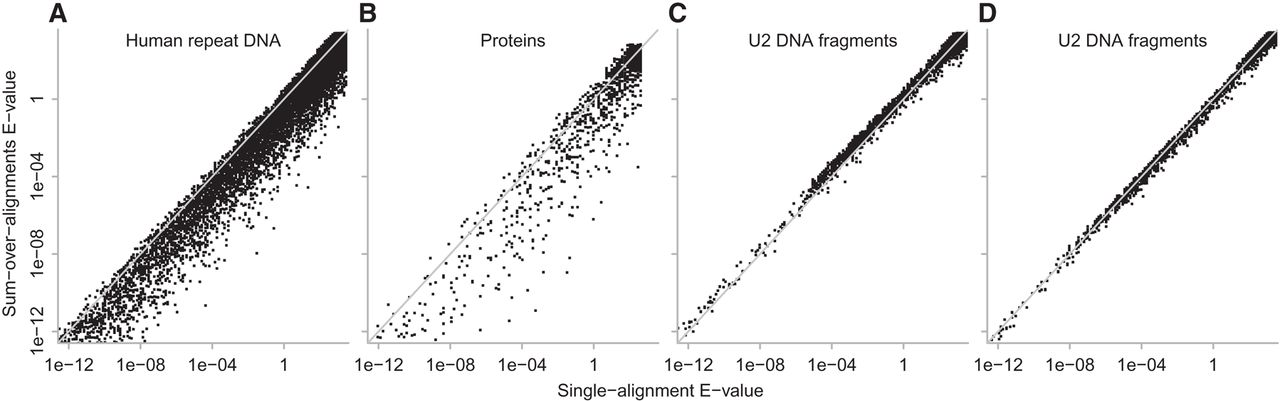
In other words, at a fixed E-value cutoff, the new method predicts more related regions. The preceding results indicate that the E-values are not overconfident. This implies that the new method finds more true positives for a given false-positive rate.
As a further test, similar regions were sought after shuffling the letters of the sequences used for Figure 6A. As expected, similarities were found with E-value ≳ 1, and the Algorithm 2 E-values did not trend lower than the Algorithm 1 E-values (Supplemental Fig. S5).
The new method can be less sensitive
Another sequence search scenario was considered: finding U2 fragments in the human genome. U2 is one of the small nuclear RNAs that splice introns. The genome has many fragmentary copies of it, which are thus considered repeats.
The human genome was searched against a U2 DNA sequence, using the match/mismatch/gap probabilities found earlier for the human genome and ancient repeat consensus sequences. This time, Algorithm 2 tended to assign higher E-values than Algorithm 1 (Fig. 6C). For example, both methods found the alignment shown in Figure 4C: The Algorithm 1 E-value is 0.00021, and the Algorithm 2 E-value is 0.0011.
The likely explanation is as follows. The genome has many short, near-exact copies of the first 30–40 bases of U2. The match/mismatch/gap probabilities, however, reflect greater sequence divergence (e.g., Fig. 4B). These mistuned probabilities pessimize both algorithms, but they pessimize Algorithm 2 more. That is because probabilities with greater divergence attach more weight to alternative alignments.
This suggests that Algorithm 2 should work better than Algorithm 1 if the probabilities are a good fit to the sequences. To test this, match/mismatch/gap probabilities were found by comparing the genome to the U2 sequence with last-train. Then, the genome was searched against U2 using these probabilities. This time, Algorithm 2 tends to assign slightly lower E-values than Algorithm 1 (Fig. 6D). Even with well-tuned probabilities, the advantage of Algorithm 2 is expected to decrease for more similar sequences because the evidence for relatedness is more concentrated in one alignment.
Discussion
We have seen how to find related parts of sequences, by summing probabilities of alternative alignments between them. This is better than standard alignment at detecting subtle relationships, at least in a few tests. It can be worse than standard alignment, when the probabilities are tuned for high divergence but the sequences have low divergence. The probabilities should be tuned for different use cases, for example, using last-train. The new method is more beneficial for higher divergence, where alternative alignments contribute more. In a spectacular example, it found gene-regulating DNA conserved in diverse animals since the Precambrian (Frith and Ni 2023).
A major further benefit is that the new method simplifies E-value calculation. This makes it easier to experiment with different alignment parameters, beyond the BLAST parameter sets. This can be useful for proteins (Ng et al. 2000), but especially for DNA, because there has been less effort to optimize DNA parameters.
Moreover, the new method can be generalized to other kinds of alignment. For example, we used the same approach for DNA-versus-protein alignment (Yao and Frith 2023). We defined probabilities similarly to Figure 2, but with extra probabilities for frameshifts. We summed probabilities of alternative alignments, and accurately predicted similarity scores between random sequences. This method was highly sensitive: It found ancient and degraded relics of Paleozoic mobile elements in vertebrate genomes. Some previous methods have used two kinds of alignment gap: short-and-frequent and long-and-rare (Allison et al. 1992; Lunter et al. 2008). This can be done by elaborating Figure 2. The approach of this paper is predicted to work in that case too. This makes it easier to try such different kinds of alignment, in particular because it provides E-values. Another alignment elaboration is position-specific match, mismatch, and gap rates (Durbin et al. 1998; Eddy 2008; Steinegger et al. 2019; Roddy et al. 2024): This was incorporated into hybrid alignment (Yu et al. 2002).
By combining Algorithm 2 with another similar algorithm (Supplemental Methods), we can calculate the probability that each alignment column is correct (Fig. 7), based on the assumed rates of matches, mismatches, and gaps (Allison et al. 1992; Durbin et al. 1998). It is also possible to make alignments based on these probabilities: For example, align letters whose alignment probability is >0.5 (Miyazawa 1995).
An alignment (the same as Fig. 4B) with colors indicating the probability that each column is correct. “∼” indicates the core (Fig. 1), whose column probabilities are assumed to be one, and “★” noncore exact matches.

For short sequences, the E-values are too high. Equation 14 assumes the number of chance similarities is proportional to mn. That becomes too high for short sequences, because a similarity must have nonzero length and fit in the sequences. There are corrections for this effect, including in hybrid alignment, but it is somewhat complex (Yu and Hwa 2001).
We should consider some basic aims and assumptions. Suppose we seek related regions between a genome of length m, and q “query” sequences with lengths n1, n2, …, nq. We could assume that related regions are equally likely to occur per query sequence or per unit sequence length. We could aim to find relationships for as many query sequences as possible or as many related regions as possible. If part of query i is similar to part of the genome, we can calculate an E-value from Equation 14 with sequence lengths m and ni. This increasingly disfavors related regions in increasingly long queries. As mentioned in the introduction, some previous methods similarly penalize related regions in longer sequences. The alternative is to use Equation 14 with sequence lengths m and . This treats related regions equally, regardless of the containing sequence's length. The aim is important when testing sequence search methods, because the test may count the number of sequences or related regions found.
Software availability
LAST is available at GitLab (https://gitlab.com/mcfrith/last) and as Supplemental Code. The last-train output files from this study are at GitLab (https://gitlab.com/mcfrith/sum-align). There also is a simple Python script to help use these ideas. It takes as input αD, αI, βD, βI, and γ; finds ωD = ωI that satisfies Equation 12; and outputs , , , , and . LAST includes code for calculating the nonheuristic similarity score (used for estimating K), in a self-contained file (mcf_alignment_path_adder.cc).
Competing interest statement
The author declares no competing interests.
Acknowledgments
I thank John Spouge for encouragement that the E-values would be simple, Yi-Kuo Yu for advice about hybrid alignment, and Travis Wheeler for advice about HMMER. This research was supported by the Japan Science and Technology Agency (JPMJCR21N6).
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279464.124.
References
- ↵Allison L, Wallace C, Yee C. 1992. Finite-state models in the alignment of macromolecules. J Mol Evol 35: 77–89. 10.1007/BF00160262
- ↵Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. 10.1093/nar/25.17.3389
- ↵Bishop M, Thompson EA. 1986. Maximum likelihood alignment of DNA sequences. J Mol Biol 190: 159–165. 10.1016/0022-2836(86)90289-5
- ↵Bucher P, Hofmann K. 1996. A sequence similarity search algorithm based on a probabilistic interpretation of an alignment scoring system. Proc Int Conf Intell Syst Mol Biol 4: 44–51.
- ↵Cameron M, Williams HE, Cannane A. 2004. Improved gapped alignment in BLAST. IEEE/ACM Trans Comput Biol Bioinform 1: 116–129. 10.1109/TCBB.2004.32
- ↵Dayhoff M, Schwartz R, Orcutt B. 1978. A model of evolutionary change in proteins. Atlas Protein Sequence Structure 5: 345–352.
- ↵Durbin R, Eddy SR, Krogh A, Mitchison G. 1998. Biological sequence analysis: probabilistic models of proteins and nucleic acids. Cambridge University Press, Cambridge, UK.
- ↵Eddy SR. 2008. A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS Comput Biol 4: e1000069. 10.1371/journal.pcbi.1000069
- ↵Frith MC. 2011a. A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Res 39: e23. 10.1093/nar/gkq1212
- ↵Frith MC. 2011b. Gentle masking of low-complexity sequences improves homology search. PLoS One 6: e28819. 10.1371/journal.pone.0028819
- ↵Frith MC. 2020. How sequence alignment scores correspond to probability models. Bioinformatics 36: 408–415. 10.1093/bioinformatics/btz576
- ↵Frith MC, Ni S. 2023. DNA conserved in diverse animals since the Precambrian controls genes for embryonic development. Mol Biol Evol 40: msad275. 10.1093/molbev/msad275
- ↵Glidden-Handgis G, Wheeler TJ. 2024. WAS IT A MATch I SAW? approximate palindromes lead to overstated false match rates in benchmarks using reversed sequences. Bioinform Adva 4: vbae052. 10.1093/bioadv/vbae052
- ↵Guidi G, Ellis M, Rokhsar D, Yelick K, Buluç A. 2021. BELLA: Berkeley efficient long-read to long-read aligner and overlapper. In SIAM Conference on Applied and Computational Discrete Algorithms (ACDA21) (ed. Bender M, et al.), pp. 123–134. SIAM (Society for Industrial and Applied Mathematics), Philadelphia.
- ↵Hamada M, Ono Y, Asai K, Frith MC. 2017. Training alignment parameters for arbitrary sequencers with LAST-TRAIN. Bioinformatics 33: 926–928. 10.1093/bioinformatics/btw742
- ↵Harris RS. 2007. “Improved pairwise alignment of genomic DNA.” PhD thesis, The Pennsylvania State University, University Park, PA.
- ↵Hein J, Wiuf C, Knudsen B, Møller M, Wibling G. 2000. Statistical alignment: computational properties, homology testing and goodness-of-fit. J Mol Biol 302: 265–279. 10.1006/jmbi.2000.4061
- ↵Hudek AK, Brown DG. 2011. FEAST: sensitive local alignment with multiple rates of evolution. IEEE/ACM Trans Comput Biol Bioinform 8: 698–709. 10.1109/TCBB.2010.76
- ↵Karlin S, Altschul SF. 1990. Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc Natl Acad Sci 87: 2264–2268. 10.1073/pnas.87.6.2264
- ↵Knudsen B, Miyamoto MM. 2003. Sequence alignments and pair hidden Markov models using evolutionary history. J Mol Biol 333: 453–460. 10.1016/j.jmb.2003.08.015
- ↵Liu D, Steinegger M. 2023. Block Aligner: an adaptive SIMD-accelerated aligner for sequences and position-specific scoring matrices. Bioinformatics 39: btad487. 10.1093/bioinformatics/btad487
- ↵Lunter G, Rocco A, Mimouni N, Heger A, Caldeira A, Hein J. 2008. Uncertainty in homology inferences: assessing and improving genomic sequence alignment. Genome Res 18: 298–309. 10.1101/gr.6725608
- ↵Ma B, Tromp J, Li M. 2002. PatternHunter: faster and more sensitive homology search. Bioinformatics 18: 440–445. 10.1093/bioinformatics/18.3.440
- ↵Miyazawa S. 1995. A reliable sequence alignment method based on probabilities of residue correspondences. Protein Eng 8: 999–1009. 10.1093/protein/8.10.999
- ↵Neyman J, Pearson ES. 1933. On the problem of the most efficient tests of statistical hypotheses. Philos Trans R Soc London, Ser A 231: 289–337. 10.1098/rsta.1933.0009
- ↵Ng PC, Henikoff JG, Henikoff S. 2000. PHAT: a transmembrane-specific substitution matrix. Bioinformatics 16: 760–766. 10.1093/bioinformatics/16.9.760
- ↵Noé L, Kucherov G. 2004. Improved hit criteria for DNA local alignment. BMC Bioinformatics 5: 149. 10.1186/1471-2105-5-149
- ↵Rivas E, Eddy SR. 2015. Parameterizing sequence alignment with an explicit evolutionary model. BMC Bioinformatics 16: 406. 10.1186/s12859-015-0832-5
- ↵Roddy JW, Rich DH, Wheeler TJ. 2024. Nail: software for high-speed, high-sensitivity protein sequence annotation. bioRxiv 10.1101/2024.01.27.577580
- ↵Roytberg M, Gambin A, Noé L, Lasota S, Furletova E, Szczurek E, Kucherov G. 2009. On subset seeds for protein alignment. IEEE/ACM Trans Comput Biol Bioinform 6: 483–494. 10.1109/TCBB.2009.4
- ↵Sheetlin S, Park Y, Frith MC, Spouge JL. 2016. ALP & FALP: C++ libraries for pairwise local alignment E-values. Bioinformatics 32: 304–305. 10.1093/bioinformatics/btv575
- ↵Steinegger M, Meier M, Mirdita M, Vöhringer H, Haunsberger SJ, Söding J. 2019. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics 20: 473. 10.1186/s12859-019-3019-7
- ↵Storer J, Hubley R, Rosen J, Wheeler TJ, Smit AF. 2021. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob DNA 12: 2. 10.1186/s13100-020-00230-y
- ↵Suzuki H, Kasahara M. 2017. Acceleration of nucleotide semi-global alignment with adaptive banded dynamic programming. bioRxiv 10.1101/130633
- ↵Thorne JL, Churchill GA. 1995. Estimation and reliability of molecular sequence alignments. Biometrics 51: 100–113. 10.2307/2533318
- ↵Thorne JL, Kishino H, Felsenstein J. 1991. An evolutionary model for maximum likelihood alignment of DNA sequences. J Mol Evol 33: 114–124. 10.1007/BF02193625
- ↵Webb BJM, Liu JS, Lawrence CE. 2002. BALSA: Bayesian algorithm for local sequence alignment. Nucleic Acids Res 30: 1268–1277. 10.1093/nar/30.5.1268
- ↵Wheeler TJ, Eddy SR. 2013. nhmmer: DNA homology search with profile HMMs. Bioinformatics 29: 2487–2489. 10.1093/bioinformatics/btt403
- ↵Yao Y, Frith MC. 2023. Improved DNA-versus-protein homology search for protein fossils. IEEE/ACM Trans Comput Biol Bioinform 20: 1691–1699. 10.1109/TCBB.2022.3177855
- ↵Yu YK, Hwa T. 2001. Statistical significance of probabilistic sequence alignment and related local hidden Markov models. J Comput Biol 8: 249–282. 10.1089/10665270152530845
- ↵Yu YK, Bundschuh R, Hwa T. 2002. Hybrid alignment: high-performance with universal statistics. Bioinformatics 18: 864–872. 10.1093/bioinformatics/18.6.864
- ↵Zhang Z, Berman P, Miller W. 1998. Alignments without low-scoring regions. J Comput Biol 5: 197–210. 10.1089/cmb.1998.5.197