
Differences in activity and stability drive transposable element variation in tropical and temperate maize
- Shujun Ou1,2,3,4,
- Armin Scheben5,
- Tyler Collins3,
- Yinjie Qiu2,
- Arun S. Seetharam1,6,
- Claire C. Menard2,
- Nancy Manchanda1,
- Jonathan I. Gent7,
- Michael C. Schatz3,
- Sarah N. Anderson6,
- Matthew B. Hufford1 and
- Candice N. Hirsch2
- 1Department of Ecology, Evolution, and Organismal Biology, Iowa State University, Ames, Iowa 50011, USA;
- 2Department of Agronomy and Plant Genetics, University of Minnesota, St. Paul, Minnesota 55108, USA;
- 3Department of Computer Science, Johns Hopkins University, Baltimore, Maryland 21218, USA;
- 4Department of Molecular Genetics, The Ohio State University, Columbus, Ohio 43210, USA;
- 5Simons Center for Quantitative Biology, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA;
- 6Department of Genetics, Development, and Cell Biology, Iowa State University, Ames, Iowa 50011, USA;
- 7Department of Plant Biology, University of Georgia, Athens, Georgia 30602, USA
Abstract
Much of the profound interspecific variation in genome content has been attributed to transposable elements (TEs). To explore the extent of TE variation within species, we developed an optimized open-source algorithm, panEDTA, to de novo annotate TEs in a pangenome context. We then generated a unified TE annotation for a maize pangenome derived from 26 reference-quality genomes, which reveals an excess of 35.1 Mb of TE sequences per genome in tropical maize relative to temperate maize. A small number (n = 216) of TE families, mainly LTR retrotransposons, drive these differences. Evidence from the methylome, transcriptome, LTR age distribution, and LTR insertional polymorphisms reveals that 64.7% of the variability is contributed by LTR families that are young, less methylated, and more expressed in tropical maize, whereas 18.5% is driven by LTR families with removal or loss in temperate maize. Additionally, we find enrichment for Young LTR families adjacent to nucleotide-binding and leucine-rich repeat (NLR) clusters of varying copy number across lines, suggesting TE activity may be associated with disease resistance in maize.
Eukaryotic genomes are largely composed of transposable elements (TEs). For example, 46% of the human genome consists of TEs (Hoyt et al. 2022). In maize, in which TEs were first discovered, the genome is 85% TE sequences, of which 75% are long terminal repeat (LTR) retrotransposons (Schnable et al. 2009). In many species, TEs are intertwined with genes and, as a result, can have functional consequences by altering transcript structure or regulation (Della Coletta et al. 2021). For example, the maize bz locus varies in length from 50 kb to 160 kb across genotypes owing to TE insertion/deletion polymorphism (Wang and Dooner 2006). TEs alter transcript abundance (Della Coletta et al. 2021), as seen with an insertion upstream of the tb1 gene (Doebley et al. 1995; Dong et al. 2019) that increases expression of the gene, enhances apical dominance, and reduces tillering in domesticated maize. Likewise, TE insertions in the promoter regions of ZmCCT9 and ZmCCT10 (Yang et al. 2013; Huang et al. 2018) alter gene expression, leading to earlier and day-length insensitive flowering in temperate maize. Expression differences can also result from insertions into intron sequences, such as a Mutator-like TE in an intron of DSX2 (Fang et al. 2020) that increases expression of the gene leading to carotenoid accumulation and yellow kernels. Total TE content in genomes has also been linked to environmental gradients such as altitude in maize (Bilinski et al. 2018) and both biotic and abiotic factors in tomato (Domínguez et al. 2020).
Despite the importance of TEs in genome evolution, there have been a limited number of pangenome studies of TE variation within species owing to challenges in assembling and annotating genomic regions containing these highly repetitive elements (Ou et al. 2019). Instead, the vast majority of studies characterizing TE content have used resequencing data mapped to a single reference genome (Quadrana et al. 2016; Carpentier et al. 2019; Domínguez et al. 2020; Wyler et al. 2020; Qiu et al. 2021). With continued advancements in long-read sequencing technologies and improved assembly algorithms, there is a growing movement in genomics toward pangenomic-based approaches (Bayer et al. 2020; Della Coletta et al. 2021; Li et al. 2022). The publication of 26 reference-quality genome assemblies for the founders of the maize nested association mapping (NAM) population (Hufford et al. 2021) provides an opportunity to explore intraspecies-level variation in TE content and to directly test the relative contribution of mechanisms underlying the abundance of individual TE families. These genomes were sequenced using Pacific Biosciences (PacBio) long-read technology, were assembled using the same set of methods, and have gold-quality assemblies as determined by the LTR Assembly Index (Ou et al. 2018; Hufford et al. 2021), and therefore, observed differences in TE content likely reflect the biology of these genomes rather than assembly artifacts. For example, structural variant analysis of the NAM lines revealed dynamic genome content linked to TEs (Munasinghe et al. 2023). The NAM panel also includes an even balance of tropical- and temperate-adapted inbred lines that exhibit marked differences in flowering time, disease resistance, plant height, and other important agronomic traits that may be driven by variation in TE content (for review, see Gage et al. 2020). Although broad differences in TE content at the superfamily level have been reported (Hufford et al. 2021), dynamics of TE amplification and removal are best captured at the family level and are presented here for the first time.
Results
LTR retrotransposons are overrepresented in pangenome TE variation
To characterize variation in TE content across 26 maize genomes, we developed panEDTA (Supplemental Code), which produces uniform TE annotations in a pangenome context (Fig. 1A). panEDTA is freely available at GitHub (https://github.com/oushujun/EDTA) and has been implemented in the browser-accessible, cloud-based platform Galaxy (The Galaxy Community et al. 2022). When compared to the original EDTA pipeline, panEDTA in maize, rice, and Arabidopsis (Supplemental Figs. S1, S2A) annotated a similar number of total bases of TE sequence, but with a substantial improvement in the consistency of element classification across individuals (Supplemental Fig. S2B–D). Using panEDTA, we annotated the 26 maize assemblies and identified 17,473 pangenome TE families and 269,847 unclassified low-copy TEs (Fig. 2A; Supplemental Fig. S3A). Together, TEs and non-TE repeats contribute an average of 88.2% of the genomes (Fig. 1B), consistent with previous reports in maize (Schnable et al. 2009; Hufford et al. 2021). The majority of maize TE families are small, with 89.7% of pangenome families comprising <100 kb per genome (Supplemental Fig. S4). Collectively, these small families comprise only 6.6% (SD = 0.1%) of total TE content (Fig. 2A). In contrast, the 1805 largest families contribute >90% (SD = 1.2%) of total TE content per genome (Fig. 2A; Supplemental Fig. S4), with the 50 largest TE families (predominantly from the Tekay, SIRE, and Retand LTR retrotransposon [LTR-RT] clades) contributing 52% (Fig. 2B). Many (72.3%) TE families that are >100 kb (Supplemental Fig. S3B,C) occur in all 26 NAM founder lines (Supplemental Fig. S5), and 91% of pangenome TE families are found when sampling as few as four lines (Fig. 2C).
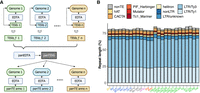
Pangenome annotation of 26 maize NAM founders using panEDTA. (A) The panEDTA workflow. The EDTA pipeline is used to annotate each genome independently, and the resulting individual TE libraries are filtered based on copy number and combined to form a nonredundant pan-TE library, which is used to reannotate each genome for a consistent pangenome TE annotation. (B) panEDTA annotation of 26 maize NAM founders. Maize lines were grouped into stiff-stalk (yellow), non-stiff-stalk (dark blue), popcorn (pink), sweet corn (red), admixed maize (gray), and tropical maize (green). Panel A was created with BioRender (https://www.biorender.com).
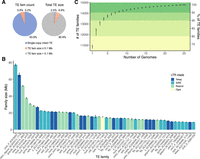
The landscape of transposable elements (TEs) in the maize NAM founder genomes. (A) Pangenome TE family number and size. Single-copy intact TEs are those not classified by the 80-80-80 rule and are mostly single-copy elements. (B) Mean size of the 50 largest TE families in the NAM founder genomes. All these families are LTR retrotransposons (LTR-RTs). The error bars denote the standard deviation among the NAM founder genomes. Clade-level classification of LTR families is denoted with different bar colors. (C) Summary of the number and percentage of pangenome TE families in the NAM founder genomes. The order of genomes was shuffled 1000 times.
Although many families are consistently observed across genomes, their abundance varies. Principal component analysis (PCA) based on family size across genomes revealed substantial divergence between temperate and tropical maize along the first PC (Fig. 3A), a finding consistent with population structure based on SNP-based PCA (Supplemental Fig. S6). Divergence between tropical and temperate genomes based on TE family size was driven by a small number of highly variable families, with only 216 families exhibiting per genome differences >0.025 Mb (Fig. 3B). The 10 families that varied most in size (Fig. 3C) were also among the 50 largest TE families in maize genomes (Fig. 2B). These families were significantly larger in tropical than temperate genomes (t-test, P < 1.0 × 10−10) (Fig. 3C), with admixed genotypes having intermediate family sizes. Those families that were larger in tropical genomes contained a combined average of 51.9 Mb more TE sequence per genome in tropical lines, whereas those larger in temperate lines contained an average of 16.8 Mb more sequence, resulting in a net difference of 35.1 Mb between tropical and temperate genomes. Structurally intact TEs contributed 50.8% (17.8 Mb) of the total TE variation, which was significantly more than the whole-genome average of 30.7% structurally intact TEs (Fisher's exact test, P = 0.02) (Supplemental Fig. S7A). LTR-RTs contributed 98.1% (34.4 Mb) of the total TE variation, with Ty3 elements showing the largest size difference (21.5 extra Mb in tropical genomes) (Fig. 3D). The remaining TE variation between tropical and temperate genomes was contributed by terminal inverted repeat (TIR) transposons (4.4%, with CACTA contributing 3.3%), Helitrons (−2.6%), and long interspersed nuclear elements (LINEs; −0.02%) (Fig. 3D).
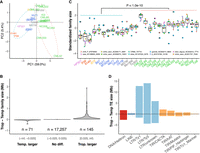
Family size variation between tropical and temperate maize genomes. (A) Principal component analysis based on pan-TE family size in the NAM founder genomes. A total of 17,473 families were included, and the size of the family was determined by the number of base pairs in each genome. Dashed ellipses indicate tropical (pink) and temperate (blue) genomes. (B) Distribution of TE family size difference between tropical and temperate lines. Families are divided into three categories with a cutoff of ±0.025 Mb difference. (C) Distribution of the top 10 TE families with the greatest size variation among the NAM founder genomes, which are all LTR families. The size of each family was standardized to have mean = 0 and standard deviation = 1 within NAM founder lines. Maize lines were grouped into temperate maize (popcorn, pink; sweet corn, red; stiff-stalk, yellow; non-stiff-stalk, dark blue) as indicated by the blue line on top of the boxes, admixed maize (gray), and tropical maize (green, as indicated by the pink line on top of the boxes). The box shows the median, upper, and lower quartiles. Whiskers indicate values ≤ 1.5× interquartile range. Black dots indicate outliers. (D) TE family size difference between tropical and temperate lines in TE superfamilies. Positive values represent families that are larger in tropical genomes, and negative values represent families that are larger in temperate genomes. (LTR/unknown) LTR families unable to be classified into Ty1/Ty3 superfamilies owing to lack of coding regions or confounding classifications.
Amplification and removal both contribute to variation in LTR family size
LTR family size can be increased or reduced through retrotransposition or illegitimate recombination, respectively. LTR sequences proliferate via reverse transcription (Fedoroff 2012), and more active proliferation could increase relative family size in some genomes. Alternatively, intact LTR elements can be reduced to solo LTRs via illegitimate recombination (Tian et al. 2009), and preferential removal could decrease relative family size. Consequently, the solo:intact ratio of each family reflects LTR removal via illegitimate recombination, with a high solo:intact ratio suggesting substantial removal (Tian et al. 2009). We evaluated the contribution of proliferation and removal to LTR family size variation between tropical and temperate genomes by comparing both family size and the solo:intact ratio of each LTR family. Amplifying families were defined as those having nonsignificant differences in removal intensities (solo:intact ratios) between tropical and temperate genomes but significant family-size differences (t-test, P ≤ 0.05) (Fig. 4A). Amplifying families were observed in both tropical (n = 145) and temperate (n = 59) genomes, with tropical amplifying families contributing 19.2 Mb of additional sequences on average in tropical genomes and temperate amplifying families contributing an average of only 0.5 Mb of additional sequence in temperate genomes (net of 18.7 Mb, 53.3% of total differences) (Fig. 4B,C; Supplemental Fig. S8A,B). Families undergoing removal were then identified as those with significantly different removal intensities (solo:intact ratios) and significantly different family sizes (Fig. 4A). These families net an extra 10.5 Mb (29.9% of total) of LTR sequences in tropical genomes (Fig. 4B,C; Supplemental Fig. S8A,B), of which 11.0 Mb is the product of stronger removal in temperate genomes. Compared to intact TEs in tropical amplification families, those in temperate removal families have shorter LTRs (median, 1226 bp vs. 1310 bp), fewer coding domains (30.4% vs. 65.0% of elements having complete sets of coding domains), and shorter overall element length (median, 7571 bp vs. 9478 bp) (Supplemental Fig. S9). In terms of LTR clade, the tropical amplification category is depleted for the Ale, Bianca, and Reina clades, and enriched for the Retand clade, whereas the temperate removal category is enriched for the Orge, Retand, and TAR clades (P < 0.05) (Supplemental Table S1). Overall, the emerging picture is that TE-induced genome size variability between tropical and temperate maize genomes is largely driven by TE families that are categorized as tropical amplification, with a lesser contribution from elevated LTR removal of specific TE families in temperate genomes.
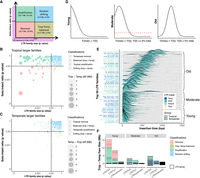
Contribution of LTR amplification and removal to genome size differentiation. (A) Classification schematic for LTR families based on solo:intact ratio and size differentiation between tropical and temperate genomes. Numbers (in Mb) indicate cumulative differences in family sizes between tropical and temperate genomes and their contribution to total TE differences. (B) LTR family classification for families that are larger in tropical genomes. (C) LTR family classification for families that are larger in temperate genomes. In both B and C, each dot represents an LTR family, and the size of each dot scales to the absolute family size difference, and x- and y-axes were log10-scaled. Removal families having an inconsistent solo:intact ratio contributed only 0.39 Mb to TE content variation and were removed from downstream analysis (Methods). (D) Classification schematic for age of LTR families based on the peak frequency of insertion time. (E) Age landscapes of the 50 largest LTR families in tropical (pink) and temperate (blue) maize genomes, with overlaps shown in green. Dots indicate family classifications using the coloring scheme shown in A. Clade-level classification of LTR families is denoted with different colors on the y-axis. The insertion time of LTR elements was estimated using the maize mutation rate µ = 3.3 × 10−8 per base pair per year (Clark et al. 2005) and assuming a constant molecular clock. (F) The accumulated TE size differentiation contributed by different LTR superfamilies (Ty3, Ty1, and unknown) in different age groups (Young, Moderate, and Old). Each box represents the contribution of an LTR family.
Differences in the abundance of TE families between tropical and temperate maize could have occurred at any time since the divergence of these two groups. The level of activity over time for an LTR family can be monitored by the age distribution of individual elements within the family, which is determined based on sequence divergence between the two terminal regions of an LTR element (SanMiguel et al. 1998; Ou and Jiang 2018). We therefore next evaluated if the per-family age of LTRs varied between tropical and temperate genomes along with their contribution to TE content variation between these groups. Families were classified as “Young” when the age of elements in the family peaked at 0 million years (MY; no intra-element divergence between LTRs) (Fig. 4D,E). “Moderate” families also have an appreciable fraction (>5%) of 0-MY-aged elements but show peak activity in the past. Finally, families were classified as “Old” if they contained few or no (≤5%) 0-MY-aged elements (Fig. 4D,E; Supplemental Table S2). Overall, Young LTR families contributed 69.8% of the TE-content difference between tropical and temperate genomes, which is significantly more than the expectation based on their genome-wide abundance of 20.2% (Fisher's exact test, P = 2.7 × 10−5). Of these, Young Ty3 LTRs preferentially amplified in tropical genomes and contributed 9.9 Mb (28.2%) of the TE content variation genome-wide (Fig. 4F), with the xilon_diguus_AC203313_7774 family (the second largest of all TE families) (Fig. 2B) contributing 3.9 Mb of the size difference. Conversely, Young Ty1 LTRs that were preferentially removed in temperate genomes contributed 4.9 Mb or 14.1% to TE-content difference (Fig. 4F), with the ji_AC215728_13156 family contributing 2.9 Mb of this size difference. Notably, the xilon_diguus family is found in DNA regions that coincide with late DNA replication during the S phase, whereas the ji family is found in early replication regions that are usually enriched with genes (Wear et al. 2017). Together, Young tropical amplifying Ty3 and Young temperate removing Ty1 LTR families contributed nearly half (42.3%) of the observed TE-content difference between tropical and temperate genomes, while only making up an average of 11.8% of all TE content in the genome. For structurally intact LTR elements, Young families contributed 91.5% of the intact LTR size differentiation (Supplemental Fig. S7B), an overrepresentation compared with the expectation of 33.1% from intact Young LTR families (Fisher's exact test, P < 1.0 × 10−10) (Supplemental Fig. S7C), suggesting structurally intact elements from Young families are driving much of the TE differences between tropical and temperate genomes. However, Old LTR families, especially Ty3 LTRs, also contributed to substantial TE size differentiation (8.9%) between tropical and temperate genomes (Fig. 4F). The removal extent (i.e., cumulative solo:intact ratio) of all the Old families was six to 10 times higher than the Moderate and Young families, respectively (Supplemental Fig. S8C,D).
Amplifying LTR families are less methylated and more highly expressed
Activity (i.e., transcription and amplification) of LTR-RTs often requires suppression of methylation (Fedoroff 2012; Rodgers-Melnick et al. 2016; Jachowicz et al. 2017; Mustafin 2019; Crisp et al. 2020; Marand et al. 2021). The lack of methylation in the CHG context, in which H = A, C, or T, is particularly informative in defining euchromatic regions. Such CHG unmethylated regions (UMRs) that originate within 5′ LTRs (UM-5′LTRs) rather than internal sequences of the element (Supplemental Fig. S10) are more likely to lead to transcription of the full-length TE. From previously reported genome-wide UMRs (Hufford et al. 2021), we identified a total of 14,074 that were UM-5′LTRs across the 26 genomes (Supplemental Table S3). These UM-5′LTRs were significantly enriched in tropical amplification families (observed 53.5% compared with the expected 38.7%; Fisher's exact test, P < 1.0 × 10−10) and significantly depleted in temperate removal families (observed 7.7% compared to the expected 13.9%; Fisher's exact test, P < 1.0 × 10−10) (Figs. 4F, 5A). The fraction of UM-5′LTRs in tropical amplification families is 2.4 times that observed in temperate removal families (1.53% vs. 0.63% of intact LTR elements). These UMRs rarely extended into the TE coding regions (Supplemental Fig. S10), and their average length ranged from 375 bp to 550 bp for Ty3 and Ty1 elements (Supplemental Fig. S11). UMRs of this length will span two to three nucleosomes (nucleosome repeat length is ∼190 bp in maize) (Chen et al. 2017), which may allow initiation of transcription. Notably, Young Ty3 UM-5′LTRs are 1.2 times more frequent in tropical than temperate genomes on average (Fisher's exact test, P = 0.04) (Fig. 5A), suggesting higher transcription potential of these LTR elements in tropical genomes.
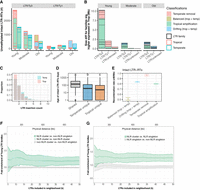
Molecular characterization of LTR families in maize. (A) The number of intact LTR-RTs carrying unmethylated regions. Data from tropical and temperate genomes are shown in side-by-side red and blue boxes, respectively. The number of families represented is indicated below each column. (B) The accumulated family size difference between tropical and temperate genome for LTR families expressed significantly higher in at least one tissue (and with consistent directionality in all tissues with expression) in tropical genomes. (A,B) The size of each box represents the number of LTR elements or effect size of each family, and only families that are larger in tropical genomes are shown. (C) LTR insertion frequency spectrum in tropical (pink) and temperate (blue) genomes. Only sites younger than 20,000 years ago were kept to increase accuracy of the polarization of the spectrum. No missing data filter was applied. (D) The age of intact LTR elements that were shared or unique in tropical and temperate genomes. The y-axis was log10-scaled. Different letters indicate significant differences in age (Tukey's HSD, P < 0.05). The box shows the median, upper, and lower quartiles. Whiskers indicate the 1.5× interquartile range. Black dots indicate outliers. (E) Mean recombination rate for genomic neighborhoods of all intact LTR-RTs. Error bars indicate the 95% confidence interval estimated from 1000 times of bootstrap resampling. (F,G) Fold enrichment of Young LTR family compared to Old LTR family neighborhoods of NLR gene clusters compared with non-NLR gene clusters (F) and NLR singletons (G). Nonsignificant comparisons shown in gray. x-axes are the number of intact LTR-RTs (k) found in the gene neighborhood. Lines indicate the mean values for all NAM lines, and the ribbon indicates the standard deviation within the population.
To evaluate the functional impact of these unmethylated LTRs on transcriptional activity, we quantified TE transcript abundance across 10 diverse plant tissues that were previously sequenced (Hufford et al. 2021). The repetitive nature of TEs makes quantification of transcript abundance challenging on a per-element basis. We therefore evaluated transcript abundance on a per-family basis within each genome, as previously described (Anderson et al. 2019), and conducted differential expression analysis between tropical and temperate genomes (Supplemental Fig. S12A,B). The total transcript abundance of each TE family is positively correlated with the size of each family (Pearson's r = 0.44, P < 1.0 × 10−10) (Supplemental Fig. S13). Particularly, the total transcript abundance of tropical amplification families was 20.8 times higher than that of temperate removal families (Supplemental Fig. S12C). When normalized with the total sequence length, the total transcript abundance of the tropical amplification families was still 4.8 times higher than that of the temperate removal families (Supplemental Fig. S12D,E), suggesting more active transcription of the tropical amplification families. We identified 1613 LTR families that had consistently higher expression in tropical genotypes across all tissues (Wald test, FDR < 0.05), and these explained 59.5% of TE family size differences between the tropical and temperate genomes. Among these 1613 LTR families, tropical amplification families contributed a significantly larger portion (5.7 times) than the random expectation (Fisher's exact test, P = 0.048), collectively explaining 32.0% of TE family size differences between the tropical and temperate genomes (Fig. 5B; Supplemental Fig. S14). In contrast, the contribution from the temperate removal families did not exceed random expectations (Fisher's exact test, P = 0.34). All but one LTR family (144/145) that possessed at least one UM-5′LTR were differentially expressed between the tropical and temperate lines (Supplemental Fig. S15A). A total of 16 tropical amplification families that had significantly and consistently higher abundance in tropical genomes also possessed at least one UM-5′LTR in a tropical genome (FDR < 0.05). These 16 families contributed 25.7% of LTR size differences between the tropical and temperate genomes (Supplemental Fig. S15B). Taken together, these results show amplification of LTR sequences in tropical maize genomes is associated with lack of methylation and increased expression of tropical amplification families in tropical maize genomes.
Dynamic LTR families are located in highly variable genomic regions
Patterns of abundance, dating of TE activity, methylation, and expression results all suggest TE content in tropical and temperate genomes has recently evolved. To further assess this possibility, we evaluated population genetic evidence for more recent TE activity in these genomes. We identified syntenic LTR loci between pairs of genomes and then summarized across genome pairs, obtaining insertion frequencies of individual TEs at the population level (Supplemental Figs. S16, S17A–D). As expected under a model of recent transposition, most LTR insertions were rare (Supplemental Fig. S17E,F). Additionally, LTR insertion frequency was positively correlated with age (Pearson's r = 0.89, P < 0.0005) and distance from genes (Pearson's r = 0.90, P < 0.0004) (Supplemental Fig. S17G,H). Because the majority of variation in genome-wide LTR content was driven by tropical amplification families (Fig. 4A), we expected and observed an excess of rare (frequency < 20%) LTR insertions in tropical genomes (Fisher's exact test, P < 1.0 × 10−10) (Fig. 5C; Supplemental Fig. S18A). This trend is also consistent with a loss of rare variants in temperate lines owing to their demographic bottleneck (Supplemental Fig. S18B,C; Romay et al. 2013), an alternative explanation of these findings. To further assess the driver of this result, we identified LTR insertions that were unique in either the tropical (n = 7790) or temperate (n = 5188) groups. The age of unique LTR insertions in tropical genomes was significantly younger than unique insertions in temperate genomes (Tukey's HSD test, P < 1.0 × 10−10) (Fig. 5D), suggesting more recent amplification activity in tropical genomes.
To explore a mechanism for temperate removal of TEs, we considered that the recombination rate might influence removal frequency and the prevalence of solo LTRs (Underwood et al. 2018). We estimated the meiotic recombination rate for genomic neighborhoods of each intact LTR element and each solo LTR using a composite recombination map from recombinant inbred lines (RILs) derived from the NAM founder parents (Ogut et al. 2015; Calfee et al. 2021). Overall, temperate removal families were located in genomic regions with a significantly higher recombination rate compared with tropical amplification families across all of the genomes (pairwise permutation test, P < 0.05) (Fig. 5E). As previously reported, illegitimate recombination is the dominating force in counteracting LTR amplification (Devos et al. 2002; Tian et al. 2009; Hu et al. 2011; VanBuren et al. 2018), and the higher level of recombination in regions that contain temperate removal families suggests that recombination is, to some extent, driving the variation in size of these families between tropical and temperate genomes.
Functional consequences of LTR family expansion and removal
Frequent insertions of LTR elements occur in genic regions, with ∼3.8% of intact LTR elements overlapping gene features (Supplemental Table S4). The relative number of intact LTR elements overlapping genes was not significantly different between the temperate removal and tropical amplifying families (chi-square test, P = 0.057). However, compared with all other LTRs, elements in both the temperate removal and tropical amplifying families overlapped significantly less with genes (chi-square test, P < 0.00001) (Supplemental Table S4).
We identified 955 unique genes with at least one intact LTR-RT from temperate removal or tropical amplifying families inserted into the coding region (Supplemental Table S5). Within these genes, the IQ calmodulin-binding motif was enriched in cinful_zeon_AC206615_9266 family insertions, a tropical amplification family in the Retand clade. We also found enrichment of insertions in the nucleotide-binding and leucine-rich repeat (NLR) motif, a key component of disease-resistance genes, and in genes containing a protein kinase domain (Supplemental Table S5).
NLR disease-resistance gene evolution is thought to be partly driven by ectopic duplication, possibly through the action of TEs (Leister 2004). There is a higher copy number of NLR disease-resistance genes in tropical maize lines compared with temperate lines (Supplemental Table S6; Hufford et al. 2021), and recent unpublished work in Brassicaceae indicates that NLR clusters may be enriched for young TEs (D. Weigel, pers. comm.). We, therefore, tested whether NLR clusters (Supplemental Fig. S19) are enriched for Young LTR families in maize. We found enrichment for Young LTR families in the closest 10 intact LTR-RTs to each NLR cluster (Fisher's exact test, Bonferroni-adjusted P = 3.84 × 10−12) (Fig. 5F,G). As a control, the enrichment of Young LTR families was tested in the neighborhoods of NLR singleton genes, non-NLR singleton genes, and non-NLR gene clusters (Fig. 5F,G), and in all cases, no significant enrichment was observed. Between NLR clusters of tropical and temperate lines, no LTR-RT enrichment was found in the NLR neighborhood (Supplemental Fig. S20). However, among the tropical lines, NLR neighborhoods were significantly enriched for the giepum, opie, and milt LTR families (Supplemental Table S7), indicating the potential functional consequence of these tropical amplifying families in tropical disease-resistance evolution. These families have also been shown to be transcriptionally active families in maize (Vicient 2010).
Discussion
TE content differs substantially across plant species. Within the genus Zea, Tenaillon et al. (2011) revealed that TEs contributed ∼70% of the genome size difference between maize and Zea luxurians (Tenaillon et al. 2011). Likewise, in the ∼900 Mb domesticated tomato genome (Solanum lycopersicum), <40% of base pairs are derived from LTRs, with a skew toward older elements (Li et al. 2023), whereas wild tomato genomes (Solanum lycopersicoides) have been shown to contain younger LTR elements, which contribute to a 35% larger genome relative to the domesticated tomato. Together, these data suggest that level of TE activity can meaningfully shape genome size at multiple evolutionary scales (e.g., population differentiation, recent domestication, speciation).
Within maize, the TE content in tropical and temperate maize genomes has been shaped by evolutionary processes during a period of rapid demographic change. Temperate maize has been subject to population bottlenecks and inbreeding (Bouchet et al. 2013; Li et al. 2017) and thus harbors less genetic variation compared with tropical maize (Romay et al. 2013). Inbreeding can result in the purging of deleterious alleles when recessive deleterious variants become homozygous. During inbreeding of maize, the removal of TEs has been previously linked to genome downsizing, potentially owing to the deleterious effects of TEs near genes (Roessler et al. 2019). Temperate removal TEs observed in our work could be linked to purging during inbreeding. In contrast, the greater diversity in tropical lines coupled with our observation of an excess of rare LTR insertions in these lines (Fig. 5C) suggests a history of population expansion. Tropical amplification TE families may be linked to recent population expansion in tropical lines. However, maize genome size may also be positively selected owing to the adaptive advantages of small genome size at high latitude and elevation (Poggio et al. 1998; Jian et al. 2017; Lai et al. 2017; Bilinski et al. 2018). Such selection would also favor temperate removal of TEs, with amplification being less constrained in tropical regions. The more abundant LTRs of tropical genomes may also contribute to increased allelic and functional variation that supports robust tropical populations in the face of biotic and abiotic challenges (Poggio et al. 1998). A better understanding of LTR amplification and purging will provide insights, not only into the genomic dynamics in maize but also into the general contribution of LTR-RTs to biodiversity in plants. The selective determinants of TE content are likely diverse, occurring at the TE family level based on insertional preference (e.g., genic vs. nongenic regions) (Stitzer et al. 2021) or at the genome level owing to factors including environmental stress and demographic change (e.g., population expansion) (Jiang et al. 2024).
In addition to the genome-wide trends we have uncovered in TE abundance, we also consider the more targeted, functional effects of TEs. The local neighborhood of genes is often influenced by TEs that contribute to gene copy number (Cerbin and Jiang 2018) and modulate gene expression (Studer et al. 2011; Huang et al. 2018). Our results show that the genomic neighborhood of NLR gene clusters is enriched for Young LTR elements. This enrichment is stronger in tropical maize, in which biotic stress is more prevalent (Tigar et al. 1994; Gong et al. 2014) and in which the NLR copy number has been shown to be higher (Hufford et al. 2021). Young, tropical amplifying LTR families may play an important role in increasing gene copy number in NLR clusters and conferring increased disease resistance. The precise mechanism underlying NLR copy-number increase in clusters is still unknown, but LTRs may induce chromosomal breakage or unequal recombination, which could lead to the growth of NLR gene clusters.
TE insertion in and around genes can have other functional consequences beyond facilitating gene cluster expansion and contraction. For example, Lai et al. (2017) used short-read resequencing to identify nonreference TEs and found gene expression differs with the presence and absence of TEs (Lai et al. 2017). We report here that ∼3.8% of intact LTR-RTs overlap genes (Supplemental Table S4). Insertion of LTR-RTs into coding regions was less common with temperate removal and tropical amplifying families compared with other types of TEs (Supplemental Table S4). These data suggest that TE families that contribute to significant differences in TE content between tropical and temperate lines are largely active in the intergenic space or have been selected against in the gene space, which is consistent with constraint on coding regions. However, this does not imply that activity of these Young LTR families lacks functional consequences given the potential regulatory effects of distal TE insertions and the substantial impact that a handful of TE insertions can have on phenotype (Butelli et al. 2012; Yokosho et al. 2016; Dong et al. 2019).
panEDTA allowed, for the first time, access to high-quality pangenome TE annotations in maize, as well as characterization of the previously undescribed TE content variability between tropical and temperate maize genomes at the family level. Characterizing the evolutionary dynamics and molecular features of TEs is still challenging owing to TEs’ repetitiveness and the lack of tools suitable for their individual analysis. We developed a new approach to identify the presence and absence of LTR elements in the maize pangenome. This approach allowed us to show that LTR elements distinguishing tropical or temperate maize lines were significantly younger than those shared across maize. Characterization of TE dynamics at the pangenome level is a new genomic frontier that has been facilitated by improvements in long-read sequencing. Future efforts will continue to clarify the evolution and functional relevance of these repetitive sequences across a broad range of species.
Methods
Development of panEDTA
We developed panEDTA to optimize pangenome TE annotation, which was incorporated into the current EDTA version (v2.1). panEDTA does not simply concatenate TE annotation sets from EDTA because this would propagate false positives in the context of multiple genomes. Instead, panEDTA initially annotates each genome individually for structural and homology annotation of TEs and then identifies and retains exemplar sequences with at least three full-length copies in a single genome. The previously proposed 80-80-80 rule is applied to determine full-length copies that meet these criteria, which requires ≥80% of the TE covered by sequences with ≥80% identity and with a minimal length of 80 bp (Wicker et al. 2007). One exemplar sequence was randomly selected from each multicopy family, including cases when there are multiple copies from different genomes to avoid a single-genome bias. By eliminating low-copy and incomplete sequences in individual libraries, panEDTA is able to filter out a large portion of potentially false TEs that will aggregate when multiple genomes are jointly annotated. Removing low-copy exemplars also keeps the pangenome library in a reasonably small size for computational efficiency, whereas the potential loss of sensitivity owing to the removal of low-copy exemplars in a single genome is offset by the compilation of multiple TE libraries into the final pangenome filtered library. Sequence redundancy of the pangenome library is removed using the 80-80-80 rule, and the remaining sequences contain a single exemplar sequence for TE families across the pangenome. Finally, this filtered pangenome library is used to reannotate all genomes in a pan-annotation, including both structurally intact and fragmented TEs, with consistent family IDs across genomes. Structurally intact TEs that were not able to be classified into a family by the 80-80-80 rule are named by their genome coordinates and regarded as single-copy intact TEs.
We have also integrated panEDTA and the original EDTA software into Galaxy (The Galaxy Community et al. 2022), a browser-accessible, cloud-based workbench for scientific computing. In brief, the Galaxy instance of panEDTA utilizes the publicly available panEDTA BioContainer and several modified scripts to render panEDTA with a graphical user interface. This implementation of panEDTA maintains the previous functionality of EDTA and first identifies structurally intact TEs in user-selected genomes. It then combines these genomes into a comprehensive library using the panEDTA algorithm and then reannotates the user-selected genomes and provides uniform TE family names across all family members in all genomes. Additionally, the Galaxy instance of panEDTA has been parallelized to run multiple instances of EDTA to decrease initial genome annotation time. We also optimized the execution of EDTA to detect Helitrons, LTRs, and TIRs in separate and simultaneous instances and then combined outputs to decrease runtime. The Galaxy integration of EDTA and panEDTA is accessible through https://usegalaxy.eu/root?tool_id=edta, which can be deployed in local servers following the general Galaxy guideline (https://galaxyproject.org/admin/get-galaxy/). panEDTA is distributed with the EDTA package (https://github.com/oushujun/EDTA), which is available through GitHub, Conda, Docker, and Singularity.
Genomes and TE annotations
Fifty Arabidopsis genomes were downloaded from NCBI (Supplemental Dataset S1) and annotated by panEDTA. The curated Arabidopsis library was obtained from Repbase 20.03 (Bao et al. 2015) and reformatted to follow the naming conventions of panEDTA. The curated Arabidopsis library was provided to panEDTA with the “‐‐curatedlib” parameter.
This study utilized previously generated genome assemblies of the 26 maize NAM founder lines (Hufford et al. 2021), and their genome assemblies and gene annotations were downloaded from https://maizegdb.org/NAM_project. This pangenome set included 13 tropical genomes (CML103, CML228, CML247, CML277, CML322, CML333, CML52, CML69, Ki3, Ki11, NC350, NC358, and Tzi8), 10 temperate genomes (dent genomes: B73, B97, Ky21, M162W, Ms71, Oh43, and Oh7B; flint genomes: HP301, P39, and Il14H), and three admixed genomes (M37W, Mo18W, and Tx303). The NAM genomes are highly contiguous within the TE space and are assembled to the chromosome level (Hufford et al. 2021). To avoid overestimation of haploid assembly size and TE content, we identified and removed alternative scaffolds in scaffold assemblies of the 26 NAM genomes (Supplemental Methods). In addition, the Zea mays ssp. mexicana, PI 566673 genome (obtained from the NCBI BioProject database [https://www.ncbi.nlm.nih.gov/bioproject/] under accession number PRJNA299874) was used as an outgroup to polarize LTR insertions (Yang et al. 2017).
Using panEDTA, we generated a new version of the TE annotation for the 26 maize NAM founder genomes. Names of sequences in the panEDTA library were ported from the original MTEC library names (Schnable et al. 2009) or generated by EDTA for novel TEs in the panEDTA library that were not previously contained in the MTEC library. Annotation and classification of TE families are available from MaizeGDB: https://ars-usda.app.box.com/v/maizegdb-public/folder/176297337613 (“EDTA.TEanno.gff3” files). The size of each TE family in base pairs was summarized from the annotation of each genome using the “buildSummary.pl” script derived from the RepeatMasker package (Smit et al. 2015).
Annotation evaluation
Repeat annotations (RepeatMasker OUT files) generated using EDTA and panEDTA libraries were evaluated for annotation inconsistency by the script “evaluation.pl” in the EDTA package. The maize B73v5, rice MSU7, and Arabidopsis TAIR10 genomes were used for the evaluation. The rice EDTA and panEDTA libraries were obtained from Qin et al. (2021), and the maize and the Arabidopsis EDTA and panEDTA libraries were generated in this study. Such evaluations are reference-free and thus do not rely on the availability of a gold-standard annotation. Briefly, annotated repeat sequences of a genome were extracted and subjected to all-versus-all BLAST within the extracted sequences, and the matching sequences covering ≥95% of the query with ≥80% identity and ≥80 bp in length were compared to the query sequence's annotation to identify inconsistently annotated entries. The annotation inconsistency was measured at the superfamily level.
Pangenome analysis
To estimate the distribution of TE families in the pan-NAM founder genomes, only pangenome families that contained at least one full-length TE (fl-TE) in at least one of the genomes were included. fl-TEs were identified using the pangenome TE annotation and the “find_flTE.pl” script in the EDTA package. The lists of fl-TE families from the NAM founder genomes were added incrementally (from one to 26 genomes) and in random order. The number of unique pangenome fl-TE families was counted after adding each genome's fl-TE list. This process was iterated 1000 times.
PCA of pan-NAM TE families (n = 17,473) was computed in R version 4.0.3 (R Core Team 2020) using the command “prcomp” and the physical size of each family in base pairs in each genome. TE family sizes were assigned as zero in the genomes that are absent. Unnormalized family sizes were used so that larger TE families will have more weight in the PCA. The SNP PCA was done using 25,000 random homozygous biallelic SNPs with no missing data that were filtered from the original NAM SNP data set (Hufford et al. 2021; https://datacommons.cyverse.org/browse/iplant/home/shared/NAM/NAM_genome_and_annotation_Jan2021_release/SUPPLEMENTAL_DATA/NAM-founder-SNPs) in R using the command “prcomp.” The reference and alternative alleles were coded as zero and one, respectively. Unnormalized values were also used to conduct the SNP PCA.
Copy number estimates for unclassified LTR-RTs
Some intact LTR-RTs could not be classified into families owing to low copy numbers in the original TE annotation and were thus named by their coordinates in the genome. To estimate the copy number of these unclassified LTR-RTs, their sequences were extracted in each genome, and redundant copies were removed using the “cleanup_nested.pl” script from the EDTA package with the parameters “-cov 0.95 -minlen 80 -miniden 80” that require at least 80 bp, 80% identity, and 95% sequence coverage. The resulting representative sequences were used to mask the original LTR sequence using RepeatMasker with the parameters “-q -no_is -norna -nolow -div 40” that allow for up to 40% of divergence. Masked sequences were annotated by the “classify_by_lib_RM.pl” script using the relaxed parameters “-cov 50 -len 70 -iden 70” that require at least 70 bp, 70% identity, and 50% coverage. After this reannotation step, the copy number of representative intact LTR-RTs was counted in the reannotated sequences.
Metadata of intact LTR-RTs
The length, classification, and divergence information of each intact LTR-RT were obtained from the original annotation of each genome (“pass.list” files) (Hufford et al. 2021). The insertion time of each intact LTR element was calculated from the LTR divergence using the maize mutation rate µ = 3.3 × 10−8 per base pairs per year (Clark et al. 2005) and assuming a constant molecular clock. For each intact LTR-RT, nested TEs were identified when other TEs were fully enclosed in the intact LTR-RT, and the copy number and length of the nested TEs were counted for each intact LTR-RT. The number of conserved coding domains in each intact LTR-RT was identified using TEsorter (v1.3) (Zhang et al. 2022) with default parameters, which were used for clade-level classifications of LTR families by the program. LTR/unknown represents LTR families unable to be classified into Ty1/Ty3 superfamilies owing to confounding classification or the lack of coding regions, which are essential to have superfamily or clade-level classification. Gene annotations used in this study were filtered from the original NAM gene data set (Hufford et al. 2021; https://datacommons.cyverse.org/browse/iplant/home/shared/NAM/NAM_genome_and_annotation_Jan2021_release/GENE_MODEL_ANNOTATIONS), with only primary isoforms retained (“T001” transcripts). TE-related genes were identified using TEsorter with default parameters and were removed. The physical distance of each intact LTR-RT to the downstream gene is calculated by “closest-features ‐‐dist” function with the BEDOPS (v2.4.39) package (Neph et al. 2012). Solo LTRs were identified using the “solo_finder.pl” script from the LTR_retriever package (Ou and Jiang 2018) and modified to adapt to the current annotation format. For each family in each genome, the solo:intact ratio was calculated using the number of solo LTRs over the number of intact LTR-RTs for the LTR family. Data of all solo and intact LTRs in each genome can be accessed from MaizeGDB (https://ars-usda.app.box.com/v/maizegdb-public/folder/176297337613).
Classification of LTR family dynamics
Family size and solo:intact ratio of each family were compared between tropical genomes (n = 13) and temperate genomes (n = 10) to determine LTR family dynamics. Comparisons between tropical and temperate genome groups were based on a Student's t-test with P ≤ 0.05 as the significance cutoff. Families with significantly different family sizes and solo:intact ratios were classified as removal families. Families with significantly different family sizes but no statistical difference in solo:intact ratios were classified as amplification families. Families with significantly different solo:intact ratios but no statistical difference in family sizes were classified as balanced families. Finally, families with neither significantly different family sizes nor solo:intact ratios were classified as drifting families. For amplification families, if the average family size was larger in tropical than in temperate genomes, the family was classified as a tropical amplification family, and conversely, if the size in temperate genomes was larger, the family was classified as a temperate amplification family. Similar assignment was carried out for removal families. A small number of removal families had inconsistent direction of removal and family size between tropical and temperate genomes; specifically, tropical removal families had a higher solo:intact ratio in temperate genomes, and temperate removal families had higher solo:intact ratios in tropical genomes. These families contributed only small effects to TE content variation (−0.20 Mb and 0.59 Mb, respectively) and were removed from downstream analysis. All family classifications can be found in Supplemental Dataset S2.
Classification of LTR families based on age
LTR families were classified into Young, Moderate, and Old using the age distribution of intact LTR-RTs in each family within the temperate and tropical genomes (Supplemental Table S2). LTR families with fewer than 10 copies were not classified owing to low confidence in inferring their age distributions. For those families with more than 10 copies among the temperate and tropical genomes, the divergence of intact LTR-RTs was binned with 0.002 identity intervals, and the frequency of intact LTR-RTs was calculated in each bin. If the first bin ([0, 0.002)) had the highest frequency, the family was classified as a “Young” family. If the first bin did not have the highest frequency but contained ≥5% total LTR-RTs of this family, the family was classified as a “Moderate” family. If the first bin contained <5% total LTR-RTs of this family, the family was classified as an “Old” family.
Determining the epigenetic status of LTR elements
UMRs were previously identified based on enzymatic methyl-seq reads (PE 150, 300 million or more reads per genotype, original data accessible from ArrayExpress [https://www.ebi.ac.uk/biostudies/arrayexpress] under accession number E-MTAB-10088) from second leaves of pooled plants with two biological replications for each genome (Hufford et al. 2021). These UMRs were defined primarily based on hypomethylation in the CHG context (H = A, T, or C), which is a strong indicator of euchromatin. Many of these CHG-defined UMRs contain high levels of methylation in the CG context (Hufford et al. 2021). Only intact LTR elements with unambiguous strand directions were used for this analysis. The coordinate of the 5′ LTR of each element was determined based on the pangenome annotation and the strand information, which was used to intersect with whole-genome UMRs using BEDTools (v2.30.0) (Quinlan and Hall 2010). UMRs that overlapped ≥200 bp with the 5′ LTRs were candidates of unmethylated LTRs. Those that started upstream of the 5′ LTR on the correct strand were removed. The remaining UMRs were determined to have originated within 5′ LTRs (UM-5′LTRs).
Sequences for UMRs originating within the centromeric retrotransposons of maize 2 (CRM2) elements were extracted from NAM founder lines, and only those located on the positive strand were retained. MAFFT (v7.487) (Katoh and Standley 2013) was used to align CRM2-UMRs to the CRM2 sequence from the TE library with default parameters. The resulting alignment was converted to the SAM format using JVarkit biostar139647 (Lindenbaum 2015) and visualized using the Integrative Genomics Viewer (IGV) (v2.4.17) (Robinson et al. 2011).
TE family expression analysis
Family-level transcript abundance estimates were computed for two replicates of 10 tissues for each genotype (Hufford et al. 2021) using a previously described method (Anderson et al. 2019) adapted for NAM TE annotations. Briefly, RNA-seq reads were downloaded from ArrayExpress E-MTAB-8633 and E-MTAB-8628 and mapped using HISAT2 (v2.1.0; parameters -p 6 -k 20) (Vaser et al. 2017), sorted by name using SAMtools sort (v1.9) (Li et al. 2009), and overlapped with features using HTseq (parameters -s no -t all -m union -a - ‐‐nonunique all). The annotation files used in HTseq were generated by first subtracting exon regions from the TE annotation for each NAM genome using BEDTools subtract (v2.27.1) (Quinlan and Hall 2010) and then concatenating this file with the full-length gene sequences before sorting. This resulting annotation file prioritizes genes over TEs in overlapping regions. Count tables for each TE family and all genes (collapsed into an entry named “Gene”) were then created with the script “te_family_mapping_ver8.2_NAM.pl.” This script counts reads toward TE families if they are uniquely mapping to a single TE or the read is multimapping and all mapping locations that intersect a feature are annotated as the same TE family. Paired-end reads were only counted once.
The table containing raw read counts for each TE family (Supplemental Dataset S3) was used to identify differentially expressed families between tropical and temperate genomes. Only families that were shared by tropical and temperate genomes were retained (n = 15,957). Libraries from the three admixed genomes (M37W, Mo18W, and Tx303) were removed. To normalize the library size effect caused by differences in sequencing depth, we first determined whether structurally intact and/or fragmented TEs are transcribable. To do this, we mapped long-read transcriptomes from six B73 tissues (ear, embryo, endosperm, pollen, root, and tassel downloaded from the NCBI Sequence Read Archive [SRA; https://www.ncbi.nlm.nih.gov/sra] under accession number SRP067440) (Wang et al. 2016) back to the B73v5 genome using minimap2 (2.17-r941) (Li 2018). For the uniquely mapped reads, we summarized mapping targets (genomic regions in B73) that overlapped at least 1 bp with either structurally intact or fragmented LTR sequences using BEDTools intersect (v2.27.1) (Quinlan and Hall 2010). We found that both structurally intact and fragmented LTR sequences contributed substantially to TE transcripts in all six B73 tissues. Thus, we used the total read counts aligned to all TE sequences to normalize the library size effect of our short-read data. The “median of ratios” method (Anders and Huber 2010) was used to estimate the normalization factor for each library (Supplemental Dataset S4). In brief, for each library, raw counts were divided by the geometric mean of each TE family. The median for nonzero ratios in a library was used as the size factor for this library. The variance stabilizing transformation (VST) was then used to transform and normalize raw counts using DESeq2 (v1.30.1) (Love et al. 2014) based on the normalization factors determined using the median of ratios method. TE families differentially expressed between tropical and temperate genomes were identified for each of the 10 tissues and for all tissues with adjusted P-values < 0.05 (Wald test, FDR) using DESeq2 (v1.30.1) (Love et al. 2014). To normalize for family size, values of fragments per kilobase of sequence per million mapped fragments (FPKM) of each family were estimated using all tissues and replicates combined. The total length of each TE family was used in the FPKM calculation, and the log2 + 1 method was used to transform raw FPKM values.
Estimation of allele frequency
Variation frequency was estimated using LTR insertional events and single-nucleotide polymorphisms (SNPs) (Supplemental Methods). LTR insertion frequency spectra were estimated on both missing-filtered and missing-unfiltered data sets using SoFoS (v2.0) (https://github.com/CartwrightLab/SoFoS) with the parameters “-r -a 1.0 -b 1.0.” The population size was rescaled to 10 (-n 10) to account for imbalanced population size between tropical and temperate groups with either folded (-f) and unfolded (-u) estimations. Folded SNP site frequency spectra (SFSs) were estimated on both missing-filtered and missing-unfiltered SNP sets for both tropical and temperate subpopulations using SoFoS with parameters “-f -r -a 1.0 -b 1.0” and population sizes rescaled to 10 (-n 10).
Estimation of recombination rate
A composite recombination map derived from all NAM RILs (backcrossed to B73) across all families was obtained from Calfee et al. (2021) and used to estimate the local recombination rate at each of the intact LTR-RTs. The genetic map was downloaded from the CyVerse data commons (https://datacommons.cyverse.org/browse/iplant/home/silastittes/parv_local_data/map/ogut_v5.map.txt) and converted to recombination rate in the unit of cM/Mb in R (v4.0.3) (R Core Team 2020) based on the B73v5 physical map. The recombination rate at each intact LTR-RT was approximated using the recombination rate data point with the nearest physical distance.
Identification of NLR genes and analyses
NLR annotations for all NAM lines were obtained from Prigozhin et al. (2024). We also identified NLR genes independently using NLR-Annotator (v2.1b) (Steuernagel et al. 2020) and intersected the resulting motif annotations with the NAM gene annotations. The identified genes were, on average, 85% consistent with the public NLR annotation (Prigozhin et al. 2024); thus, we merged the two annotations as our final NLR gene set (Supplemental Table S6). NLR clusters are generally defined by NLR genes with a physical distance of <200 kb (Van de Weyer et al. 2019). To focus on relatively tightly linked clusters and analyze their flanking regions, here we clustered genes located within 100 kb and required a minimum of two genes per cluster. To focus on genes that may have been duplicated through TE-associated mechanisms, we identified gene clusters based on tandem gene annotations for the NAM lines (Hufford et al. 2021). A background set of 5000 random non-NLR singletons and all non-NLR gene clusters was identified, with non-NLR genes clustered using the same thresholds as the NLR genes. Enrichment of LTR-RTs between NLR clusters and background genes was compared based on the k intact LTR-RTs from each cluster. Physical distances were calculated by averaging across distances in all NAM lines and for all gene types, which showed a consistent linear relationship of k, with distance increasing on average by 22,338 bp per LTR-RT added. LTR-RTs within clusters were included in the neighborhoods, and excluding internal TEs had no effect on our results. We used the Fisher's exact test to compare the frequency of Young and Old LTR families as well as tropical amplification families in tropical and temperate lines in the NLR and background neighborhoods. The Fisher's exact test was also used to test for the enrichment of TE families in the TE neighborhood of NLR clusters in tropical lines, excluding families containing fewer than 100 total elements across the NAM lines.
Statistical analyses
All statistical analyses and graphic visualizations in this paper were performed using R (v4.0.3) (R Core Team 2020) in RStudio (v1.1.442) (RStudio Team 2020). All statistical tests, when applicable, were two-sided. Aesthetic modification and compilation of plots were done using Inkscape (v1.0) (https://inkscape.org).
Software availability
All source code of panEDTA developed in this study has been released on GitHub (https://github.com/oushujun/EDTA) and as Supplemental Code. All scripts and files generated in this study are available at GitHub (https://github.com/oushujun/PopTEvo), MaizeGDB (https://ars-usda.app.box.com/v/maizegdb-public/folder/176297337613), and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank the Zeavolution online seminar series for providing critical feedback and suggestions based on an earlier version of this study. We thank Dr. Guanjing Hu for helpful guidance and discussion regarding analyses of expression data. We thank Alex Ostrovsky, Björn Grüning, and the entire Galaxy community for assistance in integrating and hosting panEDTA on the Galaxy platform. We thank MaizeGDB for hosting the data generated in this study. We thank Dr. Silas Tittes for providing a maize recombination map on version 5 coordinates of the B73 maize reference genome. This work was supported in part by the National Science Foundation (NSF), Directorate for Biological Sciences (grants IOS-1546727, IOS-1934384, IOS-1744001, IOS-1758800, IOS-2216612, and IOS-1546719), National Institutes of Health (U24HG006620), the Human Frontier Science program (RGP0025/2021), the Minnesota Agricultural Experiment Station, the Iowa State University Postdoctoral Seed Grant (PG101847), and the OSU Enterprise for Research, Innovation and Knowledge (GR130542). The computation was performed in part using the HPC platform at Iowa State University, which was purchased in part by the NSF grant MRI-1726447, and in part using the Minnesota Supercomputing Institute (MSI) at the University of Minnesota.
Author contributions: S.O., A.S.S., N.M., Y.Q., A.S., and S.N.A. generated and curated the data. N.M. performed the phylogenetic analysis. Y.Q. combined pairwise syntenic LTR information at the population level. S.N.A. generated the family-based TE expression data and assisted in analyzing them. J.I.G. assisted in planning and interpreting DNA methylation analyses. A.S.S. performed read mapping and SNP calling and assisted in recombination analyses. A.S. performed the NLR analyses. S.O. performed the pangenome TE annotations, curations, and summaries; classified and analyzed LTR family age and dynamic groups; identified and analyzed UM-5'LTRs; analyzed family-based TE expression data; identified and analyzed syntenic LTR-RTs; analyzed recombination data; and estimated SFS. C.C.M. performed quality control analyses of the TE annotations. T.C. implemented the Galaxy instance of the panEDTA module. S.O., T.C., A.S.S., N.M., Y.Q., A.S., M.B.H., and C.N.H. prepared the manuscript, and all authors revised it. M.C.S., M.B.H., and C.N.H. are senior authors who secured funding and oversaw the project.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278131.123.
- Received May 26, 2023.
- Accepted August 12, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








