
DEAD box RNA helicases are pervasive protein kinase interactors and activators
- Alexander Hirth1,2,10,
- Edoardo Fatti1,2,10,11,
- Eugen Netz3,4,10,
- Sergio P. Acebron1,12,
- Dimitris Papageorgiou5,6,
- Andrea Švorinić1,2,7,13,
- Cristina-Maria Cruciat1,14,
- Emil Karaulanov8,
- Alexandr Gopanenko8,
- Tianheng Zhu1,2,
- Irmgard Sinning7,
- Jeroen Krijgsveld5,6,
- Oliver Kohlbacher3,4,9 and
- Christof Niehrs1,8
- 1Division of Molecular Embryology, DKFZ-ZMBH Alliance, Deutsches Krebsforschungszentrum (DKFZ), 69120 Heidelberg, Germany;
- 2Faculty of Biosciences, Ruprecht-Karls University of Heidelberg, 69120 Heidelberg, Germany;
- 3Applied Bioinformatics, Department of Computer Science, University of Tübingen, 72076 Tübingen, Germany;
- 4Institute for Bioinformatics and Medical Informatics, University of Tübingen, 72076 Tübingen, Germany;
- 5Proteomics of Stem Cells and Cancer, German Cancer Research Center (DKFZ), 69120 Heidelberg, Germany;
- 6Medical Faculty, Heidelberg University, 69120 Heidelberg, Germany;
- 7Heidelberg University Biochemistry Center (BZH), 69120 Heidelberg, Germany;
- 8Institute of Molecular Biology (IMB), 55128 Mainz, Germany;
- 9Translational Bioinformatics, University Hospital Tübingen, 72076 Tübingen, Germany
-
↵10 These authors contributed equally to this work.
Abstract
DEAD box (DDX) RNA helicases are a large family of ATPases, many of which have unknown functions. There is emerging evidence that besides their role in RNA biology, DDX proteins may stimulate protein kinases. To investigate if protein kinase–DDX interaction is a more widespread phenomenon, we conducted three orthogonal large-scale screens, including proteomics analysis with 32 RNA helicases, protein array profiling, and kinome-wide in vitro kinase assays. We retrieved Ser/Thr protein kinases as prominent interactors of RNA helicases and report hundreds of binary interactions. We identified members of ten protein kinase families, which bind to, and are stimulated by, DDX proteins, including CDK, CK1, CK2, DYRK, MARK, NEK, PRKC, SRPK, STE7/MAP2K, and STE20/PAK family members. We identified MARK1 in all screens and validated that DDX proteins accelerate the MARK1 catalytic rate. These findings indicate pervasive interactions between protein kinases and DEAD box RNA helicases, and provide a rich resource to explore their regulatory relationships.
Protein phosphorylation is widespread and plays an eminent role in many cellular processes, such as cell cycle regulation, transcription, metabolism, cytoskeleton dynamics, apoptosis, and cell differentiation (Manning et al. 2002). About 2% of protein-coding genes in the human genome are protein kinases dedicated to phospho-signaling (Manning et al. 2002). Protein phosphorylation is particularly important in signal transduction, in which cascades of phosphorylation events trigger fast and reversible switches (Pearson et al. 2001; Carling 2004). By sequence homology, 478 protein kinases cluster into the eukaryotic protein kinase (ePK) superfamily, and the remaining 40 belong to the atypical protein kinases (aPKs). By substrate specificity, protein kinases are classified as serine/threonine (Ser/Thr) and tyrosine (Tyr) kinases, respectively, although there are also mixed types. Protein kinases are key targets for pharmaceutical intervention because they are frequently dysregulated in pathological states, notably cancer, and because their active site allows for inhibition by small molecules (Chaikuad et al. 2018; Roskoski 2019; Riegel et al. 2022).
To function as molecular switches, protein kinases receive input from post-translational modifications, small molecules, and protein–protein interactions that regulate the conformation and activity of these enzymes (Newton 2001; Pearson et al. 2001; Litchfield 2003; Long and Zierath 2006; Endicott et al. 2012). Examples for allosteric activation by protein cofactors include cyclin-dependent protein kinases (CDKs) stimulated by cyclins, calcium/calmodulin-dependent protein kinases (CAMK) activated by Ca2+/calmodulin, Aurora (Aur) kinases activated by TPX2 and INCENP, and G protein–coupled receptor kinases (GRKs) activated by GPCRs (Palczewski et al. 1991; Jeffrey et al. 1995; Bayliss et al. 2003; Stefan et al. 2008; Elkins et al. 2012).
An emerging class of kinase activators are DEAD-box (DDX) RNA helicases. DDX RNA helicases, and the closely related DHX RNA helicases, form a family of 44 proteins in humans (Cai et al. 2017), which play key roles in many biological processes involving RNA (Fairman-Williams et al. 2010; Linder and Jankowsky 2011; Henn et al. 2012). These helicases contain a highly conserved core domain consisting of two RecA modules exhibiting ATP and RNA-binding activity and are flanked by divergent N- and C-terminal regions (Linder and Jankowsky 2011). DDX helicases typically unwind RNA structures and dissociate RNA–protein complexes in reactions fuelled by ATP hydrolysis. Prominent cellular processes in which DDX proteins are involved include pre-mRNA splicing, ribosome biogenesis, protein biosynthesis, RNA decay, nuclear export, transcriptional regulation, and maintenance of genome stability (Jarmoskaite and Russell 2014; Cargill et al. 2021; Bohnsack et al. 2023).
One well-characterized RNA helicase is DDX3X, which has a helicase-independent “moonlighting” function during Wnt signaling, in which it acts as a specific activator of Casein kinase 1 epsilon (CSNK1E) (Cruciat et al. 2013; Dolde et al. 2018). The DDX3X function in Wnt signaling is important during Xenopus embryonic axis formation (Cruciat et al. 2013) and in Wnt-driven cancers (Bol et al. 2015; Heerma van Voss et al. 2015; Snijders Blok et al. 2015). CK1 activation by DDX3X is evolutionarily conserved because the Neurospora RNA helicase FRH activates a CK1 homolog in circadian clock regulation (Lauinger et al. 2014), and DDX3X may also stimulate IKBKE and IKBKA (Gu et al. 2013; Fullam et al. 2018). Kinase stimulation may not be limited to DDX3X and CK1, because DDX20/DP103 can directly bind to MAP3K7 (also known as TAK1) and stimulate its kinase activity toward IKBKB (Shin et al. 2014). Moreover, we recently showed that DDX1/24/41/54 bind to casein kinase 2 (CK2) and are required for full kinase activity in vitro and in vivo (Fatti et al. 2023). DDX proteins act as “V-type” activators in both CK1 and CK2 families, affecting maximal enzyme velocity (Vmax) rather than the Michaelis–Menten constant (Km). Kinase stimulation by DDX3X does not require its ATPase activity but requires the RNA-binding domain. Furthermore, data modeling of enzyme kinetics and stopped-flow spectroscopy analyses indicate that DDX proteins act as nucleotide exchange factors (NEFs) toward casein kinases. In particular, DDX proteins may stabilize a kinase conformation that favors the release of unproductive reaction intermediates to accelerate the kinase reaction (Fatti et al. 2023).
Humans have more than 500 protein kinases and 44 DDX proteins, and there is considerable evidence for regulatory interactions in which kinases phosphorylate and regulate DDX proteins and for how DDX proteins act as scaffolds for kinases (Yang et al. 2005; Gustafson and Wessel 2010; Lee et al. 2015). This study aimed to address the questions if protein kinase stimulation by DDX helicases is widespread and which other protein kinases may be regulated by DDX. To identify such protein kinase candidates, we here conducted three orthogonal large-scale screens: a proteome-wide interactome analysis with 32 DDX helicases, a protein array profiling for direct DDX binding partners, and a kinome-wide in vitro kinase activity screen. Taken together, these screens support a pervasive function of DDX proteins as kinetic modifiers of protein kinases and provide a resource to explore the underlying regulatory relationship.
Results
The DDX/DHX helicase interactome
To systematically identify protein interaction partners of DDX/DHX helicases in an unbiased manner and to shed light on biological processes and molecular functions in which DDX proteins may be involved, we performed a large-scale proteomic screen in HEK293T cells, a well-studied cell line for which kinome-wide interactomes are also available (Buljan et al. 2020). We selected 32 different human FLAG-tagged DDX/DHX helicases based on clone availability and successful expression in HEK293T cells (Supplemental Fig. S1A). Three days after transfection of HEK293T cells, FLAG-tagged DDX/DHX helicases were immunoprecipitated in triplicates and subjected to quantitative mass spectrometry to identify bound proteins. As a negative control, we used FLAG-tagged GFP.
Combined analysis of the triplicates of all transfected proteins together including GFP and using a peptide and protein FDR of 1%, identified 58,187 unique and fully tryptic peptides that represent 5012 proteins. The average number of peptides per protein was 11.58 (median, six). The 4565 identified proteins correspond to 4386 Ensembl genes. Following removal of hits represented by fewer than two peptides, 1428 proteins were retrieved that bound to one or several DDX proteins but not to GFP. As a proteome background, we pooled a published HEK293 cell proteome (Bekker-Jensen et al. 2017) with additional proteins identified in our HEK293T cell analysis, totaling around 12,000 proteins (Supplemental Table S1). Hence, the filtered DDX/DHX interactome comprises ∼12% of the HEK293T cell proteome. The data set of the 32 DDX/DHX-interacting proteomes is shown in Supplemental Table S2.
We compared the previously established interactome of DDX54 in HEK293 cells (3066 proteins) (Milek et al. 2017) with our data. Despite different methods (BioID vs. CoIP), there was a large overlap with 571 (40%) of all 1428 DDX/DHX interactors.
We also compared interactors of the 32 DDX/DHX proteins reported in the OpenCell (n = 357) and BioPlex (n = 863) databases, which showed moderate overlap between each other (24.6% of OpenCell set, 10.2% of the BioPlex set). The intersections with our own interactors are 27 of 357 (7.6%) of OpenCell interactors and 141 of 863 (16.3%) of BioPlex interactors. The moderate overlaps between all three resources may be caused by methodological differences.
Cluster representation and overrepresentation analysis of the DDX/DHX interactome
Unsupervised block-clustering (also called biclustering or two-way clustering) divided the 1428 interactors into seven clusters and the 32 DDX/DHX proteins into five clusters that were composed of between four to nine helicases (Fig. 1; Supplemental Table S3). Superimposition of the five DDX clusters with a phylogenetic tree shows little congruence (Supplemental Fig. S1B), indicating that the overall sequence similarity of DDX proteins is not a proxy for similarity of protein interactions.
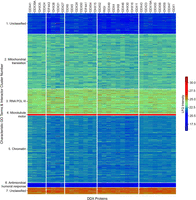
Seven clusters within the DDX/DHX interactome. Biclustering of the DDX/DHX interactome. Heatmap of the interactome (rows) against all DDX/DHX baits (columns). LFQ intensities from the IP-MS experiments were standardized row-wise to Z-scores. Rows and columns were clustered with spectral biclustering into seven row clusters and five column clusters. The heatmap was visualized using the seaborn library (https://doi.org/10.21105/joss.03021).
The seven interactome clusters were composed of two large (2, 5), two medium (1, 3) and three small clusters (4, 6, 7). Overrepresentation analysis (ORA) of clusters 1 and 7 yielded no significant terms (Figs. 1, 2; Supplemental Table S4). Cluster 2 was dominated by “mitochondrial translation”–related terms, consistent with reported activity of RNA helicases in mitoribosome biogenesis (Tu and Barrientos 2015). Cluster 2 was the second-largest cluster (433 proteins), suggesting a prominent role of DDX proteins in mitochondrial RNA processing. Enriched terms of cluster 3 (197 proteins) related besides “mitochondrial translation” to “RNA POL III” transcription, a process in which RNA helicases have not been prominently implicated. Cluster 4 consisted of only 13 proteins and featured “ATPase-dependent activity” and “microtubule motor activity.” Indeed, DDX5 (also known as p68) association with microtubule motors has been documented (Wang et al. 2013), suggesting a broader role of DDX/DHX in microtubule motors. The largest cluster was 5 (531 proteins), which showed a variety of enriched terms relating to “chromatin” but also bona fide RNA-related terms (e.g., “P-body,” “RNAi,” “poly(A)-deadenylation”). The small cluster 6 (36 proteins) showed the most unexpected term enrichment, “antimicrobial humoral response,” and “cornified envelope” with more than 30-fold enrichment and relating, for example, to secreted proteases and secreted protease inhibitors. Because many of these proteins are extracellular, they seem unlikely to encounter cytoplasmic RNA helicases. However, we note that a number of cluster 6 proteins are candidates for “unconventional protein secretion” (UPS), including kallikreins, serpins, annexin A8 (ANXA8), interleukin 36 (IL36), and crystallin beta-gamma domain containing 1 (CRYBG1) (Bendtsen et al. 2004; Noh et al. 2022), suggesting a role for certain DDX proteins in UPS. Altogether, the results suggest roles for DDX/DHX proteins in unexpected biological processes.
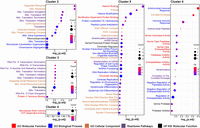
Overrepresentation analysis (ORA) of DDX/DHX interactome clusters. ORA was conducted for DDX/DHX interactome clusters 2–6. Dot plot enrichment containing results for pooled ontology categories and sorted by –log10 (P.adjusted) values. The size of the dots indicates the number of proteins in the category (“count”), and the color of the dots reflects the fold-enrichment (“FE”). Enriched ontology terms are color-coded: GO molecular function is red; GO biological process, blue; GO cellular component, orange; reactome pathway, purple; and UniProt Keyword (UP KW) molecular function, black.
DDX/DHX-interacting proteins are enriched in protein kinases
We next focused on DDX/DHX-interacting protein kinases. When we applied ORA analysis with the Gene Ontology (GO) term “molecular function” on all 1428 DDX/DHX interactors, only three terms reached significance (Supplemental Table S5). Topping the list was “RNA binding” (P = 6.3 × 10−9), consistent with the established function of DDX proteins in RNA biology. The second-best hit was “tau-protein kinase activity” (P = 6.3 × 10−4). Tau-protein kinases are a heterogenous group of enzymes that share the ability to phosphorylate microtubule-associated proteins. The 12 tau kinases retrieved were MARK1,3, GSK3A,B, DYRK1A, TAOK2, CDK5, ROCK1,2, CSNK1D, FYN, and PRKAA1. The third significant GO term was “chromatin binding” (P = 0.0076), aligning with the reported role of DDX members in chromatin organization and transcriptional regulation (Giraud et al. 2018) and GO term enrichment in interactome cluster 4 (Fig. 2). Cluster 5 also featured “Ser/Thr protein kinase” as an enriched term (Fig. 2; Supplemental Table S4).
Of the 465 protein kinases detected in the total HEK293/T cell proteome, 59 (13%) associated with one or more DDX proteins, the great majority Ser/Thr kinases (87%, n = 53, P = 0.028) (Supplemental Table S6). Projecting DDX-associated proteins on a phylogenetic kinome tree showed that hits populated all major Ser/Thr kinase families (Supplemental Fig. S2). The CDK family was most represented (five members), followed by the CK1 family (three members), echoing the function of DDX3X as CK1 regulator (Cruciat et al. 2013; Dolde et al. 2018; Fatti et al. 2023).
Unsupervised spectral clustering and heat map analysis (Fig. 3A) revealed two protein kinase C family (PKC) members (PRKCI, PRKCD) as strong, promiscuous DDX/DHX interactors, besides SCYL1, PRKAA1, and GSK3A. Four kinases (RAF1, IRAK1, PKN3, and PKN2) interacted uniquely with DDX41. There was no apparent overlap with phylogenetic relationships, not for the RNA helicases or the protein kinases. We binned the DDX/DHX kinase interactome into weak, moderate, and strong binders (Supplemental Table S7). Focusing on strong interactions only, all DDX/DHX helicases were represented by at least one “strong” kinase interaction, but only DDX41, DDX3X, DDX52 were represented four or five times. Conversely, a histogram retrieved 12 of the 59 kinases as “strongly” interacting with more than one DDX/DHX (Fig. 3B). Two kinases showed only a single strong DDX/DHX interaction: RIOK1–DDX3X and CSNK1G3–DDX24.
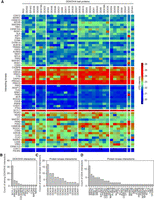
DDX/DHX proteins bind to protein kinases in HEK293T cells. (A) Heatmap of the interacting kinases (rows) against all DDX/DHX baits (columns). The empirical cumulative distribution function (eCDF) of the IP-MS data set was calculated, and the eCDF probabilities of the interacting kinases were plotted as a heatmap. Protein kinases (rows) of the heatmap were sorted by the values of the DDX/DHX baits (columns) with the highest value. The DDX/DHX baits (columns) were sorted by the values in the protein kinases (rows) with the highest value. Therefore, the highest eCDF value is in the top left corner, and the heatmap is sorted by the first row and first column. The heatmap was visualized using the seaborn library (https://doi.org/10.21105/joss.03021). (B) Bar plot indicating the number of DDX/DHX proteins interacting strongly (eCDF > 0.75) with the indicated kinase as retrieved from the DDX/DHX interactome screen. (C) Bar plot indicating the number of kinases interacting with the indicated DDX proteins as identified by Buljan et al. (2020). (D) Bar plot indicating the number of DDX proteins interacting with the indicated kinase as identified by Buljan et al. (2020).
A previously reported proteomics analysis in HEK293T cells of more than 300 protein kinases provides a data set complementary to ours (Buljan et al. 2020) and indicates 34 protein kinases with DDX/DHX partners (18 DDX and four DHX helicases). A histogram focusing on DDX proteins, retrieved 16 helicases interacting with more than one protein kinase (Fig. 3C), most prominently DDX54, DDX18, and DDX24, each interacting with 10 or more protein kinases. Conversely, the kinases that most promiscuously interacted with DDX helicases were members of the serine-arginine protein kinases (SRPK1, -2, -3) (Fig. 3D), enzymes involved in splicing regulation (Giannakouros et al. 2011). Nine Tau kinases (Cavallini et al. 2013), CSNK2A1, CSNK2A2, DYRK2, EIF2AK2, GSK3B, MARK3, NUAK1, SRPK2, and SYK were also among the DDX/DHX interactors, aligning with our observation of Tau kinase enrichment.
Collectively, these findings indicate (1) that a subset of Ser/Thr protein kinases, particularly Tau protein kinases, are prominent DDX/DHX helicase interactors, and (2) that multiple DDX proteins may interact with a given kinase and vice versa.
DDX proteins directly bind multiple protein kinases
The interactome screen (Figs. 1–3) cannot distinguish whether the DDX/DHX-protein kinase interactions are direct or indirect. To screen for direct DDX-protein kinase interactions, we used commercial protein ProtoArrays featuring about 9000 human proteins, including about 400 unique protein kinases, to identify proteins binding to purified V5-tagged DDX39A or DDX56 proteins, but not to GFP (Fig. 4A,B; Supplemental Table S8). DDX56 and DDX39A were chosen because their recombinant proteins could be obtained in sufficient quantity and because DDX56 is capable of CK1 and CK2 family stimulation in vitro (Fatti et al. 2023). Of the around 800 proteins binding to DDX39A and/or DDX56 but not to GFP at the chosen cutoff, 62 (∼8%) were protein kinases, most of them Ser/Thr kinases that map to different phylogenetic clades (Supplemental Fig. S3A). Significantly, all six CK1 and both CK2 family members represented on the array were positive hits, corroborating their reported DDX interactions.
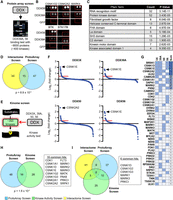
DDX proteins directly bind to protein kinases and can stimulate their enzymatic activity. (A) Outline of the protein ProtoArray binding screen. (B) Examples of DDX-interacting kinases on protein ProtoArray interrogated with V5-tagged recombinant DDX or GFP proteins. (C) Protein domain enrichment analysis (Pfam terms) of ProtoArray proteins bound specifically to DDX39A or DDX56. (D) Overlap between protein kinase hits from the DDX interactome and ProtoArray binding screen. (E) Outline of the kinome-wide activity screen. (F) Ranked results from an in vitro kinase activity screen with the indicated DDX proteins. Every point corresponds to one tested kinase. Kinases stimulated by a DDX protein with log2 fold change above 0.5 (red line) were considered positive hits. (G) Overview of all kinases identified to be stimulated by any of the four DDX proteins. (H) Venn diagram of all kinases identified in the ProtoArray and kinase activity screen showing an overlap of 16 common kinase hits. (I) Venn diagram of all kinase hits from all three screening approaches, showing six kinases being identified to be bound and regulated by DDX proteins in three independent screens.
ORA of all approximately 800 proteins expectedly yielded among the top 10 GO terms various RNA-related (e.g., “poly(A)RNA binding,” “RNA binding”) (Supplemental Table S8). Analysis of the terms of Protein Families (Pfam) once again recovered “RNA recognition motif” as the top hit (Fig. 4C; Supplemental Table S8). The second Pfam term was “protein kinase domain,” followed by other domains, which are also protein phosphorylation related (e.g., SH3, FHA, KA1 domains). In total, 62 DDX-binding protein kinases were retrieved, of which 15 (24%) overlapped with the interactome kinase hits (Fig. 4D), and significantly, these were again populated by family members of CK1, CK2, GSK3, and MARK.
Collectively, the ProtoArray results support (1) that the enrichment of Ser/Thr protein kinases in the DDX interactome reflects their ability to directly bind to RNA helicases and that this involves diverse kinase clades and (2) that besides the established CK1 and CK2 families, GSK3 and MARK families are prominent candidates for kinases susceptible to DDX regulation.
DDX proteins stimulate multiple protein kinases
To address the possibility that kinases beyond CK1 and CK2 families are susceptible to DDX stimulation, we conducted a commercial kinome-wide activity screen. Recombinant DDX3X, -39A, -50, and -56, of whom some were previously shown to stimulate CK1 or CK2 (Cruciat et al. 2013; Fatti et al. 2023), were added to in vitro kinase assays with about 350 ePKs, with matching synthetic peptide or protein substrates in the presence of radiolabeled ATP (Fig. 4E). Most kinases were little affected or unaffected by any given DDX protein, but for DDX3X, -39A, DDX50, and -56 the top hits for DDX-stimulated kinases were CK1 and CK2 family members (Fig. 4F,G; Supplemental Table S9), confirming previous findings and validating the screen. Beyond CK1 and CK2 members, 40 other kinases belonging to various kinase clades were stimulated by one or more DDX proteins (Supplemental Table S9). We also observed kinases whose activity was inhibited by DDX proteins, but because this inhibition may be owing to DDX proteins themselves acting as competitive kinase substrates, we focused on activated kinases only. Although the four DDX proteins showed many “private” hits (Fig. 4G), there were also common hits beyond the CK1 and CK2 families (e.g., MARK and CAMK family members). Other prominent hits included CDK1 and CDK2. Among the Tyr protein kinases, only MET and FLT3 were represented, and among atypical kinases, PDK1 and EEF2K were represented. Of the 44 kinome-activity hits, 16 overlapped with the 62 protein kinases recovered on the ProtoArray (P = 1.6 × 10−4), and nine overlapped with the 59 interactome-retrieved protein kinases (P = 0.11) (Fig. 4H,I). Six kinases were retrieved in all three screens (Fig. 4I), most prominently CK1 and MARK family members.
A summary of the results of the interactome, ProtoArray, and kinase activity screens is shown in Figure 5A, along with the DDX–kinase hits from the proteome screen of Buljan et al. (2020). Selecting kinase families in which at least one member was stimulated by a DDX protein and interacted both in vivo and in vitro yielded the CDK, CK1, CK2, DYRK, MARK, NEK, PRKC, SRPK, STE7/MAP2K, and STE20/PAK families. We therefore consider members of these 10 protein kinase families as strong candidates susceptible to DDX/DHX regulation. Besides, all screens identified protein kinases from all protein kinase groups (Fig. 5B; Supplemental Fig. S4).
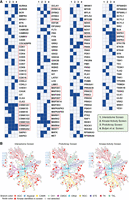
Ten candidate kinase families regulated by DDX/DHX proteins. (A) Kinases detected in one of the indicated screens are marked in blue. Kinase families are boxed in red in which at least one member was stimulated by and interacted with a DDX/DHX protein in vivo and in vitro. (B) Kinome trees depicting kinases (red) identified in the corresponding screen. For a more detailed view, see Supplemental Fig. S4.
To investigate potential commonalities among DDX-susceptible kinases identified through our screens, we analyzed the amino acid context around conservative motifs commonly found in protein kinases (Supplemental Fig. S5A). Specifically, we focused on the G-loop (GxGxxG), known for stabilizing ATP phosphates during catalysis (Barouch-Bentov et al. 2009; Steinberg 2018); the DFG motif (Hari et al. 2013; Ung and Schlessinger 2015) and APE motif (Gógl et al. 2019), governing the start and extension of the activation loop; the HRD motif (Taylor and Kornev 2011; Kanev et al. 2019) within the catalytic loop; and the DxWSxG motif identified in the top-five motifs through MEME analysis (Supplemental Fig. S5B). Sequences around these motifs were retrieved, and sequence LOGOs were generated, grouped by kinase subgroups (Supplemental Fig. S5C–G). However, inspection of these motifs did not reveal significant differences in the amino acid context around these conservative motifs for DDX-susceptible kinases. Additionally, attempts to identify motifs enriched specifically in the DDX-susceptible group or in kinases from interactome, ProtoArray, and kinome screens, using MEME with differential enrichment mode, were unsuccessful in detecting substantially enriched motifs.
DDX helicases are V-type activators of MARK1
To corroborate the validity of our prediction, we selected MARK1, member of a kinase family identified in all three screens (Fig. 5A). Mammalian microtubule affinity regulating kinase 1 (MARK1/Par-1c) belongs to the adenosine-monophosphate activated protein kinase (PRAAK) branch of the calcium/calmodulin-dependent protein kinase (CAMK) group of kinases. MARKs are Tau kinases, and they are involved in epithelial cell polarization, cell signaling, and neuronal differentiation (Drewes et al. 1997; Marx et al. 2010). Similar to CK1, MARK kinases also phosphorylate Dishevelled, a scaffold protein required in WNT signaling (Ossipova et al. 2005).
MARK1 is subject to complex regulation. First, MARK1 contains an autoinhibitory KA1 domain (Fig. 6A; Emptage et al. 2017). Second, MARKs are activated by phosphorylation by an upstream kinase, which can be mimicked by a phosphomimetic T208E substitution (Timm et al. 2003). We expressed and purified from Escherichia coli recombinant MARK1150–371 (hereafter referred to as “MARK1”) lacking the autoinhibitory domain to exclude DDX effects owing to overcoming MARK1 autoinhibition. We also produced the corresponding constitutively active MARK1T208E protein (Fig. 6B). Using a MARK1-specific peptide substrate, we established an in vitro kinase assay, in which MARK1T208E showed elevated kinase activity compared with MARK1 (Fig. 6C). Further experiments were conducted with this constitutively active MARK1T208E.
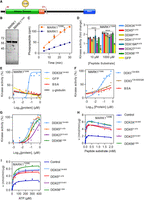
MARK1 activity is stimulated by DDX proteins in vitro. (A) Schematic of MARK1 with the boundaries indicated in red for the truncated recombinant protein used (aa 50–371). (UBA) Ubiquitin association domain, (KA1) kinase-associated domain 1, and (T208E) phosphomimetic mutation for activated MARK1. Cartoon generated using IBS v1.0.3 (Liu et al. 2015). (B) SDS-PAGE gel stained with Coomassie blue of MARK1 expressed in and purified from E. coli. (C) In vitro kinase assay linearity of MARK1 and MARK1T208E. (D) In vitro kinase assay with MARK1T208E with the addition of different DDX core domain proteins both at low (10 µM) and high (1000 µM) peptide substrate. (****) P > 0.0001. (E) In vitro kinase assay using MARK1T208E and increasing amounts of either DDX3X132–605 or different negative control proteins for EC50 determination. (F) In vitro kinase assay using MARK1T208E and increasing amounts of either DDX3X132–605, DDX3 QM, or BSA as control proteins. (G) In vitro kinase assay using MARK1T208E and increasing amounts of DDX core domain proteins for EC50 determination. (H) In vitro kinase assay using MARK1T208E with the addition of DDX core domain proteins and increasing concentrations of peptide substrate at saturating ATP concentration. (I) In vitro kinase assay using MARK1T208E with the addition of DDX core domain proteins and increasing concentrations of ATP at saturating peptide concentration.
We tested MARK1T208E activity in the presence of a panel of recombinant DDX helicases, DDX3X, -5, -27, and -56, that were found in the DDX interactome screen to bind MARK1 and that can stimulate CK1 and CK2 in vitro (Fatti et al. 2023). Because the nonconserved termini can cause protein aggregation, the helicases were produced and purified from E. coli as core domain proteins. All helicases (but not GFP) stimulated MARK1T208E at low and high peptide substrate concentration about twofold (Fig. 6D). The control proteins GFP, BSA, and γ-globulin failed to stimulate MARK1T208E, even at high concentrations (Fig. 6E), ruling out unspecific carrier protein or protein crowding effects. We previously performed alanine scanning mutagenesis of DDX3X and identified residues involved in kinase stimulation of CSNK1E and CSNK2A2 (Fatti et al. 2023). Kinase assays of MARK1T208E with increasing concentrations of the quadruple mutant DDX3X QM showed reduced kinase stimulation in comparison to DDX3X132–605 (Fig. 6F). In contrast, kinetic analysis of MARK1T208E with constant peptide substrate and ATP concentration and varying concentrations of recombinant DDX3X, -5, -27, and -56 core domain proteins all showed MARK1 kinase activity stimulation by the helicases, but with different potencies, DDX3X being the most potent (Fig. 6G). Kinetics of MARK1T208E with constant peptide substrate and ATP concentration but varying concentrations of recombinant DDX3X yielded an EC50 of 60 nM.
We carried out two-substrate steady-state kinetic analysis and measured initial reaction velocities at varying concentrations of peptide substrate and ATP. Increasing the peptide substrate concentrations at constant ATP concentration, the velocity curve rose to a maximum and then declined, indicative of substrate inhibition (Fig. 6H). Substrate inhibition frequently occurs in two-substrate enzymes owing to formation of unproductive intermediates (Cornish-Bowden 1995). The addition of DDX proteins stimulated MARK1T208E kinase activity without much affecting the substrate inhibition constant (Ki) (Supplemental Table S10). DDX proteins also caused no major change in the apparent Michaelis–Menten constant Km for the peptide substrate (Supplemental Table S10). Instead, they increased two- to 2.5-fold the apparent maximal reaction velocity Vmax (Supplemental Table S10). Increase of Vmax with little change in Km is characteristic of V-type allosteric enzyme activators and was previously observed for stimulation of both CSNK1A and CSNK2A2 by DDX helicases (Cruciat et al. 2013; Fatti et al. 2023). Of note, MARK1 stimulation by a physiological DDX/DHX partner may likely be higher.
Titrating ATP concentration at a constant high peptide concentration yielded typical Michaelis–Menten velocity curves without substrate inhibition (Fig. 6I). Addition of DDX proteins once again did not show consistent changes in Km for ATP but increased Vmax twofold to threefold (Fig. 6H; Supplemental Table S10), supporting V-type activation. We conclude that DDX helicases can function as V-type activators of MARK1.
Discussion
We present a panoramic view of the generic DDX helicase interactome, report hundreds of binary kinase–DDX interactions, and identify more than 40 protein kinases that are susceptible to DDX stimulation in vitro, including the experimentally validated MARK1. Overall, we provide evidence for a pervasive function of DDX proteins as kinetic modifiers of protein kinases. Our study provides a rich resource to explore these emerging regulatory kinase–helicase relationships.
The DDX interactome
For a few of the 44 human DEAD box helicases proteomics data are available (Copsey et al. 2017; Milek et al. 2017; Mersaoui et al. 2019), and here we report protein interactomes of 32 DDX/DHX proteins. Clustering and enrichment analyses yield biological processes, molecular functions, protein complexes, and cellular compartments generically associated with DDX/DHX proteins. RNA-binding proteins dominate the DDX/DHX interactome, notably DDX/DHX proteins themselves, suggesting that RNA helicases act in a concerted fashion. However, the discrepancy between the enriched GO terms usually associated with RNA helicases in the literature and the terms obtained in the unbiased interactome analysis was striking. The most significant terms related to “mitoribosome” (cluster 2), “Pol III transcription” (cluster 3), “microtubule motor activity” (cluster 4), “histone binding” (cluster 5), and proteins that may be secreted by UPS (cluster 6). These findings point to unexpected biological processes in which DDX/DHX proteins may play a role.
Protein kinases are widespread interactors of DEAD box RNA helicases
The key finding of this study was the widespread occurrence of protein kinases as DDX/DHX interactors. Both the interactome and the ProtoArray screen retrieved Ser/Thr protein kinases as top hits, and many kinase families overlapped between the two screens, supporting direct interactions. Recurrent families in all three screens were found among nine Ser/Thr kinase families (CDK, CK1, CK2, DYRK, MARK, NEK, PRKC, STE7/MAP2K, and STE20/PAK). In addition, among all Ser/Thr kinases analyzed in the proteome screen of Buljan et al. (2020), SRPK family members partnered most frequently with DDX proteins. Although SRPKs were not recovered in our stringently filtered DDX interactome, SRPK members were positive both in the ProtoArray and in the kinase activity screen, and they are among reported DDX54 interactors (Milek et al. 2017), suggesting that they are also strong candidates for DDX regulation. We therefore propose that members of altogether 10 kinase families represent prime candidates for DDX regulation. Among these 10 kinase families, direct, physiologically relevant interactions with RNA helicases are documented for CDK (Liu et al. 2002; Yang et al. 2015; Bush et al. 2016) and PAK (Li et al. 2021). The most recurrent was the CK1 family, members of which were retrieved in all screens, including in the previously reported kinase interactome (Buljan et al. 2020), and whose activity was most stimulated by DDX proteins. Although the stimulation of other kinases was more modest, it has to be kept in mind that the DDX proteins employed in the kinase activity screen likely are not physiological partners, and hence, they may be less potent. Identifying endogenous DDX/DHX partners of protein kinases can be guided by proteome screens, such as the here reported DDX interactome, as well as the kinase interactome (Buljan et al. 2020; Fatti et al. 2023).
Besides being Ser/Thr kinases, the 10 protein kinase families share no obvious similarities such as cellular function, protein domain, cofactor, or regulatory mode. However, one commonality in the DDX/DHX and kinase interactomes (this work and Buljan et al. 2020) were tau protein kinases, which represented five (CDK, CK1, CK2, DYRK, and MARK) of the 10 recurrent families. This raises the possibility that DDX proteins play a role in tau kinase–mediated microtubule regulation; indeed, DDX5 and DDX6 interact directly with microtubules and Tau, respectively (Wang et al. 2013; Chauderlier et al. 2018). MARK1 is one of these tau kinases, and multiple DDX proteins function as V-type activators for it in vitro, just as they do for CK1 and CK2 (Fatti et al. 2023).
The results raise the question what may be the underlying structural and kinetic mechanism whereby DDX proteins increase the Vmax of multiple Ser/Thr protein kinases. Kinetic analyses showed that DDX activation of CK1 and CK2 is strongest at high substrate concentration, when dead-end reaction intermediates dominate. Kinetic modeling predicts—and stopped flow spectroscopy confirmed—that DDX proteins may act by accelerating ADP dissociation (Fatti et al. 2023), proposed to be the rate-limiting step in many protein kinases (Adams 2001). Thereby, DDX may reduce the formation of dead-end reaction intermediates and increase Vmax. Yet, DDX proteins stimulated MARK1 without noticeably reducing substrate inhibition, suggesting that they affect steps in the reaction cycle other than ADP dissociation to increase Vmax. Clearly, structural elucidation of DDX–kinase complexes and analysis of how DDX proteins affect the rate constants of the individual MARK1 reaction steps will be illuminating in the future.
An observation that begs explanation is that multiple, arbitrarily chosen DDX proteins are more or less all able to stimulate multiple protein kinases in vitro, whereas in vivo only very specific DDX partners are required for full CK1 and CK2 activity (Cruciat et al. 2013; Fatti et al. 2023). Importantly, DDX promiscuity also pertains to DDX–RNA interactions, which are nonselective in vitro and yet show specificity in vivo. RNA specificity is thought to derive from a combination of (1) intrinsic selectivity for certain RNAs, (2) targeting of the helicases to relevant substrates and compartments via their nonconserved N and C termini, and (3) cofactors conferring specificity (Shen and Pelletier 2020).
A limitation of this study is that the interactome screen was conducted with overexpressed DDX/DHX proteins, and hence, it cannot distinguish between generic and physiological interactions. The interactome overlap with the OpenCell and BioPlex DDX/DHX interactomes was limited. The ProtoArray and kinome screens employed few selected DDX proteins, which mostly yielded generic interactions. Endogenous DDX–kinase partners and physiological relevance need to be confirmed, for which our proteomics data set and the data set of Buljan et al. (2020) will be valuable resources (see also Fig. 5). The mechanism whereby DDX proteins increase Vmax of protein kinases needs to be elaborated. The structural determinants underlying the specificity for endogenous DDX–kinase pairings remain to be determined. Our study provides the basis to explore these and other emerging questions.
Methods
Expression and purification of recombinant proteins from E. coli
MARK1, MARK1T208E, and DDX core domains were expressed in E. coli Rosetta(DE3)pLysS (Novagen) with an auto-induction medium system and were further purified as previously described (Fatti et al. 2023).
Immunoprecipitation of FLAG-DDX proteins
HEK293T cells of three to five 15 cm dishes transfected with the respective DDX protein were harvested for immunoprecipitation using FLAG beads. A detailed protocol can be found in the Supplemental Methods.
Protein digestion
DDX proteins captured on FLAG beads were digested at 37°C overnight using 1 µg of trypsin–LysC mix. A detailed protocol can be found in the Supplemental Methods.
LC-MS/MS analysis
Peptides were resolved using the easy NanoLC1200 fitted with a trapping (Acclaim Pepmap C18, 5 µm, 100 Å, 100 µm × 2 cm) and an analytical column (nanoEase M/Z Peptide BEH C18 column, 130 Å, 1.7 µm, 75 µm × 250 mm). The outline of the analytical column was coupled directly to an Orbitrap fusion (Thermo Fisher Scientific) mass spectrometer. Solvent A was 0.1% formic acid (vol/vol), and solvent B was 80% acetonitrile (vol/vol), 0.1% formic acid (vol/vol). The peptides were loaded on the trap column with a constant flow of solvent A at a maximum pressure of 800 bar. Peptides were eluted from the analytical column at a constant flow rate of 300 nL/min and a temperature of 55°C. During the elution, the percentage of solvent B was increased in a linear gradient from 3% to 8% in 4 min, then from 8% to 10% in 2 min, then from 10% to 32% in 68 min, and then from 32% to 50% in another 12 min. At the end of the gradient, solvent B was kept at 100% for 7 min followed by re-equilibration of the analytical column for 10 min at 97% solvent A. The peptides were introduced to the mass spectrometer via a Sharp singularity emitter (365 µm OD × 20 µm ID, 8 cm long; Fossiliontech) and a nano-source spray voltage of 2.5 kV. The ion transfer tube temperature was set to 320°C. Full scan MS2 spectra were acquired within the range (m/z) of 375–1500 in the Orbitrap detector with a resolution of 120,000. The maximum injection time was set to 50 msec and automatic gain control target (AGC) to 106 ions with the normalized AGC of 250%. The most abundant ions within a 3 sec cycle time window were selected for fragmentation. Ions with unassigned charges and charges of one or above four were excluded. Dynamic exclusion was set to 20 sec with a mass tolerance of ±10 ppm. For the MS2 scans, the quadrupole was used with an isolation window of 1.6 m/z. For peptide fragmentation, higher-energy collisional dissociation (HCD) was used at 33%. MS2 scans were acquired in the linear ion trap that was operated in the rapid ion scan rate with an AGC target of 1 × 104 ions or a maximum injection of 50 msec. MS2 scans were acquired as centroid data type. MS data were then analyzed using MaxQuant software (version 2.0.3.0) (Tyanova et al. 2016); for details, see the Supplemental Methods.
HEK293T proteome
To define the proteome background for the enrichment analyses, we used data from a deep proteome study of multiple human cell lines including HEK293 (Bekker-Jensen et al. 2017). A protein was accepted into the set if it was identified in at least one of the two replicates for the HEK293 cell line. Additionally, all proteins from our own filtered IP-MS data were added. Thus, our background proteome contained 12,226 proteins. For the full list of proteins included in this proteome data set, see Supplemental Table S1.
GO molecular function enrichment on DDX/DHX bait interactome
We retrieved GO terms (The Gene Ontology Consortium et al. 2000) in the molecular function category using the REST API service from the UniProt web server (release 2022_02) (UniProt Consortium 2023a) and performed an enrichment analysis. To define the DDX interactome in the IP-MS data, we selected all proteins that were quantified in any experiment with any of the DDX/DHX baits and removed all proteins that were also found in any of the three GFP controls. The set of remaining interactors contained 1428 proteins. The enrichment analysis for each GO molecular function term was performed using a one-sided binomial test as implemented in SciPy v1.7.1 (Virtanen et al. 2020). Multiple testing correction was performed using the Benjamini–Hochberg FDR approach as implemented in the Python package statsmodels v0.12.2 (https://doi.org/10.25080/MAJORA-92BF1922-011).
Biclustering of DDX/DHX baits and interactome
The IP-MS matrix of all interactors against the DDX/DHX baits was clustered using spectral biclustering (Kluger et al. 2003) as implemented in scikit-learn v0.24.2 (Pedregosa 2011). The cluster numbers for both axes were estimated using the elbow method and yielded five DDX clusters (columns) and seven interactome clusters (rows). The same method was used for biclustering of the interacting kinases and yielded seven DDX clusters (columns) and six kinase clusters (rows). The visualization of the clustered heatmaps was done using the seaborn library (Waskom 2021).
Heatmap of DDX/DHX baits and interacting protein kinases
For the heatmap in Fig. 3A, the empirical cumulative distribution function (eCDF) of the IP-MS data excluding the GFP controls was calculated and plotted in a heatmap for the 59 protein kinases interacting with any DDX/DHX bait. Protein kinases (rows) of the heatmap were sorted by the values of the DDX/DHX baits (columns) with the highest value. The DDX/DHX baits (columns) were sorted by the values in the protein kinases (rows) with the highest value. Therefore, the highest eCDF value is in the upper left corner, and the heatmap is sorted by the first row and first column. Kinases with an eCDF of > 0.75 were considered strong binders; moderate binders showed an eCDF of >0.5 and <0.75.
Phylogeny of DDX proteins
To compute a phylogenetic tree for the DDX/DHX baits used in this study, a multiple sequence alignment was generated using the Clustal Omega (v1.2.4) (Sievers et al. 2011) server provided by the EMBL-EBI (Li et al. 2015) with the default settings for protein sequences. The tree was constructed using the Bio.Phylo.TreeConstruction module (Talevich et al. 2012) of Biopython v1.78 (Cock et al. 2009) and employing neighbor-joining using the BLOSUM62 distance matrix. It was visualized using NetworkX v2.6.3 (https://conference.scipy.org/proceedings/SciPy2008/paper_2/).
Over-representational analysis (ORA)
ORA was conducted with clusterProfiler (v.4.4.4) for GO terms (molecular function, biological process, cellular component) and ReactomePA (v.1.40.0) for reactome pathways. The DAVID 2021 web tool (Dec. 2021, https://david.ncifcrf.gov/) was used for ORA for UniProt keyword molecular function terms (UP KW MF). For GO and reactome pathway analysis, org.Hs.eg.db (v. 3.15.0) was used as a database. As gene sets (for each proteome cluster), the UniProt Accessions IDs were utilized, and as the universe (background), the HEK293T-related background gene set (UniProt Accessions IDs; number of genes = 12.194) was used. For reactome pathway analysis, UniProt Accessions IDs were transformed to entrez gene IDs using the bitr function from clusterProfiler. FoldEnrichment values were calculated as a GeneRatio/aBgRation ratio. Simplification of redundant terms (only for GO) was carried out using simplify function from clusterProfiler with defaults (cutoff = 0.7). Enrichment dot plots were generated with ggplot2 (v. 3.3.6, Wickham 2016) and arranged with the ggarange function from ggpubr (v. 0.4.0) (https://rpkgs.datanovia.com/ggpubr/). Only categories passing P.adjusted < 0.05 and q.value < 0.05 cutoffs were retained.
Short motif analysis
The protein kinase sequences were collected from https://ftp.Ensembl.org/pub/release-109/fasta/homo_sapiens/pep/ based on the human kinase list provided at KinHub (http://www.kinhub.org). The “main” isoform of the kinase (for each gene among isoforms that exist owing to the existence of different transcripts' isoforms) was selected based on Ensembl annotation of the canonical transcripts. The classification of kinases was added based on interactome screen (Supplemental Table S6), ProtoArray screen (Supplemental Table S8), kinome screen (Supplemental Table S9), and Figure 5A (red box labeled), DDX-susceptible. The protein kinase sequences were scanned with MEME (v. 5.4.1; parameters: -mod oops -nmotifs 5 -minw 3 -maxw 7) (Bailey and Elkan 1994). The top five identified motifs corresponding to G-loop (GxGxxG), HRD motif, DFG motif, APE motif, and DxWSxG were used as input for FIMO (v. 5.4.1) (Grant et al. 2011) to locate these motifs in protein kinase sequences. Coordinates of located motifs were used to retrieve the ±5 amino acids context around the motifs. The locations of the motifs were identified by FIMO (q-values < 0.05). Kinases with the motif order G-loop, HRD, DFG, APE, DxWSxG (from N- to C-terminus) were retained. The ggseqlogo package (v. 0.1) (Wagih 2017) was utilized to produce sequencing logos.
Protein ProtoArray analysis
HEK293T cells were grown in 15 cm dishes; transfected with 15 µg of V5-tagged DDX39A, DDX56, or GFP; and harvested in ice-cold PBS. Cells were lysed with lysis buffer C (50 mM HEPES-KOH at pH 7.7, 1 mM MgCl2, 300 mM NaCl, 0.8% Triton X-100, 5 mM NaF, 0.1% sodium deoxycholate, 2 mM β-mercaptoethanol, 1× protease inhibitor mixture tablet; Roche). Cleared lysates were immunoprecipitated using anti-V5 agarose beads (Roche) for 3 h at 4°C. Beads were washed in TBS-T supplemented with 1 mM MgCl2, 2 mM β-mercaptoethanol, and 1× protease inhibitor mixture tablet (Roche), followed by elution in the same buffer using 200 µg/mL V5 peptide. Eluted proteins were analyzed in a Coomassie gel to assess their purity, supplemented with 10% glycerol, and stored at –80°C. Purified proteins were supplemented with 1% BSA before incubation on the protein arrays.
Human protein ProtoArrays (ProtoArray v5.1, Invitrogen) containing more than 9000 unique proteins were incubated for 1 h at 4°C in blocking buffer I (PBS, 0.1% Tween 20, 1% BSA). The arrays were washed three times at 4°C in blocking buffer II (TBS supplemented with 0.5% Triton X-100, 1 mM β-mercaptoethanol, 5% glycerol, 2 mM MgCl2, 1% BSA). Arrays were placed at RT and incubated for 1 h with 600 µL of 0.5 µM of immunopurified V5-DDX39A, V5-DDX56, or V5-GFP (see above). Treated arrays were washed twice in blocking buffer II at 4°C and incubated with 1:1000 a-V5 tag monoclonal antibody (Thermo Fisher Scientific R960-25) for 2 h at 4°C. After three additional washes in blocking buffer II, arrays were incubated with antimouse secondary antibody, Alexa Fluor 680 (Thermo Fisher Scientific A28183) for 1 h at 4°C and washed again three times. ProtoArrays were rinsed three times in water, dried by spinning at 300g for 5 min, and scanned in an Odyssey scanner (LI-COR) using the following parameters: channel 700, grid = 8 × 3, 21.17, intensity = 5, off-set = 0. ProtoArray signal data were determined with GenePix Pro v5.0.0.49 (Molecular Devices) and then processed following the manufacturer's recommendations using ProtoArray Prospector v5.2 (Invitrogen). Candidate interactors were called with a Z-score > 1, coefficient of variation below 0.5, and at least eightfold higher background-subtracted fluorescence intensity in DDX39A and/or DDX56 compared with the GFP control. ProtoArray Database IDs from GEO platform annotation GPL27459 were converted using bioDBnet (https://biodbnet-abcc.ncifcrf.gov/db/db2db.php) to current official gene/protein symbols and their corresponding Entrez gene IDs. Hit enrichment analysis was carried out with DAVID v6.8 using Entrez gene IDs of all ProtoArray proteins as a specific background. Up-to-date human protein kinase annotation was derived from UniProt release “2020_02” (UniProt Consortium 2023; https://ftp.uniprot.org/pub/databases/uniprot/).
Kinome-wide activity screen
Kinome-wide screen and kinase candidate retests were carried out by a commercial provider, Reaction Biology (RBC). DDX3X (Origene), DDX39A (Abnova), and DDX56 (Origene) were screened at a concentration of 100 nM, and DDX50 (Origene) at a concentration of 100–125 nM, against a panel of 350 protein kinases using RBCs proprietary HotSpotSN Technology (Anastassiadis et al. 2011). In brief, kinase assays were carried out in a buffer containing 20 mM HEPES (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, and 1% DMSO. Kinase concentrations were between 0.1 and 500 nM, and those of substrates were between 0.2 and 100 µM. Each kinase and its substrate and the DDX protein were added to the reaction buffer, and the kinase reaction was initiated by the addition of 50 µM 33P-γATP. Following incubation for 2 h at RT, kinase activity was detected by a filter-binding method. For kinase hit validation, assays were performed using three different conditions, that is, substrate alone ± DDX protein, kinase alone ± DDX protein, and substrate with kinase ± DDX protein. Protein kinase official symbols were updated using UniProt IDs with bioDBnet to allow unbiased comparisons with the ProtoArray hits.
Statistical analysis
The statistical testing of the overlap significance between kinase sets identified by interactome, ProtoArray, and kinome screens was performed utilizing the GeneOverlap package (v. 1.32.0) using the Fisher's exact test (alternative = greater). The kinase sets identified by interactome screen (IS; n = 59), ProtoArray screen (PS; n = 62), and kinome screen (KS; n = 44) were retrieved from Supplemental Tables S6, S8, and S9, respectively. The background (bg) for each pair of comparisons was estimated as the intersection of all kinases available for the platform (“common” background). Only hits that belong to “common” backgrounds were used for statistical testing.
In vitro kinase assay
Kinase filter binding assays were performed with 32P-γATP (PerkinElmer NEG502A001MC; 10 m Ci/mL, 3000 Ci/mmol) and specific peptide substrate (CHKtide: KKKVSRSGLYRSPSMPENLNRPR). Preliminary experiments were performed to determine optimal assay conditions for MARK1 and MARK1T208E. All kinase reactions were carried out in the linear phase of the reaction progress curve (initial rate). Kinase assays were performed in 30 µL in 1.5 mL tubes for 15–30 min at 30°C in kinase buffer (30 mM HEPES-KOH at pH 7.7, 10 mM MgCl2, 0,02 µg/mL BSA, 1 mM DTT). Control proteins or DDX proteins were incubated in MARK1 and the peptide substrate. The DDX3X QM mutant used was DDX3X132–605(K255A/R262A/R263A/R264A/K452A) (Fatti et al. 2023). Assay components were premixed, and reactions were started by the addition of 50–100 µM ATP containing 1 µCi of radiolabeled 32P-γATP and stopped by pipetting 10 µL onto p81 phosphocellulose filter paper (St. Vincent's Institute). P81 filter paper was washed four times in 0.5% ortho-phosphoric acid and dried, and radioactivity was detected by Cherenkov radiation in a scintillation counter (Packard Tri-Carb 2100 TR). Kinase-specific activity was expressed as nmol of phosphate transferred from ATP to peptide substrate per minute per microgram of kinase (nmol/min/µg).
For determination of Km[S], Km[ATP], KI, and Vmax, the reaction velocity data were fitted using nonlinear regression to the Michaelis–Menten equation or to the substrate inhibition
equation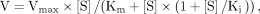 where v is the reaction rate, Vmax is the maximal reaction rate, Km is the Michaelis–Menten constant, [S] is the substrate concentration, and Ki is the dissociation constant of the inhibitory enzyme–substrate (SES) complex.
where v is the reaction rate, Vmax is the maximal reaction rate, Km is the Michaelis–Menten constant, [S] is the substrate concentration, and Ki is the dissociation constant of the inhibitory enzyme–substrate (SES) complex.
For EC50 and Hill coefficient determination, data were fitted to the corresponding dose-response equation. All data were analyzed with GraphPad Prism 7.0 (GraphPad software).
Data access
The ProtoArray data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE150859. The mass spectrometry proteomics data generated in this study have been submitted to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.proteomexchange.org) with the data set identifier PXD043289.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank C. Reinhard and U. Fenger for expert technical assistance, J. Oakhill (University of Melbourne) for advice and providing P81 paper, and H. Lee for graphical illustration support. Funding was from the German Research Council (DFG) via the Collaborative Research Center 1324 (TP B1 to C.N., TP B7 to I.S., TP Z4 to J.K and D.P.). E.N. was supported by the Ministry of Science, Research, and Arts, Baden Württemberg.
Author contributions: A.H., E.F., and E.N. contributed equally to this work. A.H. carried out DDX/DHX binding experiments. E.F. and T.Z. analyzed MARK1 kinetics. E.N., O.K., and A.G. conducted bioinformatic analysis of mass spectrometry data. D.P. and J.K. conducted proteomic analysis. S.P.A. conducted ProtoArray binding assays. C.-M.C. coordinated kinome activity screening. E.K. conducted bioinformatic analysis of the ProtoArray and kinome activity assay. A.Š. purified recombinant proteins for kinetic analyses. I.S. supervised A.Š. All authors analyzed and discussed the data. C.N. conceived and coordinated the study and wrote the paper with contribution from A.H.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278264.123.
- Received July 10, 2023.
- Accepted June 12, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








