
Abstract
Eusocial Hymenoptera have the highest recombination rates among all multicellular animals studied so far, but it is unclear why this is and how this affects the biology of individual species. A high-resolution linkage map for the ant Cardiocondyla obscurior corroborates genome-wide high recombination rates reported for ants (8.1 cM/Mb). However, recombination is locally suppressed in regions that are enriched with TEs, that have strong haplotype divergence, or that show signatures of epistatic selection in C. obscurior. The results do not support the hypotheses that high recombination rates are linked to phenotypic plasticity or to modulating selection efficiency. Instead, genetic diversity and the frequency of structural variants correlate positively with local recombination rates, potentially compensating for the low levels of genetic variation expected in haplodiploid social Hymenoptera with low effective population size. Ultimately, the data show that recombination contributes to within-population polymorphism and to the divergence of the lineages within C. obscurior.
Meiotic recombination is a key feature of sexually reproducing organisms (Maynard Smith 1978; Bell 1982; Crow 1994), in which homologous chromosomes form a bivalent structure through physical linkage involving one to two crossovers per chromosome (White 1973; Kleckner 1996; Roeder 1997). These crossovers are essential for chromosome stability and proper segregation and can promote genotypic variation, thereby enhancing the adaptive potential of organisms (Kleckner 1996; Roeder 1997; Jones and Franklin 2006; Ritz et al. 2017). Recombination rates vary between and within species and are strongly influenced by genome architecture (Lynn et al. 2004; Stapley et al. 2017). Recombination itself can drive genome evolution, both through its mutagenic effects (Langley et al. 1988; Montgomery et al. 1991; Webster and Hurst 2012; Arbeithuber et al. 2015; Halldorsson et al. 2019) and by counteracting the diversity-reducing effects of linked selection (Cutter and Payseur 2013; Ellegren and Galtier 2016).
Genetic linkage maps across several Apocrita species (ants, wasps, and bees) (Hunt and Page 1995; Sirviö et al. 2006, 2011a,b; Wilfert et al. 2006; Shi et al. 2013; Liu et al. 2015; Rueppell et al. 2016; Waiker et al. 2021) indicate that social Hymenoptera have particularly high recombination rates compared with other animals studied so far (Wilfert et al. 2007; Stapley et al. 2017). Studies in Nasonia, a haplodiploid solitary parasitoid wasp, revealed low recombination rates (∼1.5–2.5 cM/Mb) (Gadau et al. 1999; Niehuis et al. 2010), indicating that the haplodiploid system alone does not explain the high recombination rates observed in social Hymenoptera. The prevailing hypotheses suggesting that the extreme recombination rates in social insects are related to the complex needs of social life have found only ambiguous support so far and are exclusively from studies on the honey bee Apis mellifera.
First, by increasing the efficiency of natural selection (Felsenstein 1974; Felsenstein and Yokoyama 1976; Comeron et al. 2008), high recombination rates could promote the independent evolution of caste-specific genes (Kent et al. 2012; Kent and Zayed 2013). However, studies have shown conflicting results regarding the association between recombination rates and selection efficiency (Kent et al. 2012; Jones et al. 2019). Second, by increasing colony-level genotypic diversity, these elevated rates could facilitate social life (Oldroyd and Fewell 2007; Liu et al. 2015; Kozomara et al. 2019). For example, elevated recombination rates around genes with a caste-biased expression pattern suggest a link with division of labor (Kent et al. 2012; Liu et al. 2015; Wallberg et al. 2015). However, this pattern was also observed for orthologous sequences in a solitary bee, suggesting that the high recombination rates in the vicinity of these genes preceded the evolution of sociality in the bee lineage (Jones et al. 2019). Lastly, increasing diversity could also help resist the high pathogen pressure of social life (Kraus and Page 1998; Schmid-Hempel 1998; Hughes and Boomsma 2004; Liu et al. 2015), but immune-related genes did not coincide with regions of high recombination rates (Liu et al. 2015). In summary, there is no consensus regarding possible causes and consequences of high recombination in honey bees, let alone in other social Hymenoptera. In ants, two studies conducted in Acromyrmex echinatior (Sirviö et al. 2006) and Pogonomyrmex rugosus (Sirviö et al. 2011b) using a limited number of markers (145–215 markers) corroborated elevated recombination rates in social insects. However, these studies did not explore the genomic features associated with the genome-wide variation in recombination rates.
Here, we use genetic linkage mapping, a chromosome-level genome assembly, and extensive population-genomic data of the model species Cardiocondyla obscurior (Oettler 2021) for first analyses of the causes and consequences of recombination rate variation in an ant. In particular, we explore putative factors associated with local recombination rate variation in the C. obscurior genome. Furthermore, using published gene expression data of larvae of queens, workers, and winged and wingless males (Schrader et al. 2015, 2017) and the ratio between nonsynonymous and synonymous substitutions (dN/dS), we critically address the hypotheses linking high recombination rates to the evolution or elaboration of sociality in Hymenoptera.
Results
Initial linkage mapping and genome assembly refinement and annotation
To improve the current genome assembly (Cobs2.1) to chromosome resolution, we produced an initial ordering of 57,557 markers (out of 342,508 raw biallelic SNPs) into 29 LGs (Supplemental Fig. S1) using whole-genome data from 100 F2 offspring of a single F1 mother obtained from a cross between a male and a queen belonging to two Brazilian populations of C. obscurior. These populations were specifically chosen to maximize the number of heterozygous markers and minimize runs of homozygosity in the experimental cross. Although some small LGs may be physically connected to each other or to larger LGs, they were not combined owing to large genetic distances (∼50 cM), indicating that they segregate independently from other LGs.
The new genome (Cobs3.1) spans 193.05 Mb and is an improvement over the previous version (Cobs2.1) with ∼96% (184.4 Mb) of the physical assembly placed into pseudomolecules and a scaffold N50 of 7.38 Mb compared with 6.29 Mb of Cobs2.1 (Supplemental Table S1). Using the Liftoff tool (v.1.6.3) (Shumate and Salzberg 2021), we converted the coordinates of 20,944 genes (36,599 transcripts) annotated in Cobs2.1 to Cobs3.1.
Transposable elements (TEs) exhibit a bimodal distribution in the genome of C. obscurior with regions of high TE content (“TE islands”) in a genomic background of low TE content (low-density regions [LDRs]) (Supplemental Fig. S2A), as described previously (Schrader et al. 2014; Errbii et al. 2021). A total of 30 TE islands are located at the center (e.g., LGs 1, 2, 3, and 5) and the ends (e.g., LGs 4, 8, and 9) of LGs. We also observed secondary TE accumulations on several LGs (e.g., 1, 5, and 10).
Tandem repeat (TR) sequences are commonly associated with centromeres across various species, including ants (Melters et al. 2013; Huang et al. 2016). Upon annotating TRs in C. obscurior, we observed an overlap of these sequences with TE islands (Supplemental Fig. S2B). Candidate centromeric repeats from five different ant species (Melters et al. 2013; Huang et al. 2016) were not enriched in TE islands, suggesting a distinct repertoire of repeats in centromeres of C. obscurior, consistent with prior findings of rapid divergence among centromeric TRs in ants (Huang et al. 2016). Together, these results suggests that TE islands may serve as potential centromeres for the chromosomes of C. obscurior.
The recombination landscape of C. obscurior
To estimate recombination rates, we used the updated assembly (Cobs3.1) for a second round of linkage mapping with 57,309 high-quality markers (out of 342,616 raw biallelic SNPs) covering 84% (162.9 Mb) of the physical assembly and revealing 1061 effectively recombining positions. It is important to note that the number of recombining positions is independent of marker density and is instead determined by the occurrences of crossover events in the experiment. The final map (1315.4 cM in 29 LGs) (Supplemental Fig. S3) showed a positive correlation between genetic and physical positions of markers, indicating a high-quality map (Supplemental Fig. S4, correlation coefficients are given in the figure). Based on the proportion of the physical assembly covered by markers, the average recombination rate in C. obscurior is 8.1 cM/Mb. Although higher than observed in other nonsocial insects and animal species (Supplemental Table S2), this estimate falls within the range of other ants (Sirviö et al. 2006, 2011b) and is lower than those of the honey bee (Beye et al. 2006; Shi et al. 2013; Liu et al. 2015; Rueppell et al. 2016).
Recombination rate varied considerably among the 29 LGs (Table 1) with LGs <5 Mb having a higher recombination rate (median = 14.2 cM/Mb, 95% CI [8.52, 18.06]) compared with LGs >10 Mb (median = 7.5 cM/Mb, 95% CI [3.65, 10.29]) and intermediate LGs (≥5 Mb and ≤10 Mb; median = 7.5 cM/Mb, 95% CI [4.60, 9.55]). Physical and genetic distances correlated positively (Spearman's ρ = 0.57, P = 0.0014) (Supplemental Fig. S5A), whereas average recombination rate per LG showed a significant negative correlation (Spearman's ρ = −0.39, P = 0.039) with physical lengths of LGs (Supplemental Fig. S5B).
Physical length (Mb), map length (cM), recombination rate (cM/Mb), and number of crossovers per LG in C. obscurior
| Linkage group | Physical length (Mb) | Map length (cM) | Recombination rate (cM/Mb) | Crossover events |
|---|---|---|---|---|
| LG1 | 13.82 (13.28) | 91.57 | 6.90 | 89 |
| LG2 | 13.23 (12.87) | 104.21 | 8.09 | 96 |
| LG3 | 11.56 (11.10) | 97.77 | 8.81 | 96 |
| LG4 | 11.12 (10.29) | 41.89 | 4.07 | 41 |
| LG5 | 9.85 (8.94) | 93.37 | 10.44 | 94 |
| LG6 | 9.25 (7.55) | 2.00 | 0.27 | 1 |
| LG7 | 9.20 (8.34) | 111.04 | 13.31 | 109 |
| LG8 | 7.92 (7.88) | 60.82 | 7.72 | 56 |
| LG9 | 7.39 (7.25) | 50.34 | 6.95 | 49 |
| LG10 | 7.38 (7.09) | 45.62 | 6.43 | 44 |
| LG11 | 7.36 (6.10) | 42.17 | 6.91 | 41 |
| LG12 | 6.60 (5.37) | 5.99 | 1.12 | 7 |
| LG13 | 5.99 (5.02) | 37.76 | 7.52 | 38 |
| LG14 | 5.90 (5.70) | 46.27 | 8.12 | 46 |
| LG15 | 5.63 (5.04) | 50.03 | 9.94 | 46 |
| LG16 | 5.48 (4.72) | 45.43 | 9.62 | 45 |
| LG17 | 5.21 (5.07) | 5.01 | 0.99 | 4 |
| LG18 | 5.20 (4.90) | 8.02 | 1.64 | 8 |
| LG19 | 5.03 (2.97) | 44.99 | 15.13 | 44 |
| LG20 | 4.86 (4.40) | 49.62 | 11.28 | 47 |
| LG21 | 4.44 (4.32) | 54.65 | 12.64 | 54 |
| LG22 | 4.34 (2.76) | 33.62 | 12.18 | 33 |
| LG23 | 3.81 (3.12) | 49.24 | 15.78 | 47 |
| LG24 | 3.70 (3.01) | 49.21 | 16.34 | 47 |
| LG25 | 3.14 (2.66) | 45.56 | 17.14 | 44 |
| LG26 | 2.30 (0.33) | 1.00 | 3.06 | 1 |
| LG27 | 1.93 (1.70) | 39.65 | 23.26 | 36 |
| LG28 | 1.68 (0.72) | 1.50 | 2.08 | 1 |
| LG29 | 1.08 (0.36) | 7.00 | 19.19 | 7 |
| Total | 184.4 (162.87) | 1315.33 | 8.08 | 1271 |
[i] Numbers in brackets indicate distances (Mb) between the first and last marker on each LG, used to calculate average recombination rate.
Eight LGs showed very little or no recombination because of a reduced number of crossovers (LGs 4, 6, 12, 17, 18, 26, 28, 29) (Table 1). Although the number of markers varied between LGs (Supplemental Fig. S6), there was no correlation between average recombination rates and marker count (Spearman's ρ = −0.087, P = 0.65) per LG (Supplemental Fig. S5C). In the following, we investigated potential factors contributing to the variation in the number of crossovers and, consequently, recombination rate in C. obscurior.
Recombination rate distribution and genome architecture in C. obscurior
To investigate the genomic determinants of recombination in C. obscurior, we estimated the local recombination rate in 250 kb nonoverlapping windows and analyzed these with respect to various sequence parameters (Fig. 1A; Supplemental Figs. S7, S8). Because crossovers can only be detected between two markers, recombination rate estimates were restricted to regions between the first and last markers on each chromosome, excluding the chromosome ends.
Recombination rate variation and genome architecture in C. obscurior. (A) Circos plot showing the following inward: (SV) the location of identified structural variants (dark green), (RR) recombination rates across the genome (cM/Mb) with introgressions depicted with purple bars, (TE) TE content (“LDRs”, blue; “TE islands”, orange) with the location of TE islands shown as black bars, (Ex) exon content, and (π) the log10-transformed nucleotide diversity in two natural populations for C. obscurior from Itabuna (gold) and Una (maroon). C. obscurior queen with brood (photo: L. Schrader). (B) Recombination rates between TE-poor regions (“LDRs” in blue) and TE-rich regions (“TE islands” in orange). (C) Recombination rates in introgressed regions (“LDRs introgr.” In purple) compared with the remainder of LDRs (“LDRs other” in blue). (D) Levels of nucleotide diversity (π) in nonrecombining (“LDRs nonrec.”) compared with recombining regions LDRs (“LDRs rec.”) in two populations of C. obscurior from Itabuna and Una. (****) P < 0.0001, (**) P < 0.01.
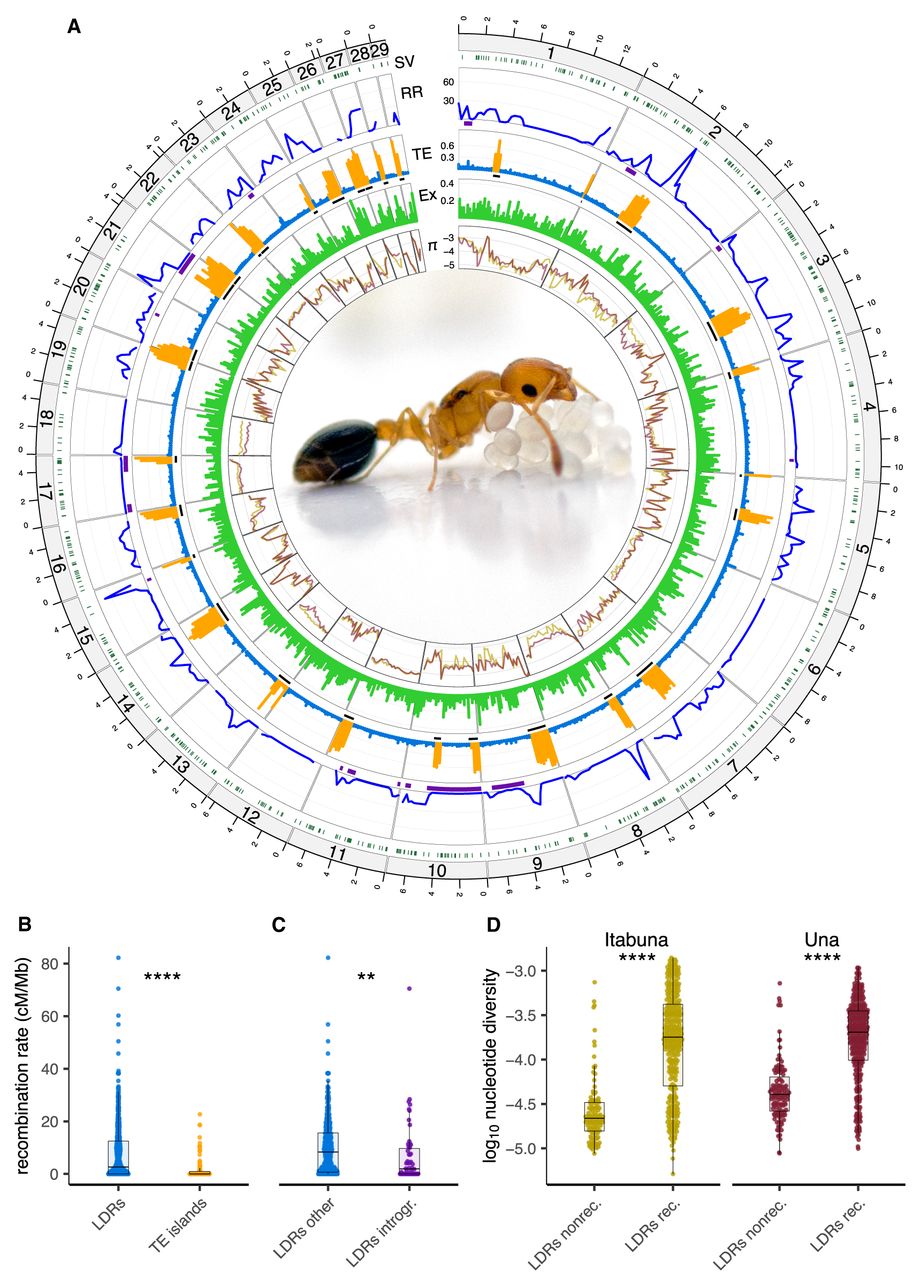
The recombination rate in C. obscurior ranged from 0 to 82.2 cM/Mb, with ∼38% (∼70 Mb) of the analyzed sequence showing minimal to no recombination (e.g., on LGs 6, 12, 17, and 18) (Fig. 1A; Supplemental Fig. S7A). Note that the recombination rate estimates were not affected by marker density. Although a correlation was observed between marker density and recombination rate in 250 kb windows (Spearman's ρ = 0.075, P = 0.049) (Supplemental Fig. S9A), this correlation disappeared when considering only recombining windows (Spearman's ρ = 0.045, P = 0.37) (Supplemental Fig. S9B).
Markers were unevenly distributed across the LGs, with 23 out of the 29 LGs having median intermarker distances below 0.64 kb (range: 0.12 to 256.97 kb) (Supplemental Table S3). Three LGs 6, 12, and 18 showed minimal recombination and displayed particularly large intermarker distances (medians = 210.27, 37.84, and 256.97 kb, respectively), 90% of which were <750 kb (Supplemental Table S3). Because of chiasma interference (Sturtevant 1915; Muller 1916), in which the occurrence of one crossover disrupts the formation of another nearby, these intervals should still be sufficient to capture crossovers. Unless exceptionally rare double crossovers occur within these short intervals, we expect an average of one crossover every 2.7 Mb (Liu et al. 2015) to 7.14 Mb (Sirviö et al. 2011b), according to studies in the honey bee and harvester ants, respectively.
Recombination rates are significantly reduced in TE islands (median = 0 cM/Mb), which are gene-poor, compared with LDRs (median = 3.11 cM/Mb) in which gene density is higher (Wilcoxon test: W = 43644, P = 5.25 × 10−13) (Fig. 1B). Note that the decrease in recombination rate within TE islands is not because of a lack of markers (Supplemental Fig. S6; Supplemental Table S4). LDRs on their own show no significant correlation between recombination rate and TE content (Spearman's ρ = 0.016, P = 0.74) or exon content (Spearman's ρ = −4 × 10−3, P = 0.93) (Supplemental Fig. S8).
Beside TE islands, several other regions in LDRs also showed little or no recombination (Fig. 1A). Some of these nonrecombining regions are characterized by high sequence divergence between grandparental haplotypes (gp-dXY) (Supplemental Fig. S7B). In these regions, one of the two grandparental haplotypes showed high sequence similarity to Asian and European populations of C. obscurior (Supplemental Fig. S7C), indicating introgression from the Old-World lineage (Errbii et al. 2021). Based on a cutoff of threefold mean gp-dXY (∼0.001), these introgressions occupy a large fraction (>40%) of LGs 9, 10, 17, and 21 and smaller regions on LGs 1 and 2. After accounting for differences in the number of introgressed (53) and nonintrogressed (330) windows, we found that regions with high gp-dXY recombine significantly less (median = 1.8 cM/Mb) than the rest of the genome (median = 8.52 cM/Mb) (Wilcoxon test: W = 1043.024, P = 0.046) (Fig. 1C). Several inversions overlap with these introgressions (on LGs 10, 17, and 21) (Supplemental Table S5). Together, this indicates that strong sequence divergence among homologous chromosomes impairs recombination in C. obscurior, at least in part caused by polymorphic inversions.
Other nonrecombining regions on LGs 1, 4, 6, 8, and 12 showed no signs of introgression and contained no large-scale inversions that could explain local suppression of recombination (Fig. 1A; Supplemental Fig. S7A,B; Supplemental Table S5). Considering that meiotic recombination disrupts the linkage between segregating loci, we thus hypothesized that the absence of recombination in these regions could result from epistatic selection acting to reduce recombination in regions harboring coadapted loci (Turner 1967; Charlesworth 2016). To explore this possibility, we screened these regions for signs of reduced genetic variation and Tajima's D, measures commonly used in population genetics to infer signatures of selection (Nielsen 2005). The nonrecombining regions on LGs 1, 4, 6, 8, and 12 showed significantly reduced levels of nucleotide diversity in natural populations (Itabunamedian = 2.18 × 10−5; Unamedian = 4.04 × 10−5) compared with the remainder of the LDRs (excluding introgressions; Itabunamedian = 1.79 × 10−4; Unamedian = 2.03 × 10−4; Wilcoxon test; Itabuna: W = 5559.5, P < 2.2 × 10−16; Una: W = 6289.5, P < 2.2 × 10−16) (Fig. 1D). Similarly, Tajima's D was also significantly reduced in these nonrecombining regions (Itabunamedian = −1.86; Unamedian = −2.1) compared with the remainder of the LDRs (excluding introgressions; Itabunamedian = −0.99; Unamedian = −1.37; Wilcoxon test; Itabuna: W = 9547, P = 1.32 × 10−15; Una: W = 9860, P = 1.07 × 10−14) (Supplemental Figs. S7D, S10).
We further applied a generalized linear model with recombination as the response variable and found that TE content, sequence divergence, and epistasis were all significant explanatory variables (Supplemental Table S6).
Finally, there was no correlation between recombination rate and GC content (overall: Spearman's ρ = 0.044, P = 0.35; intergenic: Spearman's ρ = 0.025, P = 0.6) (Supplemental Figs. S7E, S8) in C. obscurior.
Meiotic recombination contributes to genetic variation in natural populations and promotes intraspecific divergence
In support of a substantial effect of recombination on patterns of genetic variation, we found a positive correlation of recombination rates with measures of nucleotide diversity (Itabuna: Spearman's ρ = 0.28, P = 2.1 × 10−9; Una: Spearman's ρ = 0.25, P = 9.9 × 10−8) and Tajima's D (Itabuna: Spearman's ρ = 0.21, P = 7.1 × 10−6; Una: Spearman's ρ = 0.3, P = 5.6 × 10−11) calculated in 250 kb windows in LDRs (Fig. 2).
Relationship between recombination rates and measures of diversity in populations of C. obscurior. (Left) Correlation between recombination rates and nucleotide diversity and Tajima's D. (Middle) Correlation between recombination rates and nucleotide diversity at synonymous (πS) and nonsynonymous (πNS) sites. (Right) Correlation between recombination rates and levels of genetic differentiation (FST) and absolute divergence (dXY) between lineages of C. obscurior. The x-axis represents binned recombination rates, with the upper limit of each bin indicated and “TEI” referring to TE islands (orange boxplot). (ρ) Spearman's rank correlation coefficient, with significance shown by asterisks: (****) P < 0.0001, (***) P < 0.001, (**) P < 0.01. All correlations were performed using the raw data. Lines and shaded areas are linear regressions and 95% confidence intervals.
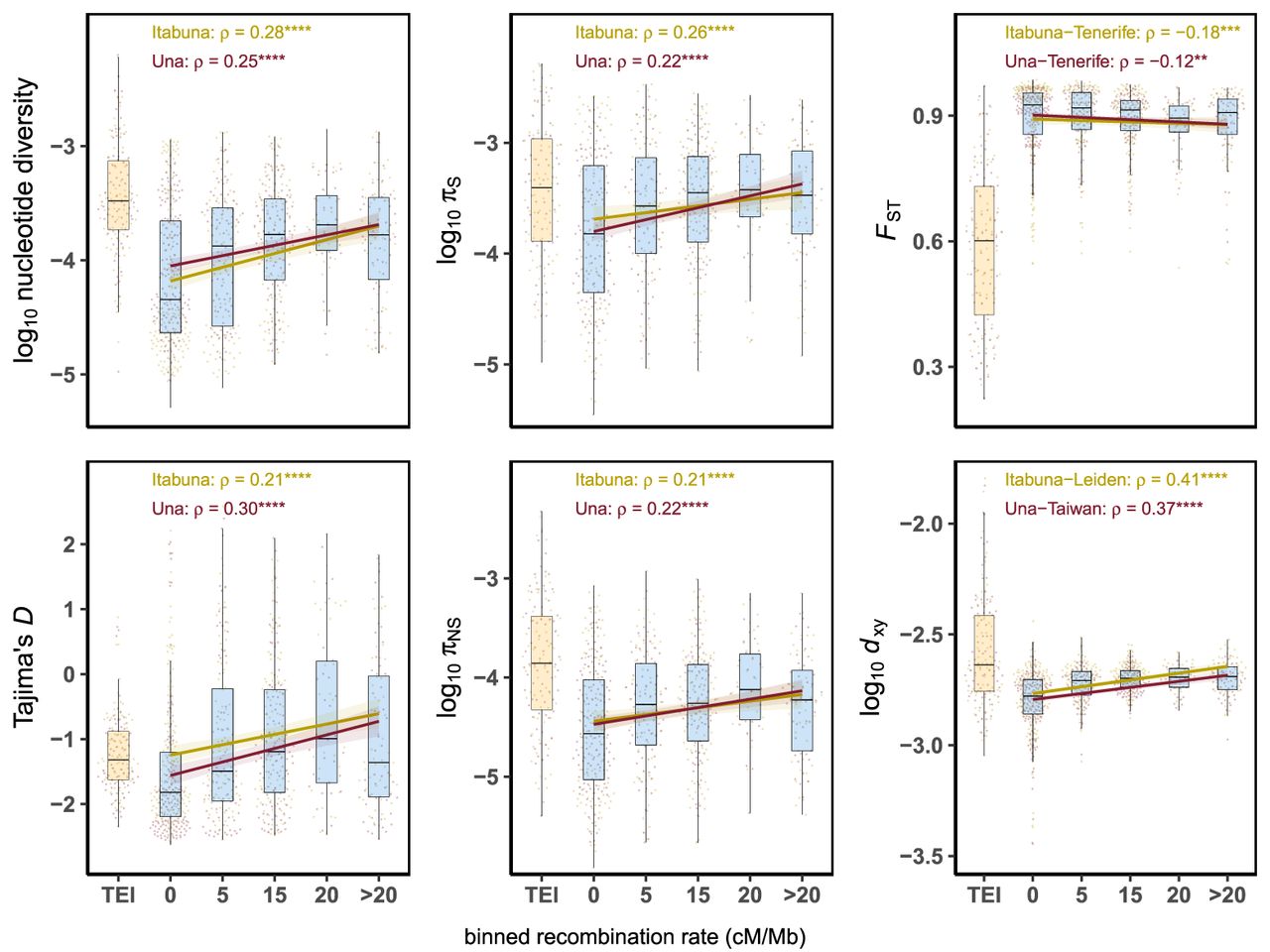
Recombination rates were also positively correlated with levels of diversity at both nonsynonymous (πNS; Itabuna: Spearman's ρ = 0.21, P = 1 × 10−5; Una: Spearman's ρ = 0.22, P = 3.7 × 10−6) and synonymous sites (πS; Itabuna: Spearman's ρ = 0.26, P = 3.9 × 10−8; Una: Spearman's ρ = 0.22, P = 2.3 × 10−6) (Fig. 2), but not with their ratio (πNS/πS; Itabuna: Spearman's ρ = 0.0062, P = 0.91; Una: Spearman's ρ = 0.0018, P = 0.97) (Supplemental Fig. S11), indicating that meiotic recombination does not correlate with positive or purifying selection.
Consistent with models of linked selection in which diversity is reduced in regions of low recombination (Cutter and Payseur 2013; Burri 2017), there was a negative correlation between FST and recombination in two different population pairs (FST [Itabuna-Tenerife]: Spearman's ρ = −0.18, P = 1.7 × 10−4; FST [Una-Tenerife]: Spearman's ρ = −0.12, P = 9.9 × 10−3) (Fig. 2). However, there was also a significant positive association between recombination and dXY in two additional population pairs (dXY [Itabuna-Leiden]: Spearman's ρ = 0.41, P < 2.2 × 10−16; dXY [Una-Taiwan]: Spearman's ρ = 0.37, P = 8.6 × 10−16) (Fig. 2). Together, this indicates that recombination contributes to within-population polymorphism and to the divergence of C. obscurior lineages. Future studies should explore whether the same patterns persist when using recombination rates estimated for a population of the Old World lineage.
Recombination is associated with small and large genome rearrangements
Ectopic recombination (i.e., crossovers between nonallelic homologous loci), particularly in repeat-rich regions, has been shown to produce structural variants (SVs) (Liu et al. 2012; Savocco and Piazza 2021). To investigate to what extent the high recombination rates in C. obscurior are correlated with SVs, we used whole-genome sequencing data generated previously for 16 individuals from five populations (Errbii et al. 2021).
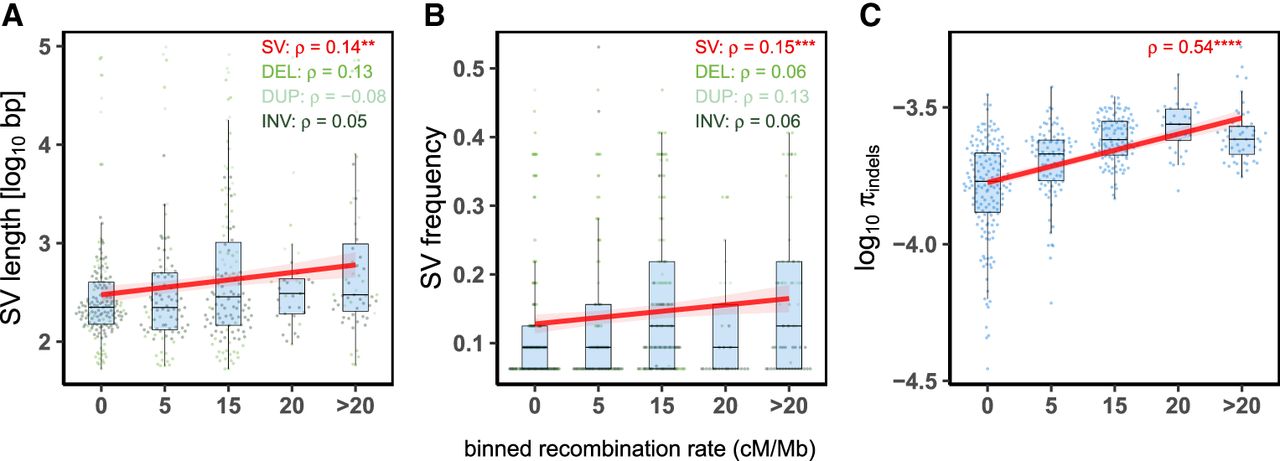
Analysis of 481 SVs (129 deletions, 54 duplications, and 298 inversions) showed a positive correlation between recombination rates and SV frequencies and their lengths in LDRs (SVs length: Spearman's ρ = 0.14; P = 0.0025; SVs frequency: Spearman's ρ = 0.15, P = 8.1 × 10−4) (Fig. 3A,B). When analyzed separately, none of the individual SV types correlate significantly with recombination (Supplemental Fig. S12). Additionally, genetic diversity at 95,841 short insertions and deletions (indels) showed a strong and significant positive association with recombination (Spearman's ρ = 0.54, P < 2.2 × 10−16) (Fig. 3C). A similar pattern was observed when genic and intergenic indels were analyzed separately (Supplemental Fig. S13).
Association between recombination rates and genome dynamics in C. obscurior. Correlations between recombination rates and length (A) and frequency (B) of SVs and indel polymorphism (C) in 16 individual workers. The x-axis represents binned recombination rates, with the upper limit of each bin indicated. (ρ) Spearman's rank correlation coefficient is indicated for all structural variants (SVs) and separately for deletions (DEL), duplications (DUP), and inversions (INV), with significance shown by asterisks: (****) P < 0.0001, (***) P < 0.001, (**) P < 0.01. All correlations were performed using the raw data. Red lines and shaded areas are linear regressions and 95% confidence intervals.
No association between recombination and evolutionary rates
Meiotic recombination is expected to enhance the efficacy of selection by uncoupling linkage between targets of selection and mitigating the impact of Hill–Robertson interference (HRI) (Hill and Robertson 1966; Betancourt et al. 2009; Campos et al. 2014). To examine this relationship, we used omega (ω) as a proxy, that is, the ratio of nonsynonymous (dN) to synonymous (dS) substitutions (Webster and Hurst 2012). In regions of low recombination, increased genetic interference should compromise both positive selection for advantageous variants and purifying selection against deleterious ones (Betancourt et al. 2009). Given that the majority of nonsynonymous mutations are deleterious, a negative correlation between recombination rate and ω is expected (Haddrill et al. 2007; Betancourt et al. 2009; Bolívar et al. 2016). However, we found no significant correlation between recombination and dN (Spearman's ρ = 0.008, P = 0.61), dS (Spearman's ρ = −0.015, P = 0.35), or ω (Spearman's ρ = 0.019, P = 0.24) for 3916 single-copy ortholog genes in LDRs (Supplemental Fig. S14).
No association between recombination and gene expression bias
In honey bees, genes with caste-biased expression in brains were enriched in regions of high recombination, suggesting that caste-specific transcriptional plasticity is facilitated by the effects of recombination (Kent et al. 2012; Liu et al. 2015; Wallberg et al. 2015; Jones et al. 2019). In C. obscurior genes showing caste-biased brain expression are as yet unknown. However, plastic gene expression has been studied in this species in the context of caste differentiation (Schrader et al. 2015, 2017). To assess a potential association between recombination and developmental gene expression plasticity, we reanalyzed this RNA-seq data set of third instar larvae of different castes and sexes (Supplemental Fig. S15). Based on 6807 expressed genes, we did not find a significant association between recombination and gene expression plasticity over multiple contrasts (Spearman's ρ = 0.014, P = 0.26) (Supplemental Fig. S16).
Functional analysis of genes in regions of high recombination
To explore whether regions of high recombination rates in C. obscurior show signatures of functional specialization, we conducted Gene Ontology (GO) enrichment analyses on genes contained in the 10% most frequently recombining regions in the genome. These regions showing recombination rates >22.3 cM/Mb and containing 1541 genes were significantly enriched for fatty acid elongase and reductase genes (Table 2; Supplemental Table S7). Although this implies a potential association between recombination and cuticular hydrocarbon (CHC) biosynthesis, it is important to acknowledge the limitations of these findings, particularly the failure to determine the specific genes directly affected by recombination.
GO terms enriched in regions of high recombination rates in C. obscurior genome (top 10%; recombination rate > 22.3 cM/Mb)
| GO.ID | Term | Annotated | Significant | Expected | P value |
|---|---|---|---|---|---|
| GO:0009922 | Fatty acid elongase activity | 15 | 10 | 1.22 | 4.6 × 10−6 |
| GO:0080019 | Fatty-acyl-CoA reductase (alcohol-forming) activity | 19 | 9 | 1.54 | 6.17 × 10−4 |
[i] P-values are false-discovery rate (FDR)-corrected.
Discussion
Linkage mapping and recombination rate for C. obscurior
The high recombination rates reported for the social Hymenoptera are not easily explained and have not been thoroughly investigated in ants. To start filling this gap, we produced a high-resolution genetic map. At least 26 of the 29 LGs represent full chromosomes, whereas the remaining small LGs are most likely unlinked fragments of larger chromosomes or extra ones in the mapping population. The updated chromosome-level genome of C. obscurior (Cobs3.1) confirms the architecture of the previous assembly-based versions and that it is the smallest ant genome described so far (Schrader et al. 2014; Errbii et al. 2021).
Average recombination rate in C. obscurior is 8.1 cM/Mb, which is within rates reported for two ants (Sirviö et al. 2006, 2011b) and is lower compared to several honey bee species (Hunt and Page 1995; Beye et al. 2006; Liu et al. 2015; Rueppell et al. 2016). This value is likely an underestimate because of two reasons. First, we excluded unlikely double-crossover events as false positives. Although such double crossovers are highly improbable owing to chiasma interference (Sturtevant 1915; Muller 1916), they may occur at low frequencies. Second, recombination in our experiment was suppressed in a large fraction of the genome (∼38%), partly owing to introgressed haplotypes in the grandparental cross. These regions are likely to recombine freely in the absence of introgression, which should increase average recombination rates quite significantly.
Two factors can affect the resolution of the map. First is the number of markers and their distribution across the genome. Despite the clustering of nonrecombining markers, which reduced the effective number of markers available for recombination rate estimation (from 57,309 markers to 1061 clusters), the map maintained its density. The average intermarker distance in this study stands at 1.24 cM and is relatively lower than the other two available ant maps (A. echinatior: 145 markers or an intermarker distance of 14.32 cM [Sirviö et al. 2006] and P. rugosus: 215 markers or an intermarker distance of 16.55 cM [Sirviö et al. 2011b]). A second factor that affects map resolution is the size of the mapping population. Here, we used data from 100 F2 individuals from a single queen, comparable to previous maps in ants (Sirviö et al. 2006, 2011b) and bees (Liu et al. 2015, 2017; Rueppell et al. 2016) and generally considered sufficient for recombination rate estimation and the construction of a well-resolved linkage map.
Recombination rates varied among the 29 LGs, showing a negative correlation with chromosome size, a feature shared among many species (Hunt and Page 1995; Jensen-Seaman et al. 2004; Pessia et al. 2012; Haenel et al. 2018). Several LGs in our map were ∼50 cM long, aligning with the expectation under the one obligate crossover rule. This raises the question whether the high recombination rates observed in eusocial species can be attributed to an increase in the number of chromosomes with obligate crossovers. However, chromosome numbers of eusocial and solitary species of Hymenoptera do not differ significantly (Ross et al. 2015). Furthermore, these high rates can also not be linked to genome size reductions in social Hymenoptera, as smaller genomes do not generally exhibit higher recombination rates in animals (Stapley et al. 2017). Thus, neither chromosome number nor genome size is sufficient to explain the high recombination rates of social Hymenoptera.
The factors affecting recombination rates in C. obscurior
Recombination rates, calculated in 250 kb windows, varied considerably (0 to 82.2 cM/Mb), resulting in a complex recombinational landscape. Such regions of extremely high recombination rates have also been observed in the honey bee (Beye et al. 2006; Liu et al. 2015). Consistent with previous observations indicating higher recombination rates in gene-dense regions and open chromatin (Tiley and Burleigh 2015; Marand et al. 2017), gene-rich LDRs displayed higher recombination rates compared with gene-poor TE islands (Schrader et al. 2014; Errbii et al. 2021). This supports the interpretation that TE islands are (peri-)centromeric regions, where recombination is usually suppressed (Beadle 1932; Lambie and Roeder 1986; Talbert and Henikoff 2010). TE insertions can accumulate in regions with reduced recombination because ectopic recombination as a mechanism to remove TEs is not available (Dolgin and Charlesworth 2008; Kent et al. 2017). In accordance, centromere structure and location in heterochromatin can locally suppress recombination (Talbert and Henikoff 2010), allowing accumulations of TEs in these regions to evolve secondarily. Future cytological examinations are necessary to confirm that these regions are indeed centromeres.
Sequence divergence resulting from introgression between distant populations of C. obscurior (Errbii et al. 2021) also inhibit recombination, in line with studies in other taxa (Liharska et al. 1996; Opperman et al. 2004; Li et al. 2006; Delame et al. 2019). Particularly, inversions that occur in some of these introgressions can significantly impact recombination (Noor et al. 2001; Rieseberg 2001; Stevison et al. 2011) by disrupting the pairing of homologous chromosomes and synapsis formation during meiosis (Chakraborty et al. 2019).
Recombination was also suppressed in regions that showed low TE content and no signs of introgression, as well as significantly reduced levels of genetic diversity in two populations. Such patterns can emerge under epistatic selection (Otto and Lenormand 2002; Hadany and Comeron 2008; Takahasi 2009; Fuller et al. 2020; Navarro-Domínguez et al. 2022), which acts to reinforce linkage between coadapted and/or essential loci through recruitment of recombination modifiers (Otto and Feldman 1997). These modifiers become associated with high-fitness allele combinations favored by selection, rendering any alternative allelic combination deleterious and making recombinant offspring less viable (Altenberg and Feldman 1987; Barton 1995; Singhal et al. 2019). Future studies examining the functional role of these regions are necessary to resolve whether they indeed evolved because of epistatic selection of coevolved loci.
Associations between GC content and recombination were described in the honey bee (Wallberg et al. 2015; Jones et al. 2019) and other organisms (Meunier and Duret 2004; Pessia et al. 2012), most likely as a result of GC-biased gene conversion (Webster and Hurst 2012). However, our investigation in C. obscurior, consistent with recent findings in butterflies (Torres et al. 2023), revealed no significant correlation between recombination rates and GC content. This mirrors patterns seen in selfing plants like Arabidopsis thaliana, in which it has been suggested to result from increased inbreeding and a low Ne (Marais et al. 2004).
No support for hypotheses regarding the role of recombination in social evolution
It has been suggested that recombination contributes to diversity around genes involved with social life. By increasing the efficiency of natural selection (Felsenstein 1974; Comeron et al. 2008), high recombination rates may promote the evolution of caste-specific genes (Kent et al. 2012; Kent and Zayed 2013). Studies in Drosophila have suggested a link between recombination and the efficacy of selection (Betancourt and Presgraves 2002; Haddrill et al. 2007; Campos et al. 2014), whereas other reports found no compelling evidence for such an association (Webster and Hurst 2012), in line with data from humans (Bullaughey et al. 2008). Consistent with a recent study in honey bees (Jones et al. 2019), in C. obscurior we found no association between recombination rate and rates of dN/dS, used as a proxy for selection efficiency. Hence, our finding does not support the hypothesis that high recombination rates promoted the evolution of sociality by reducing HRI effects (Hill and Robertson 1966) and allowing the evolution of genes important for social life.
Alternative models suggested that elevated rates of recombination in social Hymenoptera may increase resistance to pathogens and facilitate behavioral and morphological differentiation by increasing genetic diversity at the colony level (Shykoff and Schmid-Hempel 1991; Hughes and Boomsma 2004; Wilfert et al. 2007; Liu et al. 2015; Wallberg et al. 2015; Jones et al. 2019). We found no enrichment of genes involved in immunity or behavioral plasticity in regions of high recombination. Additionally, there was no correlation between recombination rates and genes showing plastic expression in larvae of C. obscurior, indicating that high recombination rates are also not related to caste differentiation in this species.
GO term enrichment analyses revealed a significant enrichment of elongase and reductase genes in regions of high recombination rates. These genes are primarily known for their roles in regulating fatty acids' elongation and their conversion into fatty alcohols during the biosynthesis of CHCs (Holze et al. 2021), which play an important role in nestmate recognition in ants (Hefetz 2007; Sprenger and Menzel 2020), including C. obscurior (Drakula et al. 2023). Gene family expansions of elongases and reductases have been suggested to contribute to the evolution of CHC complexity (Finck et al. 2016; Hartke et al. 2019) and pheromone communication (Tupec et al. 2019) in ants. To what extent high recombination rates could contribute to the function or evolution of this key component of ant communication has so far not been explored. Although the direct impact of recombination on these genes or their surrounding regulatory sequences remains unclear, it is plausible that frequent recombination at these loci could facilitate the production of unique colony odors by increasing genotypic variation between colonies in genes involved in the CHC biosynthesis pathway (Holze et al. 2021). Future studies should investigate whether this pattern can be found in other ants and whether ectopic recombination might have facilitated the expansion of these gene families.
Recombination promotes genetic diversity of haplodiploid social insects with low Ne
In haplodiploids, Ne is expected to be three-fourths that of diploid species, theoretically reducing genetic variation and constraining adaptation to environmental variation (Wright 1931; Kimura and Crow 1964). In eusocial haplodiploids, Ne and, consequently, genetic variation are reduced further (Metcalf et al. 1975; Pamilo et al. 1978; Pamilo and Crozier 1997; Romiguier et al. 2014), as only a fraction of the individuals of a population reproduces (but for other potential determinants of Ne in Hymenoptera, see Weyna and Romiguier 2021). Studies in taxa beyond Hymenoptera, such as social shrimp (Chak et al. 2022) and termites (Roux et al. 2024), corroborate the association between eusociality and reduced Ne. So, how is genetic variation as the substrate for natural selection and species evolution maintained in eusocial haplodiploids?
Our study reveals a positive correlation between genetic diversity in populations of C. obscurior and recombination rates, consistent with previous findings (Begun and Aquadro 1992; Sirviö et al. 2006; Spencer et al. 2006; Roesti et al. 2013; Jones et al. 2019). Recombination rates also correlated positively with SV frequency and indel polymorphisms, likely owing to ectopic recombination—a significant mechanism driving SV emergence in various organisms, including honey bees (Rueppell et al. 2016), Drosophila (Montgomery et al. 1991; Delprat et al. 2009), and humans (Liu et al. 2012; Robberecht et al. 2013; Startek et al. 2015). Thus, high recombination rates may function as a compensatory mechanism for the reduced genetic diversity observed in haplodiploid social Hymenoptera with low Ne. Although there is some reasoning that increased recombination may evolve in small populations (Otto and Barton 2001), further investigations are necessary to elucidate the relationship between Ne and recombination rate evolution in social and nonsocial haplodiploids, particularly using empirical approaches.
Recombination is associated with genome dynamics and divergence in C. obscurior
The correlation between genetic diversity and recombination in C. obscurior has important implications for its genome dynamics and evolutionary processes. Through hitchhiking and background selection, linked selection in regions with low recombination leads to lower diversity and a skew toward rare alleles (Charlesworth et al. 1993; Comeron et al. 2008; Sella et al. 2009; Smukowski and Noor 2011; Ellegren and Galtier 2016). Conversely, the divergence of populations by genetic drift is expected to be stronger in regions where recombination rates and, thus, genetic diversity are high (Hellmann et al. 2003; Ellegren and Galtier 2016). Accordingly, in C. obscurior, recombination rates are positively correlated with genetic diversity and levels of dXY. However, there was no correlation with ω, consistent with a role of recombination in driving within-species divergence but not between-species divergence. Moreover, C. obscurior is a highly inbred tramp species and its populations experience genetic bottlenecks, reducing diversity in this species (Heinze 2017). Along with genetic hybridization and an increased TE activity (Errbii et al. 2021), introduced populations of this species may benefit from the effects of recombination on genetic diversity, providing yet another possible resolution to the “genetic paradox of invasive species” (Frankham 2005).
Methods
Mapping population
C. obscurior originates from Southeast Asia, and colonies across the globe belong to two (or more) genetically distinct lineages (Errbii et al. 2021). In this study, we performed a cross involving a male and a queen collected from two New World populations separated by ∼50 km (Itabuna and Una, respectively, in Brazil). These samples were collected in 2018 under permit 63371-1 issued by the Brazilian Ministério do Meio Ambiente. In haplodiploid species, haploid males develop from unfertilized eggs that represent the direct product of meiosis in females—the only recombining sex—making them ideal for linkage mapping. However, C. obscurior queens are not very productive (Jaimes-Nino et al. 2022) and cannot produce sufficient males for linkage mapping. To compensate, the F1 queen was mated with her brother (herein F1 male) to sample a total of 100 F2 individuals (12 males and 88 workers) at the pupal stage.
DNA extraction and sequencing
DNA was extracted from the two grandparents, the F1 parents, and 100 F2 individuals using a modified CTAB (0.75 M NaCl, 50 mM Tris/HCl at pH 8.0, 10 mM EDTA, 1% hexadecyltrimethylammonium bromide) protocol (Doyle and Doyle 1987). Libraries were prepared using the Illumina DNA prep kit with five to six PCR cycles, and 150 bp paired-end read sequencing was performed on an Illumina NovaSeq platform at the Cologne Center for Genomics. The grandmother and the F1 queen were sequenced to an average coverage of ∼60×, the males to an average coverage of ∼27×, and the workers to an average coverage of ∼42×.
Read mapping and variant calling
The quality of the raw reads was assessed using FastQC (v.0.11.7) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and Trimmomatic (v.0.38) (Bolger et al. 2014) was used to remove short and low-quality reads as well as adapter sequences (options: ILLUMINACLIP:NexteraPE-PE.fa:2:30:10 SLIDINGWINDOW:4:20 MINLEN:40). The resulting paired reads were mapped to the Cobs2.1 assembly (Errbii et al. 2021) using BWA-MEM (v.0.7.17) (Li and Durbin 2009) with default parameters. The mapping quality was checked using Qualimap (v.2.2.2-dev) (Supplemental Table S8; Okonechnikov et al. 2016), and duplicate reads were marked using Picard's MarkDuplicates (v.2.20.0). Joint-variant calling was performed using GATK's HaplotypeCaller (v.4.1.2.0) (McKenna et al. 2010) with default parameters.
Variant filtering and phasing
After removing indels, the remaining 342,508 biallelic single-nucleotide polymorphisms (SNPs) were screened for high-quality markers using the following criteria: First, markers must be genotyped in both F1 parents and the grandparents. Second, in the F1 queen, markers must be heterozygous, whereas in the grandparents, they must be homozygous for different alleles or heterozygous in the grandmother. Because males are haploid, each SNP is expected to be hemizygous in males. Third, consequently, heterozygous markers in at least one male, likely owing to copy number variations, were discarded. Fourth, in BCFtools (v.1.14) (Danecek et al. 2021), genotypes with Mendelian errors were removed. Markers were further filtered using VCFtools (v.0.1.16) (Danecek et al. 2011) to only include those with a minor allele frequency (MAF) > 0.2, genotyped across >80% of samples, a mean coverage between 19× and 50×, and at least five reads per genotype. In total, we identified 57,558 high-quality markers that were heterozygous in the F1 queen and were used as the basis for phasing (Supplemental Fig. S17).
The mapping population included 88 diploid F2 workers, each carrying the F1 father's haplotype and one of the two recombining haplotypes from the F1 queen (Supplemental Fig. S17). Using a custom script (Supplemental Code), we excluded the F1 male contribution from each of the F2 workers, retaining only the maternal contribution.
Scaffold anchoring into chromosomes
We improved the current version of the genome (Cobs2.1) by correcting misassembled (i.e., chimeric) and misoriented scaffolds. We used MSTmap (v.1.0) (Wu et al. 2008), with parameters specified in Supplemental Table S9, to order all 57,558 markers into LGs according to the method of Gadau (2009). We then generated a BED file with the corrected genomic coordinates and orientation of scaffolds (Supplemental Table S10) based on the initial genetic map (Supplemental Fig. S1). Using BEDTools’ getfasta (v.2.30.0) (Quinlan and Hall 2010) and Cobs2.1, we extracted and concatenated genomic sequences belonging to the same LG.
To accurately split chimeric scaffolds, we first checked for the existence of gaps separating the parts belonging to different LGs. When no gap was found, scaffolds were split after the last marker of the first part. For misoriented scaffolds, we inspected the correlation between the physical and genetic positions of markers. When the correlation was positive, the original orientation was kept, and when negative, the reverse complementary sequence was used. Scaffolds lacking markers and those that could not be mapped to any of the LGs were kept unplaced. The final chromosome-level assembly contained 29 LGs and 84 unplaced sequences.
Gene and (tandem) repeat annotation
For gene annotation, Liftoff (v.1.6.3) (Shumate and Salzberg 2021) was used to transfer the Cobs2.1 annotation (Errbii et al. 2021) to Cobs3.1 with the default parameters and the -polish option. For repeat annotation, we used RepeatMasker (v.4.0.7) (https://www.repeatmasker.org) and a previous TE library (Errbii et al. 2021). TE islands were defined as previously described (Errbii et al. 2021).
The tandem repeats finder tool (v.4.09.1) (Benson 1999) was used with the recommended parameters (2 5 7 80 10 50 2000) to identify TRs in the genome. Additionally, TRs from five ant species (Melters et al. 2013; Huang et al. 2016) were used with RepeatMasker to explore their enrichment in the genome of C. obscurior. Further details on annotation procedures are provided in the Supplemental Methods.
Estimation of recombination rate
To calculate local recombination rates, Cobs3.1 was used as a reference following the pipeline described above for read mapping, variant calling, filtering, and phasing.
We visually inspected all markers and discarded erroneous genotypes and those that indicated unlikely double-crossover events. These events were primarily identified as single-locus markers as well as blocks of two to three adjacent markers, tens to hundreds of base pairs long (Supplemental Figs. S18–S25), showing genotypic differences from immediately adjacent markers. Our reasoning to exclude these markers, was based on the expectations that double crossovers should be exceedingly rare due the suppression via chiasma interference (Sturtevant 1915; Muller 1916), in which the occurrence of one crossover disrupts the formation of another on the same chromosome. The effect of this interference remains robust over long distances, ranging from hundreds of kilobases to tens of megabases (Hillers 2004; Fowler et al. 2018). We also excluded regions where we could not identify crossovers unambiguously owing to low mapping accuracy and/or copy number variations (e.g., Supplemental Fig. S26).
Because all 84 unplaced scaffolds had insufficient markers (two or fewer), we only used markers on the 29 refined LGs. The final map was based on 57,532 high-quality markers that were clustered into 29 LGs (Supplemental Fig. S3) using MSTmap (Supplemental Table S9). Crossovers were defined as genotype changes between the maternal haplotypes, with most crossover tracks spanning >100 kb (Supplemental Figs. S22, S24, S25).
For local recombination rates, markers on each LG were ordered according to their physical distance, and we removed 223 markers showing discrepancies between their physical and genetic positions. Subsequently, nonrecombining markers were clustered according to their shared genetic positions, resulting in 1061 effectively recombining marker clusters. Local recombination rates were then inferred by calculating the ratio of the genetic (in cM) to the physical (Mb) distances between adjacent clusters. Average distance between crossover events was 149 kb (95% CI [122, 177]). To ensure that genomic regions had a proportional influence on estimates of recombination rate and to account for the nonuniform distribution of crossovers along the LGs, we calculated a weighted mean of recombination rate in 250 kb nonoverlapping windows. To this end, each window was assigned the recombination rate of the corresponding intercluster interval. When a window matched multiple intervals, the mean recombination rate of those intervals, weighted by their physical length, was assigned (Supplemental Code).
Estimating genetic diversity and differentiation
Nucleotide diversity (π) and Tajima's D were calculated in 250 kb nonoverlapping windows using two Pool-seq data sets of C. obscurior, a pool of 30 workers from Itabuna, Brazil (Errbii et al. 2021), and a pool of 50 workers from a ∼50 km distant location in Una, Brazil. Following the method of Errbii et al. (2021), we filtered paired reads, mapped them to Cobs3.1, and generated an indel-free mpileup file for each population using SAMtools (v.1.15.1) (Li et al. 2009). We used the PoPoolation (Kofler et al. 2011a) Variance-sliding.pl script to calculate π and Tajima's D. For π at synonymous (πS) and nonsynonymous (πNS) sites, we used the PoPoolation Syn-nonsyn-sliding.pl script, the mpileup files, and the codon and nonsynonymous codon length tables from PoPoolation.
For genetic differentiation (FST) between lineages of C. obscurior, we combined the alignment files of the two pools with a third alignment file of a pool of 16 workers from Tenerife, Spain (Errbii et al. 2021), to create a single mpileup file using SAMtools. We then converted this file into a synchronized file and used the fst-sliding.pl script under PoPoolation2 (Kofler et al. 2011b) to calculate FST in 250 kb nonoverlapping windows.
Calculation of absolute divergence
To explore genetic divergence between lineages of C. obscurior (dXY) and the grandparental haplotypes (gp-dXY), we calculated absolute divergence in 250 kb windows using scripts available at GitHub (https://github.com/simonhmartin/genomics_general).
Published paired-end short reads data from single workers of the Old World (Taiwan and Leiden) and New World (Itabuna and Una) lineages of C. obscurior (Errbii et al. 2021) were filtered and mapped to Cobs3.1 as described above. Note that reads from both lineages can be aligned to the reference genome with high confidence (Supplemental Fig. S27). Further details on SNP calling and filtering are given in the Supplemental Methods.
SV identification
To identify SVs, we adopted an approach by Mérot et al. (2023) using published paired-end short read data from 16 workers from five populations (Errbii et al. 2021). Reads were filtered and mapped to Cobs3.1, and read duplicates were removed using Picard's MarkDuplicates. SVs were identified using three different tools: Manta (v.1.6.0) (Chen et al. 2016); smoove (v.0.2.8) (https://github.com/brentp/smoove), which is based on LUMPY (Layer et al. 2014); and DELLY (v.0.8.1) (Rausch et al. 2012), all run with default parameters. To identify genomic inversions potentially affecting recombination in the F1 queen, we used the three tools and followed the same approach. Subsequently, both sets of variants were filtered following protocols detailed in the Supplemental Methods.
For indels, we used the raw variant calls generated by GATK's HaplotypeCaller for the 16 workers. Indels were extracted and subjected to GATK's recommended hard filtering criteria (QD < 2.0; QUAL < 30.0; FS > 200, and ReadPosRankSum < −20.0). For further details on the filtering process and diversity calculation, see the Supplemental Methods.
Genome parameters estimation
BEDTools and BEDOPS (v.2.4.37) (Neph et al. 2012), along with the gene annotation, were used to calculate gene content. Overall and intergenic GC contents were calculated using the BEDTools nuc function in 250 kb nonoverlapping windows.
Estimating substitution rates
To explore the correlation between recombination and the efficiency of selection, we calculated omega (ω) for single-copy ortholog genes between C. obscurior and three ant species (Monomorium pharaonis [GCA013373865v2], Solenopsis invicta [UNIL_Sinv_3.0], and Ooceraea biroi [Obir_v5.4]) and the parasitoid wasp Nasonia vitripennis (Nvit_psr_1.1). Genomic and protein sequences, and gene annotations, were downloaded from Ensembl (release-54). To estimate the branch-wide rate ω and rates of synonymous (dN) and synonymous mutations (dS), we applied HyPhy's aBSREL (v.2.5.48; adaptive Branch-Site Random Effects Likelihood) (Smith et al. 2015) and the FitMG94 model, implemented in HyPhy. Further details on single-copy ortholog retrieval and ω calculation are given in the Supplemental Methods.
Gene expression bias
To investigate the relationship between local recombination rate and developmental gene expression bias, we calculated the plasticity index (Schrader et al. 2017), capturing gene expression bias across multiple caste/morph contrasts. High values indicate caste-specific gene expression, whereas low values suggest uniform expression across castes. RNA-seq data of worker, queen, wingless male, and winged male third instar larvae (n = 7 individuals each) were retrieved from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA237579 (Schrader et al. 2015, 2017). For detailed information on read filtering and mapping, differential expression analysis, and computation of plasticity indice, see Supplemental Methods.
GO term enrichment analysis
To test whether genes found in regions of high recombination rates are enriched for specific functions, we ran a GO enrichment analysis. We defined these regions as windows with recombination rates falling within the top 10% (recombination rate > 22.3 cM/Mb). We used BEDTools’ intersect to extract 1541 genes located in these regions and ran topGO (v.2.46.0) (https://bioconductor.org/packages/release/bioc/html/topGO.html) to identify biological processes enriched in these regions. TE islands and regions affected by introgression were excluded from the analyses.
Correlation analyses and plotting
All correlations using the raw data and plots were produced in R (v.4.1.3) (R Core Team 2020). To explore the associations of TE content, sequence divergence, and epistasis with recombination rates per 250 kb, a generalized linear model was fitted using the glm function in R. We used Circos (Krzywinski et al. 2009) for graphical visualization in R. LGs 9, 10, 11, 17, and 21 were excluded from all correlation analyses, as introgression is evident in a significant fraction of these LGs. Further, LGs 26 and 29 were excluded because of low marker coverage (Supplemental Fig. S6).
Data access
The genome assembly, annotation, and raw sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA934066. Intermediate data required to reproduce the analyses are provided as Supplemental Data and are also available on figshare (https://doi.org/10.6084/m9.figshare.22178717.v2). A detailed description of the bioinformatic analysis pipelines is available as Supplemental Code and on GitHub (https://github.com/merrbii/CobsLinkagemapping).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This research was funded by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation)–403813881 with grants to L.S. (SCHR 1554/2-1) and J.O. (OE 549/4-1) under the priority program “Rapid evolutionary adaptation: Potential and constraints” (SPP 1819). We thank the DFG Research Training Group 2220 “Evolutionary Processes in Adaptation and Disease” and the CORE environment (cardiocondyla.org) for supporting M.E. J.G. acknowledges support from the DFG as part of SFB TRR 212 (NC³)–TP C04 project numbers 316099922 and 396780988. We further thank Tobias van Elst, Jacques Delabie, and Eva Schultner for help collecting ants in the field. We thank Xim Cerda for his support to collect ants in Spain under permission of Real Decreto 124/2017. We thank Simon H. Martin for his valuable advice on the analysis of sequence divergence. We thank three anonymous reviewers for their constructive comments.
Author contributions: J.O. and L.S. conceptualized the study and acquired funding. M.E., J.G., J.O., and L.S. designed the experiments. M.E. produced the data and performed the analyses. K.B. was responsible for Illumina sequencing. M.E., L.S., and J.O. wrote the manuscript, all authors contributed to the final version.
Notes
[3] Supplementary material [Supplemental material is available for this article.]
[4] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278392.123.
References
- ↵Altenberg L, Feldman MW. 1987. Selection, generalized transmission and the evolution of modifier genes. I. The reduction principle. Genetics 117: 559–572. 10.1093/genetics/117.3.559
- ↵Arbeithuber B, Betancourt AJ, Ebner T, Tiemann-Boege I. 2015. Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc Natl Acad Sci 112: 2109–2114. 10.1073/pnas.1416622112
- ↵Barton NH. 1995. A general model for the evolution of recombination. Genet Res (Camb) 65: 123–144. 10.1017/S0016672300033140
- ↵Beadle GW. 1932. A possible influence of the spindle fibre on crossing-over in Drosophila. Proc Natl Acad Sci 18: 160–165. 10.1073/pnas.18.2.160
- ↵Begun DJ, Aquadro CF. 1992. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356: 519–520. 10.1038/356519a0
- ↵Bell G. 1982. The masterpiece of nature: the evolution and genetics of sexuality, 1st ed. University of California Press, Berkeley, CA.
- ↵Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580. 10.1093/nar/27.2.573
- ↵Betancourt AJ, Presgraves DC. 2002. Linkage limits the power of natural selection in Drosophila. Proc Natl Acad Sci 99: 13616–13620. 10.1073/pnas.212277199
- ↵Betancourt AJ, Welch JJ, Charlesworth B. 2009. Reduced effectiveness of selection caused by a lack of recombination. Curr Biol 19: 655–660. 10.1016/j.cub.2009.02.039
- ↵Beye M, Gattermeier I, Hasselmann M, Gempe T, Schioett M, Baines JF, Schlipalius D, Mougel F, Emore C, Rueppell O, 2006. Exceptionally high levels of recombination across the honey bee genome. Genome Res 16: 1339–1344. 10.1101/gr.5680406
- ↵Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170
- ↵Bolívar P, Mugal CF, Nater A, Ellegren H. 2016. Recombination rate variation modulates gene sequence evolution mainly via GC-biased gene conversion, not Hill–Robertson interference, in an avian system. Mol Biol Evol 33: 216–227. 10.1093/molbev/msv214
- ↵Bullaughey K, Przeworski M, Coop G. 2008. No effect of recombination on the efficacy of natural selection in primates. Genome Res 18: 544–554. 10.1101/gr.071548.107
- ↵Burri R. 2017. Interpreting differentiation landscapes in the light of long-term linked selection. Evol Lett 1: 118–131. 10.1002/evl3.14
- ↵Campos JL, Halligan DL, Haddrill PR, Charlesworth B. 2014. The relation between recombination rate and patterns of molecular evolution and variation in Drosophila melanogaster. Mol Biol Evol 31: 1010–1028. 10.1093/molbev/msu056
- ↵Chak STC, Harris SE, Hultgren KM, Duffy JE, Rubenstein DR. 2022. Demographic inference provides insights into the extirpation and ecological dominance of eusocial snapping shrimps. J Hered 113: 552–562. 10.1093/jhered/esac035
- ↵Chakraborty U, Mackenroth B, Shalloway D, Alani E. 2019. Chromatin modifiers alter recombination between divergent DNA sequences. Genetics 212: 1147–1162. 10.1534/genetics.119.302395
- ↵Charlesworth D. 2016. The status of supergenes in the 21st century: recombination suppression in Batesian mimicry and sex chromosomes and other complex adaptations. Evol Appl 9: 74–90. 10.1111/eva.12291
- ↵Charlesworth B, Morgan MT, Charlesworth D. 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303. 10.1093/genetics/134.4.1289
- ↵Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, Saunders CT. 2016. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32: 1220–1222. 10.1093/bioinformatics/btv710
- ↵Comeron JM, Williford A, Kliman RM. 2008. The Hill–Robertson effect: evolutionary consequences of weak selection and linkage in finite populations. Heredity (Edinb) 100: 19–31. 10.1038/sj.hdy.6801059
- ↵Crow JF. 1994. Advantages of sexual reproduction. Dev Genet 15: 205–213. 10.1002/dvg.1020150303
- ↵Cutter AD, Payseur BA. 2013. Genomic signatures of selection at linked sites: unifying the disparity among species. Nat Rev Genet 14: 262–274. 10.1038/nrg3425
- ↵Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. 10.1093/bioinformatics/btr330
- ↵Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM. 2021. Twelve years of SAMtools and BCFtools. GigaScience 10: giab008. 10.1093/gigascience/giab008
- ↵Delame M, Prado E, Blanc S, Robert-Siegwald G, Schneider C, Mestre P, Rustenholz C, Merdinoglu D. 2019. Introgression reshapes recombination distribution in grapevine interspecific hybrids. Theor Appl Genet 132: 1073–1087. 10.1007/s00122-018-3260-x
- ↵Delprat A, Negre B, Puig M, Ruiz A. 2009. The transposon Galileo generates natural chromosomal inversions in Drosophila by ectopic recombination. PLoS One 4: e7883. 10.1371/journal.pone.0007883
- ↵Dolgin ES, Charlesworth B. 2008. The effects of recombination rate on the distribution and abundance of transposable elements. Genetics 178: 2169–2177. 10.1534/genetics.107.082743
- ↵Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19: 11–15.
- ↵Drakula M, Buellesbach J, Schrader L. 2023. The role of cuticular hydrocarbons in intraspecific aggression in the invasive ant Cardiocondyla obscurior. Myrmecol News 33: 187–196. 10.25849/myrmecol.news_033:187
- ↵Ellegren H, Galtier N. 2016. Determinants of genetic diversity. Nat Rev Genet 17: 422–433. 10.1038/nrg.2016.58
- ↵Errbii M, Keilwagen J, Hoff KJ, Steffen R, Altmüller J, Oettler J, Schrader L. 2021. Transposable elements and introgression introduce genetic variation in the invasive ant Cardiocondyla obscurior. Mol Ecol 30: 6211–6228. doi: 10.1111/mec.16099
- ↵Felsenstein J. 1974. The evolutionary advantage of recombination. Genetics 78: 737–756. 10.1093/genetics/78.2.737
- ↵Felsenstein J, Yokoyama S. 1976. The evolutionary advantage of recombination. II. Individual selection for recombination. Genetics 83: 845–859. 10.1093/genetics/83.4.845
- ↵Finck J, Berdan EL, Mayer F, Ronacher B, Geiselhardt S. 2016. Divergence of cuticular hydrocarbons in two sympatric grasshopper species and the evolution of fatty acid synthases and elongases across insects. Sci Rep 6: 33695. 10.1038/srep33695
- ↵Fowler KR, Hyppa RW, Cromie GA, Smith GR. 2018. Physical basis for long-distance communication along meiotic chromosomes. Proc Natl Acad Sci 115: E9333–E9342. 10.1073/pnas.1801920115
- ↵Frankham R. 2005. Resolving the genetic paradox in invasive species. Heredity (Edinb) 94: 385. 10.1038/sj.hdy.6800634
- ↵Fuller ZL, Koury SA, Leonard CJ, Young RE, Ikegami K, Westlake J, Richards S, Schaeffer SW, Phadnis N. 2020. Extensive recombination suppression and epistatic selection causes chromosome-wide differentiation of a selfish sex chromosome in Drosophila pseudoobscura. Genetics 216: 205–226. 10.1534/genetics.120.303460
- ↵Gadau J. 2009. Phase-unknown linkage mapping in ants. Cold Spring Harb Protoc 2009: pdb.prot5251. 10.1101/pdb.prot5251
- ↵Gadau J, Page RE, Werren JH. 1999. Mapping of hybrid incompatibility loci in Nasonia. Genetics 153: 1731–1741. 10.1093/genetics/153.4.1731
- ↵Hadany L, Comeron JM. 2008. Why are sex and recombination so common? Ann N Y Acad Sci 1133: 26–43. 10.1196/annals.1438.011
- ↵Haddrill PR, Halligan DL, Tomaras D, Charlesworth B. 2007. Reduced efficacy of selection in regions of the Drosophila genome that lack crossing over. Genome Biol 8: R18. 10.1186/gb-2007-8-2-r18
- ↵Haenel Q, Laurentino TG, Roesti M, Berner D. 2018. Meta-analysis of chromosome-scale crossover rate variation in eukaryotes and its significance to evolutionary genomics. Mol Ecol 27: 2477–2497. 10.1111/mec.14699
- ↵Halldorsson BV, Palsson G, Stefansson OA, Jonsson H, Hardarson MT, Eggertsson HP, Gunnarsson B, Oddsson A, Halldorsson GH, Zink F, 2019. Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science 363: eaau1043. 10.1126/science.aau1043
- ↵Hartke J, Schell T, Jongepier E, Schmidt H, Sprenger PP, Paule J, Bornberg-Bauer E, Schmitt T, Menzel F, Pfenninger M, 2019. Hybrid genome assembly of a neotropical mutualistic ant. Genome Biol Evol 11: 2306–2311. 10.1093/gbe/evz159
- ↵Hefetz A. 2007. The evolution of hydrocarbon pheromone parsimony in ants (Hymenoptera: Formicidae): interplay of colony odor uniformity and odor idiosyncrasy: a review. Myrmecological News 10: 59–68.
- ↵Heinze J. 2017. Life-history evolution in ants: the case of Cardiocondyla. Proc R Soc B Biol Sci 284: 20161406. 10.1098/rspb.2016.1406
- ↵Hellmann I, Ebersberger I, Ptak SE, Pääbo S, Przeworski M. 2003. A neutral explanation for the correlation of diversity with recombination rates in humans. Am J Hum Genet 72: 1527–1535. 10.1086/375657
- ↵Hill WG, Robertson A. 1966. The effect of linkage on limits to artificial selection. Genet Res 8: 269–294. 10.1017/S0016672300010156
- ↵Hillers KJ. 2004. Crossover interference. Curr Biol 14: R1036–R1037. 10.1016/j.cub.2004.11.038
- ↵Holze H, Schrader L, Buellesbach J. 2021. Advances in deciphering the genetic basis of insect cuticular hydrocarbon biosynthesis and variation. Heredity (Edinb) 126: 219–234. 10.1038/s41437-020-00380-y
- ↵Huang YC, Lee CC, Kao CY, Chang NC, Lin CC, Shoemaker D, Wang J. 2016. Evolution of long centromeres in fire ants. BMC Evol Biol 16: 189. 10.1186/s12862-016-0760-7
- ↵Hughes WOH, Boomsma JJ. 2004. Genetic diversity and disease resistance in leaf-cutting ant societies. Evolution (N Y) 58: 1251–1260. 10.1554/03-546
- ↵Hunt GJ, Page RE. 1995. Linkage map of the honey bee, Apis mellifera, based on RAPD markers. Genetics 139: 1371–1382. 10.1093/genetics/139.3.1371
- ↵Jaimes-Nino LM, Heinze J, Oettler J. 2022. Late-life fitness gains and reproductive death in Cardiocondyla obscurior ants. eLife 11: e74695. 10.7554/eLife.74695
- ↵Jensen-Seaman MI, Furey TS, Payseur BA, Lu Y, Roskin KM, Chen CF, Thomas MA, Haussler D, Jacob HJ. 2004. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res 14: 528–538. 10.1101/gr.1970304
- ↵Jones GH, Franklin FCH. 2006. Meiotic crossing-over: obligation and interference. Cell 126: 246–248. 10.1016/j.cell.2006.07.010
- ↵Jones JC, Wallberg A, Christmas MJ, Kapheim KM, Webster MT. 2019. Extreme differences in recombination rate between the genomes of a solitary and a social bee. Mol Biol Evol 36: 2277–2291. 10.1093/molbev/msz130
- ↵Kent CF, Zayed A. 2013. Evolution of recombination and genome structure in eusocial insects. Commun Integr Biol 6: e22919. 10.4161/cib.22919
- ↵Kent CF, Minaei S, Harpur BA, Zayed A. 2012. Recombination is associated with the evolution of genome structure and worker behavior in honey bees. Proc Natl Acad Sci 109: 18012–18017. 10.1073/pnas.1208094109
- ↵Kent TV, Uzunović J, Wright SI. 2017. Coevolution between transposable elements and recombination. Philos Trans R Soc Lond B Biol Sci 372: 20160458. 10.1098/rstb.2016.0458
- ↵Kimura M, Crow JF. 1964. The number of alleles that can be maintained in a finite population. Genetics 49: 725–738. 10.1093/genetics/49.4.725
- ↵Kleckner N. 1996. Meiosis: How could it work? Proc Natl Acad Sci 93: 8167–8174. 10.1073/pnas.93.16.8167
- ↵Kofler R, Orozco-terWengel P, de Maio N, Pandey RV, Nolte V, Futschik A, Kosiol C, Schlötterer C. 2011a. PoPoolation: a toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS One 6: e15925. 10.1371/journal.pone.0015925
- ↵Kofler R, Pandey RV, Schlötterer C. 2011b. PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (pool-seq). Bioinformatics 27: 3435–3436. 10.1093/bioinformatics/btr589
- ↵Kozomara A, Birgaoanu M, Griffiths-Jones S. 2019. miRBase: from microRNA sequences to function. Nucleic Acids Res 47: D155–D162. 10.1093/nar/gky1141
- ↵Kraus B, Page RE. 1998. Parasites, pathogens, and polyandry in social insects. Am Nat 151: 383–391. 10.1086/286126
- ↵Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. 10.1101/gr.092759.109
- ↵Lambie EJ, Roeder GS. 1986. Repression of meiotic crossing over by a centromere (CEN3) in Saccharomyces cerevisiae. Genetics 114: 769–789. 10.1093/genetics/114.3.769
- ↵Langley CH, Montgomery E, Hudson R, Kaplan N, Charlesworth B. 1988. On the role of unequal exchange in the containment of transposable element copy number. Genet Res (Camb) 52: 223–235. 10.1017/S0016672300027695
- ↵Layer RM, Chiang C, Quinlan AR, Hall IM. 2014. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15: R84. 10.1186/gb-2014-15-6-r84
- ↵Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324
- ↵Li L, Jean M, Belzile F. 2006. The impact of sequence divergence and DNA mismatch repair on homeologous recombination in Arabidopsis. Plant J 45: 908–916. 10.1111/j.1365-313X.2006.02657.x
- ↵Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352
- ↵Liharska T, Koornneef M, Van Wordragen M, Van Kammen A, Zabel P. 1996. Tomato chromosome 6: effect of alien chromosomal segments on recombinant frequencies. Genome 39: 485–491. 10.1139/g96-062
- ↵Liu P, Carvalho CMB, Hastings PJ, Lupski JR. 2012. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev 22: 211–220. 10.1016/j.gde.2012.02.012
- ↵Liu H, Zhang X, Huang J, Chen J-Q, Tian D, Hurst LD, Yang S. 2015. Causes and consequences of crossing-over evidenced via a high-resolution recombinational landscape of the honey bee. Genome Biol 16: 15. 10.1186/s13059-014-0566-0
- ↵Liu H, Jia Y, Sun X, Tian D, Hurst LD, Yang S. 2017. Direct determination of the mutation rate in the bumblebee reveals evidence for weak recombination-associated mutation and an approximate rate constancy in insects. Mol Biol Evol 34: 119–130. 10.1093/molbev/msw226
- ↵Lynn A, Ashley T, Hassold T. 2004. Variation in human meiotic recombination. Annu Rev Genomics Hum Genet 5: 317–349. 10.1146/annurev.genom.4.070802.110217
- ↵Marais G, Charlesworth B, Wright SI. 2004. Recombination and base composition: the case of the highly self-fertilizing plant Arabidopsis thaliana. Genome Biol 5: R45. 10.1186/gb-2004-5-7-r45
- ↵Marand AP, Jansky SH, Zhao H, Leisner CP, Zhu X, Zeng Z, Crisovan E, Newton L, Hamernik AJ, Veilleux RE, 2017. Meiotic crossovers are associated with open chromatin and enriched with Stowaway transposons in potato. Genome Biol 18: 203. 10.1186/s13059-017-1326-8
- ↵Maynard Smith J. 1978. The evolution of sex. Cambridge University Press, Cambridge
- ↵McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. 10.1101/gr.107524.110
- ↵Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby JG, Sebra R, Peluso P, Eid J, Rank D, 2013. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol 14: R10. 10.1186/gb-2013-14-1-r10
- ↵Mérot C, Stenløkk KSR, Venney C, Laporte M, Moser M, Normandeau E, Árnyasi M, Kent M, Rougeux C, Flynn JM, 2023. Genome assembly, structural variants, and genetic differentiation between lake whitefish young species pairs (Coregonus sp.) with long and short reads. Mol Ecol 32: 1458–1477. 10.1111/mec.16468
- ↵Metcalf RA, Marlin JC, Whitt GS. 1975. Low levels of genetic heterozygosity in Hymenoptera. Nature 257: 792–794. 10.1038/257792a0
- ↵Meunier J, Duret L. 2004. Recombination drives the evolution of GC-content in the human genome. Mol Biol Evol 21: 984–990. 10.1093/molbev/msh070
- ↵Montgomery EA, Huang SM, Langley CH, Judd BH. 1991. Chromosome rearrangement by ectopic recombination in Drosophila melanogaster: genome structure and evolution. Genetics 129: 1085–1098. 10.1093/genetics/129.4.1085
- ↵Muller HJ. 1916. The mechanism of crossing-over. Am Nat 50: 193–221. 10.1086/279534
- ↵Navarro-Domínguez B, Chang CH, Brand CL, Muirhead CA, Presgraves DC, Larracuente AM. 2022. Epistatic selection on a selfish segregation distorter supergene: drive, recombination, and genetic load. eLife 11: 78981. 10.7554/eLife.78981
- ↵Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S, 2012. BEDOPS: high-performance genomic feature operations. Bioinformatics 28: 1919–1920. 10.1093/bioinformatics/bts277
- ↵Niehuis O, Gibson JD, Rosenberg MS, Pannebakker BA, Koevoets T, Judson AK, Desjardins CA, Kennedy K, Duggan D, Beukeboom LW, 2010. Recombination and its impact on the genome of the haplodiploid parasitoid wasp Nasonia. PLoS One 5: e8597. 10.1371/journal.pone.0008597
- ↵Nielsen R. 2005. Molecular signatures of natural selection. Annu Rev Genet 39: 197–218. 10.1146/annurev.genet.39.073003.112420
- ↵Noor MAF, Gratos KL, Bertucci LA, Reiland J. 2001. Chromosomal inversions and the reproductive isolation of species. Proc Natl Acad Sci 98: 12084–12088. 10.1073/pnas.221274498
- ↵Oettler J. 2021. Cardiocondyla: heart node ants. In Encyclopedia of social insects (ed. Starr CK). Springer, Cham, Switzerland. https://doi.org/10.1007/978-3-030-28102-1_19
- ↵Okonechnikov K, Conesa A, García-Alcalde F. 2016. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32: 292–294. 10.1093/bioinformatics/btv566
- ↵Oldroyd BP, Fewell JH. 2007. Genetic diversity promotes homeostasis in insect colonies. Trends Ecol Evol 22: 408–413. 10.1016/j.tree.2007.06.001
- ↵Opperman R, Emmanuel E, Levy AA. 2004. The effect of sequence divergence on recombination between direct repeats in Arabidopsis. Genetics 168: 2207–2215. 10.1534/genetics.104.032896
- ↵Otto SP, Barton NH. 2001. Selection for recombination in small populations. Evolution (N Y) 55: 1921–1931. 10.1111/j.0014-3820.2001.tb01310.x
- ↵Otto SP, Feldman MW. 1997. Deleterious mutations, variable epistatic interactions, and the evolution of recombination. Theor Popul Biol 51: 134–147. 10.1006/tpbi.1997.1301
- ↵Otto SP, Lenormand T. 2002. Resolving the paradox of sex and recombination. Nat Rev Genet 3: 252–261. 10.1038/nrg761
- ↵Pamilo P, Crozier RH. 1997. Population biology of social insect conservation. Mem Museum Victoria 56: 411–419. 10.24199/j.mmv.1997.56.32
- ↵Pamilo P, Varvio-Aho S, Pekkarinen A. 1978. Low enzyme gene variability in Hymenoptera as a consequence of haplodiploidy. Hereditas 88: 93–99. 10.1111/j.1601-5223.1978.tb01607.x
- ↵Pessia E, Popa A, Mousset S, Rezvoy C, Duret L, Marais GAB. 2012. Evidence for widespread GC-biased gene conversion in eukaryotes. Genome Biol Evol 4: 675–682. 10.1093/gbe/evs052
- ↵Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033
- ↵Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. 2012. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28: i333–i339. 10.1093/bioinformatics/bts378
- ↵R Core Team. 2020. R: a language and environment for statistical computing, 4.0.2. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/.
- ↵Rieseberg LH. 2001. Chromosomal rearrangements and speciation. Trends Ecol Evol 16: 351–358. 10.1016/S0169-5347(01)02187-5
- ↵Ritz KR, Noor MAF, Singh ND. 2017. Variation in recombination rate: adaptive or not? Trends Genet 33: 364–374. 10.1016/j.tig.2017.03.003
- ↵Robberecht C, Voet T, Esteki MZ, Nowakowska BA, Vermeesch JR. 2013. Nonallelic homologous recombination between retrotransposable elements is a driver of de novo unbalanced translocations. Genome Res 23: 411–418. 10.1101/gr.145631.112
- ↵Roeder GS. 1997. Meiotic chromosomes: it takes two to tango. Genes Dev 11: 2600–2621. 10.1101/gad.11.20.2600
- ↵Roesti M, Moser D, Berner D. 2013. Recombination in the threespine stickleback genome: patterns and consequences. Mol Ecol 22: 3014–3027. 10.1111/mec.12322
- ↵Romiguier J, Lourenco J, Gayral P, Faivre N, Weinert LA, Ravel S, Ballenghien M, Cahais V, Bernard A, Loire E, 2014. Population genomics of eusocial insects: the costs of a vertebrate-like effective population size. J Evol Biol 27: 593–603. 10.1111/jeb.12331
- ↵Ross L, Blackmon H, Lorite P, Gokhman VE, Hardy NB. 2015. Recombination, chromosome number and eusociality in the Hymenoptera. J Evol Biol 28: 105–116. 10.1111/jeb.12543
- ↵Roux C, Weyna A, Lodé M, Romiguier J. 2024. Social evolution in termites reduces natural selection efficacy. bioRxiv 10.1101/2024.04.26.591327
- ↵Rueppell O, Kuster R, Miller K, Fouks B, Correa SR, Collazo J, Phaincharoen M, Tingek S, Koeniger N. 2016. A new metazoan recombination rate record and consistently high recombination rates in the honey bee genus Apis accompanied by frequent inversions but not translocations. Genome Biol Evol 8: 3653–3660. 10.1093/gbe/evw269
- ↵Savocco J, Piazza A. 2021. Recombination-mediated genome rearrangements. Curr Opin Genet Dev 71: 63–71. 10.1016/j.gde.2021.06.008
- ↵Schmid-Hempel P. 1998. Parasites in social insects. Princeton University Press, Princeton, NJ.
- ↵Schrader L, Kim JW, Ence D, Zimin A, Klein A, Wyschetzki K, Weichselgartner T, Kemena C, Stökl J, Schultner E, 2014. Transposable element islands facilitate adaptation to novel environments in an invasive species. Nat Commun 5: 5495. 10.1038/ncomms6495
- ↵Schrader L, Simola DF, Heinze J, Oettler J. 2015. Sphingolipids, transcription factors, and conserved toolkit genes: developmental plasticity in the ant Cardiocondyla obscurior. Mol Biol Evol 32: 1474–1486. 10.1093/molbev/msv039
- ↵Schrader L, Helantera H, Oettler J. 2017. Accelerated evolution of developmentally biased genes in the tetraphenic ant Cardiocondyla obscurior. Mol Biol Evol 34: 535–544. 10.1093/molbev/msw240
- ↵Sella G, Petrov DA, Przeworski M, Andolfatto P. 2009. Pervasive natural selection in the Drosophila genome? PLoS Genet 5: e1000495. 10.1371/journal.pgen.1000495
- ↵Shi YY, Sun LX, Huang ZY, Wu XB, Zhu YQ, Zheng HJ, Zeng ZJ. 2013. A SNP based high-density linkage map of Apis cerana reveals a high recombination rate similar to Apis mellifera. PLoS One 8: e76459. 10.1371/journal.pone.0076459
- ↵Shumate A, Salzberg SL. 2021. Liftoff: accurate mapping of gene annotations. Bioinformatics 37: 1639–1643. 10.1093/bioinformatics/btaa1016
- ↵Shykoff JA, Schmid-Hempel P. 1991. Parasites and the advantage of genetic variability within social insect colonies. Proc R Soc London Ser B Biol Sci 243: 55–58. 10.1098/rspb.1991.0009
- ↵Singhal S, Gomez SM, Burch CL. 2019. Recombination drives the evolution of mutational robustness. Curr Opin Syst Biol 13: 142–149. 10.1016/j.coisb.2018.12.003
- ↵Sirviö A, Gadau J, Rueppell O, Lamatsch D, Boomsma JJ, Pamilo P, Page RE. 2006. High recombination frequency creates genotypic diversity in colonies of the leaf-cutting ant Acromyrmex echinatior. J Evol Biol 19: 1475–1485. 10.1111/j.1420-9101.2006.01131.x
- ↵Sirviö A, Johnston JS, Wenseleers T, Pamilo P. 2011a. A high recombination rate in eusocial Hymenoptera: evidence from the common wasp Vespula vulgaris. BMC Genet 12: 95. 10.1186/1471-2156-12-95
- ↵Sirviö A, Pamilo P, Johnson RA, Page RE, Gadau J. 2011b. Origin and evolution of the dependent lineages in the genetic caste determination system of Pogonomyrmex ants. Evolution (N Y) 65: 869–884. 10.1111/j.1558-5646.2010.01170.x
- ↵Smith MD, Wertheim JO, Weaver S, Murrell B, Scheffler K, Kosakovsky Pond SL. 2015. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol 32: 1342–1353. 10.1093/molbev/msv022
- ↵Smukowski CS, Noor MAF. 2011. Recombination rate variation in closely related species. Heredity (Edinb) 107: 496–508. 10.1038/hdy.2011.44
- ↵Spencer CCA, Deloukas P, Hunt S, Mullikin J, Myers S, Silverman B, Donnelly P, Bentley D, McVean G. 2006. The influence of recombination on human genetic diversity. PLoS Genet 2: e148. 10.1371/journal.pgen.0020148
- ↵Sprenger PP, Menzel F. 2020. Cuticular hydrocarbons in ants (Hymenoptera: Formicidae) and other insects: how and why they differ among individuals, colonies, and species. Myrmecol News 30: 1–26. 10.25849/myrmecol.news_030:001
- ↵Stapley J, Feulner PGD, Johnston SE, Santure AW, Smadja CM. 2017. Variation in recombination frequency and distribution across eukaryotes: patterns and processes. Philos Trans R Soc Lond B Biol Sci 372: 20160455. 10.1098/rstb.2016.0455
- ↵Startek M, Szafranski P, Gambin T, Campbell IM, Hixson P, Shaw CA, Stankiewicz P, Gambin A. 2015. Genome-wide analyses of LINE–LINE-mediated nonallelic homologous recombination. Nucleic Acids Res 43: 2188–2198. 10.1093/nar/gku1394
- ↵Stevison LS, Hoehn KB, Noor MAF. 2011. Effects of inversions on within- and between-species recombination and divergence. Genome Biol Evol 3: 830–841. 10.1093/gbe/evr081
- ↵Sturtevant AH. 1915. The behavior of the chromosomes as studied through linkage. Z Indukt Abstamm Vererbungsl 13: 234–287.
- ↵Takahasi KR. 2009. Coalescent under the evolution of coadaptation. Mol Ecol 18: 5018–5029. 10.1111/j.1365-294X.2009.04424.x
- ↵Talbert PB, Henikoff S. 2010. Centromeres convert but don't cross. PLoS Biol 8: e1000326. 10.1371/journal.pbio.1000326
- ↵Tiley GP, Burleigh G. 2015. The relationship of recombination rate, genome structure, and patterns of molecular evolution across angiosperms. BMC Evol Biol 15: 194. 10.1186/s12862-015-0473-3
- ↵Torres AP, Höök L, Näsvall K, Shipilina D, Wiklund C, Vila R, Pruisscher P, Backström N. 2023. The fine-scale recombination rate variation and associations with genomic features in a butterfly. Genome Res 33: 810–823. 10.1101/gr.277414.122
- ↵Tupec M, Buček A, Janoušek V, Vogel H, Prchalová D, Kindl J, Pavlíčková T, Wenzelová P, Jahn U, Valterová I, 2019. Expansion of the fatty acyl reductase gene family shaped pheromone communication in Hymenoptera. eLife 8: e39231. 10.7554/eLife.39231
- ↵Turner JRG. 1967. On supergenes. I. The evolution of supergenes. Am Nat 101: 195–221. 10.1086/282485
- ↵Waiker P, de Abreu FCP, Luna-Lucena D, Freitas FCP, Simões ZLP, Rueppell O. 2021. Recombination mapping of the Brazilian stingless bee Frieseomelitta varia confirms high recombination rates in social hymenoptera. BMC Genomics 22: 673. 10.1186/s12864-021-07987-3
- ↵Wallberg A, Glémin S, Webster MT. 2015. Extreme recombination frequencies shape genome variation and evolution in the honeybee, Apis mellifera. PLoS Genet 11: e1005189. 10.1371/journal.pgen.1005189
- ↵Webster MT, Hurst LD. 2012. Direct and indirect consequences of meiotic recombination: implications for genome evolution. Trends Genet 28: 101–109. 10.1016/j.tig.2011.11.002
- ↵Weyna A, Romiguier J. 2021. Relaxation of purifying selection suggests low effective population size in eusocial Hymenoptera and solitary pollinating bees. Peer Community J 1: e2. 10.24072/pcjournal.3
- ↵White MJD. 1973. Animal cytology and evolution. Cambridge University Press, London
- ↵Wilfert L, Gadau J, Schmid-Hempel P. 2006. A core linkage map of the bumblebee Bombus terrestris. Genome 49: 1215–1226. 10.1139/g06-075
- ↵Wilfert L, Gadau J, Schmid-Hempel P. 2007. Variation in genomic recombination rates among animal taxa and the case of social insects. Heredity (Edinb) 98: 189–197. 10.1038/sj.hdy.6800950
- ↵Wright S. 1931. Evolution in Mendelian populations. Genetics 16: 97–159. 10.1093/genetics/16.2.97
- ↵Wu Y, Bhat PR, Close TJ, Lonardi S. 2008. Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet 4: e1000212. 10.1371/journal.pgen.1000212