
Measuring X-Chromosome inactivation skew for X-linked diseases with adaptive nanopore sequencing
- Sena A. Gocuk1,2,3,7,
- James Lancaster4,7,
- Shian Su4,5,
- Jasleen K. Jolly6,
- Thomas L. Edwards2,3,
- Doron G. Hickey2,
- Matthew E. Ritchie4,5,
- Marnie E. Blewitt4,5,
- Lauren N. Ayton1,2,3,8 and
- Quentin Gouil4,5,8
- 1Department of Optometry and Vision Sciences, The University of Melbourne, Parkville Victoria 3010, Australia;
- 2Centre for Eye Research Australia, East Melbourne, Victoria 3002, Australia;
- 3Ophthalmology, Department of Surgery, University of Melbourne, Melbourne, Victoria 3010, Australia;
- 4Epigenetics and Development Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria 3052, Australia;
- 5Department of Medical Biology, The University of Melbourne, Melbourne, Victoria 3010, Australia;
- 6Vision and Eye Research Institute, Anglia Ruskin University, Cambridge CB1 2LZ, United Kingdom
Abstract
X-linked genetic disorders typically affect females less severely than males owing to the presence of a second X Chromosome not carrying the deleterious variant. However, the phenotypic expression in females is highly variable, which may be explained by an allelic skew in X-Chromosome inactivation. Accurate measurement of X inactivation skew is crucial to understand and predict disease phenotype in carrier females, with prediction especially relevant for degenerative conditions. We propose a novel approach using nanopore sequencing to quantify skewed X inactivation accurately. By phasing sequence variants and methylation patterns, this single assay reveals the disease variant, X inactivation skew, and its directionality and is applicable to all patients and X-linked variants. Enrichment of X Chromosome reads through adaptive sampling enhances cost-efficiency. Our study includes a cohort of 16 X-linked variant carrier females affected by two X-linked inherited retinal diseases: choroideremia and RPGR-associated retinitis pigmentosa. As retinal DNA cannot be readily obtained, we instead determine the skew from peripheral samples (blood, saliva, and buccal mucosa) and correlate it to phenotypic outcomes. This reveals a strong correlation between X inactivation skew and disease presentation, confirming the value in performing this assay and its potential as a way to prioritize patients for early intervention, such as gene therapy currently in clinical trials for these conditions. Our method of assessing skewed X inactivation is applicable to all long-read genomic data sets, providing insights into disease risk and severity and aiding in the development of individualized strategies for X-linked variant carrier females.
X-Chromosome inactivation (XCI) is a dosage-compensation mechanism equalizing the expression of the majority of X-linked genes between males and females (Van den Veyver 2001). In 46, XX females, one X Chromosome is chosen stochastically as the inactive X (Xi) in a small number of cells early in embryonic development and remains silenced throughout life (Lyon 1961). Females are thus mosaics of cells in which either one of the two X Chromosomes is inactive. Skewed X inactivation, whereby allelic Xi status in a pool of cells departs from the predicted 50:50 ratio, may explain the phenotypic variability in female carriers of X-linked conditions, when the gene in question is subject to X inactivation (Morleo and Franco 2008). Skew may be organism-wide (global skew observable in all or most tissues if the skew is owing to biased X choice early in development), tissue-specific (when a skewed pool of cells gives rise to a tissue, a common occurrence in highly clonal tissues like blood), or geographic (spatial heterogeneity within a tissue owing to ontological patches of cells with the same Xi, like the coat of calico cats). XCI skew may be protective when the disease-causing variant is on the preferentially silenced X or may be deleterious when on the preferentially active X. Skewed X inactivation is common, with estimates that 50% of females display skews of 65:35 or greater in blood (Tukiainen et al. 2017; Shvetsova et al. 2019).
X-linked inherited retinal diseases, such as choroideremia and X-linked retinitis pigmentosa (XLRP), most strongly affect males, with night blindness and peripheral vision loss during early adolescence that progress to central vision loss at later stages of the condition. Female carriers of X-linked inherited retinal disorders present with a spectrum of disease severities, ranging from near normal retinae to severe retinal degeneration, the latter known as “male-pattern” retinal degeneration (Edwards et al. 2015; Gocuk et al. 2023). This variation and the observed prevalences are compatible with skewed X inactivation as a major factor of disease severity in females (Fry and MacLaren 2023). Retinal phenotypes of female carriers can present as a mosaic pattern comprising regional patches of degeneration owing to retinal mottling and/or pigmentary changes, which would again be compatible with a geographic XCI skew. Although it would be ideal to investigate the role of X inactivation in inherited retinal disorders directly in the affected retinal epithelium and photoreceptor cells, retinal biopsy from living patients is not justifiable owing to the risk of vision loss directly and indirectly relating to the procedure, as well as the costs and resources involved in obtaining samples (Gocuk et al. 2023).
To date, the evidence for the importance of skewed XCI for X-linked inherited retinal disorders remains limited (Fahim and Daiger 2016; Fry and MacLaren 2023). As a proxy for the retina, X inactivation skew of female carriers of X-linked inherited retinal disorders has been previously reported in blood samples, based on DNA methylation measurements. This works because the inactive X Chromosome has hypermethylated CpG islands (Tribioli et al. 1992). These studies used conventional methods based on methylation-sensitive restriction enzymes and PCR fragment-length analysis (Fahim et al. 2020). However, such methods provide incomplete information owing to the lack of informative polymorphisms found in >20% of individuals (Onozawa et al. 2015), quantitatively limited analysis of just a few CpG sites and one or two genomic loci with the potential for PCR bias, the requirement for parental DNA to determine the origin of the variant and direction of the skew, and additional genetic testing to ascertain the deleterious variant. A sequencing-based version of this test has recently been proposed (Johansson et al. 2023), directly reading the methylation of the AR and RP2 CpG islands by nanopore sequencing of Cas9-targeted native DNA. DNA modifications trigger recognizable variation in the electrical current measured at the nanopore, enabling base-level modification analysis in long reads. Although this targeted test improves quantitation accuracy, it suffers from most of the same limitations outlined above. Furthermore, blood samples have been found to increase in XCI skew with age and may therefore not accurately reflect the XCI status of the retina and other tissues (Sharp et al. 2000; Bittel et al. 2008).
We set out to develop a new method to measure X inactivation skew based on long-read whole-genome sequencing to simultaneously identify variants and investigate the contribution of skew to X-linked diseases in females. We used the Oxford Nanopore Technologies PromethION 24 platform and its adaptive sampling capacity to enrich X Chromosome reads. We sequenced peripheral samples (blood, saliva, and buccal mucosa) for a cohort of female carriers of X-linked inherited retinal disorders (choroideremia and RPGR-associated XLRP) to determine X inactivation skew in these tissues, infer the retinal skew, and correlate with disease severity and retinal phenotype. We also include a female individual who did not have an inherited retinal disorder but for whom we were able to retrieve fresh retinal tissue (following enucleation for an invasive orbital tumor), allowing a direct comparison of skew in the retina and peripheral samples. Recent advances in the development of gene therapy for retinal diseases in males have sparked interest to determine whether female carriers, particularly those at risk of severe disease, should be considered for upcoming therapeutic intervention. Accurate measurement of skewed X inactivation may assist in determining the most suitable candidates.
Results
Optimizing X Chromosome enrichment with adaptive sampling
To increase the number of samples that could be run on one PromethION flow cell while still reaching a target coverage of 15×–20× per sample, we made use of the adaptive sampling capacity of the PromethION P24 (beta feature in MinKNOW). The first few hundred bases of each read are live-basecalled and mapped to the genome reference. If the sequence maps to the X Chromosome, sequencing of that read is allowed to continue, whereas if the sequence does not map or maps elsewhere, the read is deemed off-target and its sequencing is interrupted by voltage reversal to make the pore available for another fragment (Fig. 1A). To guide protocol optimization, we estimated the relationship between read length and target coverage (see Methods). When targeting the entire X Chromosome (154 Mb), maximizing read length is beneficial, up to the point at which pore blocking and decreased pore occupancy (longer fragments mean fewer molecules) become the dominant factors in reducing the output.
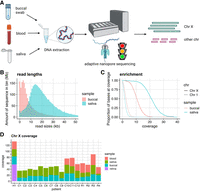
X inactivation skew sequencing workflow. (A) Adaptive nanopore sequencing of peripheral samples (blood, saliva, and buccal mucosa) to maximize coverage of the X Chromosome. Sequencing of fragments that do not map to the X Chromosome is terminated early. Degraded buccal DNA results in shorter read lengths (B) and reduced X Chromosome enrichment performance (C). The read lengths for (accepted) reads mapping to the X Chromosome are plotted for patient C9 for illustrative purposes. Rejected reads are typically <1 kb. (D) X Chromosome coverage for each patient, split by sample. At least 30× combined X Chromosome coverage was obtained for each patient. (H1) Healthy individual, (C1–C12) female carriers of choroideremia, and (R1–R4) female carriers of RPGR-associated X-linked retinitis pigmentosa.
Our sequencing results corroborated this estimate, as libraries dominated by small fragments enriched poorly for X Chromosome reads (Fig. 1B,C). Two factors contributed to small fragments dominating the sequencing: Either the DNA samples were degraded (buccal swabs had consistently worse DNA integrity than did blood or saliva) or in intact samples the unsheared, high-molecular-weight fragments could not be efficiently sequenced. Therefore, we strived to increase the average fragment lengths in the libraries by a combination of clean-ups (selective precipitation when DNA amounts allowed; bead-based clean-ups in the case of low input DNA) and shearing to 10–20 kb fragments (Fig. 1B). With optimized libraries, we obtained up to 90× coverage of the X Chromosome per PromethION flow cell, equivalent to a threefold to fourfold enrichment over nonadaptive sequencing. We obtained at least 30× total coverage of the X Chromosome for each patient, across two to four samples per patient, providing sufficient data for analysis of alleles and DNA methylation (Fig. 1D).
Calculating X inactivation skew by phasing alleles and epialleles
Next, we developed a method using the phasing of long reads into alleles (maternal and paternal, or haplotype 1 and haplotype 2 when the parent of origin is unknown) and their clustering based on DNA methylation (Xi being hypermethylated and Xa being hypomethylated) to determine skewed X inactivation (Fig. 2A). We generated a data set in which the maternal and paternal alleles are distinct by crossing two different mouse strains and in which X inactivation is genetically skewed through the inheritance of a mutated Xist (Royce-Tolland et al. 2010). We made neural stem cells from the F1 animals and sequenced the DNA by nanopore according to the ultralong sequencing protocol.

Bioinformatic workflow to measure X inactivation skew with long-read genomics, demonstrated on ground-truth data. (A) Five-step bioinformatic pipeline: basecalling and mapping reads including modified bases, SNV calling, phasing alleles and reads based on SNVs, clustering reads based on their methylation profile, and tabulating the allele/epiallele combinations to calculate the skew. (B) Validation on mouse data with genetically engineered skewed X inactivation: A deletion of the Xist A-repeat (haplotype 2) triggers its constitutive choice as the Xi, as confirmed by the DNA methylation profile of the Xist promoter (red indicates methylated CpG; blue, unmethylated CpG). (C) Example of read clustering by methylation profile for the Phf6 CpG island: reads split into two clusters, one with low methylation (CpG sites with low probability of methylation in yellow; cluster assigned as Xa) and one with high methylation (CpG sites with high probability of being methylated in blue; cluster assigned as Xi). (D) Cumulative sum of CpG islands overlapped by haplotype blocks. Large haplotype blocks overlap many CpG islands, providing additional power to estimate the X inactivation skew in those haplotype blocks. (E) CpG island methylation clustering results for one haplotype block. Each point represents a read. CpG islands with gray reads failed to split into Xi/Xa groups. (F) Nonfolded and folded histograms of the haplotype block-wise skews, binned in 10% bins and scaled by the underlying number of reads. Skew is measured as the proportion of reads supporting haplotype 1 as Xa. Because skewed X inactivation is genetically engineered in this experiment, haplotype blocks are expected to yield skews close to either zero or one. In the case of perfectly balanced X inactivation, haplotype blocks should return skews of 0.5. Folding is done around the x = 0.5 axis, to account for the random assignment of haplotype 1/2 labels to the two alleles in each block. Folding would not be necessary if we could assign haplotype 1 to a single parental haplotype consistently. (G) From the block skews, the sample skew P is calculated by maximizing the joint probability of observing the data. q = 1 − P; for each haplotype block, k is the number of haplotype 1 reads that are Xa and haplotype 2 reads that are Xi (“successes”), and n is the total number of haplotyped and clustered reads (“trials”).
After small variant calling with DeepVariant (Poplin et al. 2018) and read haplotyping with WhatsHap (Martin et al. 2016), visualization of the Xist locus confirmed the correct SNV-based separation of haplotypes with the presence of the XistΔA mutation as an ∼750-base deletion ∼120 nucleotides (nt) downstream from the Xist transcriptional start site on haplotype 2 (from the 129/Sv strain background). It also illustrated the contrasting DNA methylation patterns between the active and inactive X, correlating with the Xist gene expression: highly methylated haplotype 2 with mCpGs in red (silenced, nonfunctional Xist) and methylation-depleted haplotype 1 with CpGs in blue, expressing a functional Xist (Fig. 2B). Of note, the methylation pattern at the Xist CpG island is reversed compared with the rest of the X Chromosome CpG islands: the Xi, which expresses Xist, is unmethylated (haplotype 1), whereas the active X (Xist silenced) is the methylated allele (haplotype 2). For all other CpG islands on the X Chromosome, methylation-based clustering of reads assigns Xa to the group of low-methylation reads and Xi to the group of high-methylation reads (Fig. 2C). This clustering is performed using the density-based clustering algorithm DBSCAN within NanoMethViz (Su et al. 2021). The promoter CpG island of the X-linked Phf6 gene illustrated how reads clustered into two well-separated groups (Fig. 2C).
Independent of their methylation profile, reads were also assigned to either haplotype 1 or haplotype 2. Regions of low SNV density or regions that are highly repetitive break up the haplotype phasing into haplotype blocks. As a result, haplotype 1 in one block may be the paternal allele, whereas haplotype 1 in the next block may be the maternal allele (phase switching). Haplotype blocks are separated by genomic segments that cannot be haplotyped. In the mouse neural stem cell data set, out of 469 annotated CpG islands (UCSC mm10 annotation) in which we can expect DNA methylation on the Xi, 454 were haplotyped (Fig. 2D; Supplemental Table S1), allowing the assignment of reads to haplotype 1 and haplotype 2.
Of these haplotyped CpG islands, 105 CpG islands clustered into two methylation-level clusters, amounting to 2300 reads assigned
as Xa or Xi (Fig. 2E; Supplemental Table S1). The remaining haplotyped CpG islands were not split into Xi and Xa methylation patterns for one of several reasons: similarly
high or low methylation across all reads, lack of separation owing to continuous variation in methylation, or insufficient
read numbers to form a cluster (minimum set as five reads) (Fig. 2E). We then tallied up the reads that were both haplotypes and clustered as Xa/Xi to calculate the X inactivation skew for
each haplotype block. Large haplotype blocks may overlap multiple CpG islands, which allows pooling the counts of haplotype
1/haplotype 2 and Xa/Xi combinations for all CpG islands across the block. The haplotype block overlapping the most CpG islands
covered 80 CpG islands (Fig. 2D). The haplotype-block skew for each block i is expressed as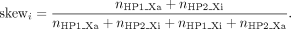 This represents the bias for haplotype 1 to be the active allele.
This represents the bias for haplotype 1 to be the active allele.
Because phase switching may occur between blocks, there is a symmetry in outcomes: preferential haplotype 1 silencing in one
block is equivalent to preferential haplotype 2 silencing in another block. A folded binomial model allows to represent this
symmetry in outcomes. In a series of binomial trials, a folded binomial model arises when the two categories of outcomes (successes
and failures) can be distinguished, but we cannot say which were the successes and which were the failures (Mantel 1970). Therefore, we fold the skew: 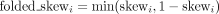 , equivalent to mirroring the skew that is above 0.5 down to below 0.5 with the symmetry axis x = 0.5 (Fig. 2F). As expected in the genetically skewed mouse NSC data set, skews for the haplotype blocks distributed around the extremes
at zero and one, and the folded distribution peaked around zero (Fig. 2F).
, equivalent to mirroring the skew that is above 0.5 down to below 0.5 with the symmetry axis x = 0.5 (Fig. 2F). As expected in the genetically skewed mouse NSC data set, skews for the haplotype blocks distributed around the extremes
at zero and one, and the folded distribution peaked around zero (Fig. 2F).
The global skew P for the sample is derived from the maximum likelihood estimate of the observed read counts in each block under the folded binomial model (Fig. 2G; Gart 1970; Mantel 1970). The numerical optimization for P for the mouse NSC data yielded a global skew of 0.031, or a 97:3 ratio allele1_Xa:allele2_Xi, close to the expected 100:0.
Measuring X inactivation skew in human retinal and peripheral samples
Having established the skew analysis pipeline on ground-truth data, we next applied it to a patient for whom we could collect retina, blood, saliva, and buccal swab samples. This allowed us to investigate the correlation in skew between tissues, as well as the spatial heterogeneity of the retina by collecting discs across four quadrants of the retina (Fig. 3A).
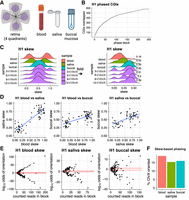
X inactivation skew across blood, saliva, buccal swab, and retina in a healthy patient. (A) Collected samples: four quadrants of the retina from one eye (purple discs), blood, saliva, and buccal swab. (B) Cumulative sum of CpG islands overlapped by haplotype blocks. (C) Nonfolded and folded distributions of haplotype blocks’ skews in the seven samples. Blood and saliva show a clear skew, whereas the buccal swab and all four retina samples are not highly skewed. (D) Positive correlation of the nonfolded haplotype blocks’ skews across samples shows that the Xa allelic bias is consistent across tissues (the same X Chromosome in the preferentially active X in all skewed tissues). (E) Skewed X inactivation allows scaffolding haplotype blocks into alleles. Log-likelihood ratio of haplotype 1 being the preferential Xa over Xi for each block. A red line is plotted at y = 1 (haplotype 1 is 10 times more likely than haplotype 2 to be the preferential Xa) and y = −1 (haplotype 2 is 10 times more likely than haplotype 1 to be the preferential Xa). (F) Up to 18% of the X Chromosome for patient H1 can be arranged in a consistent haplotype (higher-order phasing) based on haplotype block skew.
The 10–20 kb reads optimized for adaptive sampling of the X Chromosome resulted in shorter haplotype blocks than with the ultra-long read mouse data, but overall, 579 CpG islands (of 879 CpG islands) were phased into alleles (Fig. 3B; Supplemental Table S1).
The distribution of block skews revealed a strong X inactivation skew in blood (Pblood = 0.16), a slightly smaller skew in saliva (Psaliva = 0.24), and a more balanced X inactivation profile in the buccal swab and retinal discs (0.39 to 0.43) (Fig. 3C). Each quadrant of the retina had a similarly balanced X inactivation profile, suggesting a well-mixed pattern of cells with the maternal or paternal X inactivated. The lack of correlation between the blood and retinal skews cautions against the ready extrapolation of retinal skew from blood skew, although it is common practice. Here the sampling of multiple tissues enabled us to rule out an organism-wide (global) skew in favor of a tissue-specific skew.
Among the samples collected, the buccal swab is the most likely to correlate with the retina. It is less susceptible to high clonal expansion than are the cells present in blood and saliva (Tukiainen et al. 2017; Mengel-From et al. 2021). Although in our experience it was more challenging to extract high-quality DNA from it, the buccal swab may be the most informative for evaluating skew in the retina. In this particular patient's case, it was the one that most closely matched the nonskewed retinal X inactivation profile.
Using phase to orient skew and using skew to phase
Although from block to block the identity of haplotypes can switch, for a single patient the haplotype for a block is the same across tissues. Therefore, the correlation in unfolded skew between blocks informs on the direction of the skew: A positive correlation indicates the skew is in the same direction in both samples, whereas a negative correlation would indicate a reversal of skew between samples.
We observed a strong positive correlation between the blood and saliva block skews (Fig. 3D), indicating that the skew direction was the same in both highly skewed samples. The correlation with the more mildly skewed buccal swab sample remained positive, again pointing to the same direction of skew.
We then asked whether we could improve phasing of the X Chromosome by using the skew information: For a highly skewed sample, if in one block haplotype 1 is the predominantly active allele and in the next block haplotype 2 is the predominantly active allele, this would indicate that there was a phase switch between these two blocks. Therefore, the unfolded, haplotype-block skews inform which haplotypes H1/H2 belong to the same allele across haplotype blocks, and this allows “scaffolding” of the haplotypes across the chromosome into parent1/parent2 alleles. Even though the haplotype blocks remain disjointed, they can be oriented so that haplotype 1 is consistently the same parental haplotype across haplotype blocks (higher-order phasing). Strong skews gave very high certainty in orienting blocks (Fig. 3E), but even for the buccal sample, ∼16% of the X Chromosome could be phased across haplotype blocks by using skew (Fig. 3F). Orienting the skew in each block is also helpful to assess whether a particular variant is likely to be on the predominantly active or inactive X.
One outlier haplotype block with a high number of phased and clustered reads could not be confidently oriented (outlier in Fig. 3E, blood panel). Further inspection revealed that this was because of an error in variant phasing in the difficult region harboring duplicated RHOXF2 and RHOXF2B: Ambiguous read mapping over a 48.9 kb segmental duplication (Niu et al. 2011) led to a phase switch within the haplotype block (Supplemental Fig. S1). Haplotype 1 was predominantly the Xa upstream of the ambiguous region, whereas it became predominantly the Xi downstream. Thus, the skew analysis enabled the identification and correction of a variant phasing error across this complex region.
X inactivation skew in female carriers of inherited retinal disorders
Next, we applied our method of measuring X inactivation skew to a cohort of patients with inherited retinal disorders: 12 carrying variants in the CHM gene causing choroideremia and four carrying variants in the RPGR gene, a major cause of XLRP (Table 1). There were two patient pairs carrying the same variants: a mother and daughter pair, and two sisters. Seven carriers had grade 4 male-pattern degeneration (four with variants in the RPGR gene and three with variants in the CHM gene). All other carriers had variants in the CHM gene and had relatively milder retinal phenotypes: one carrier with grade 1 (fine, near normal), four with grade 2 (coarse), and four with grade 3 (geographic) (Edwards et al. 2015).
Variants in affected patients
Long-read sequencing identified disease-causing variants for all patients, including for those for whom there was no prior genetic diagnostic available. The long reads and chromosome-wide sequencing approach resolved multiple types of events: exonic SNVs, intronic SNVs predicted to affect splicing, and large structural variants removing parts of the gene (Table 1).
The availability of longer read lengths for certain samples had a strong effect on increasing the total haplotype block length and the haplotype block N50 (Supplemental Table S1). For example, for patient C9 saliva (read N50 of 18 kb), 81% of the X Chromosome and 692 CpG islands could be haplotyped. In contrast, samples with more fragmented reads and lower coverage had only 55%–58% of the X haplotyped. Higher coverage was associated with a higher proportion of haplotyped CpG islands that could be clustered into Xa and Xi reads and, thus, a higher number of informative reads for the skew calculation.
All but one of the 16 patients presented at least one sample with a skew below 0.4 (60:40 or greater) (Fig. 4A–C). For some patients, the extent of the skew varied between samples (Fig. 4A,C). For example, C3 demonstrated a highly skewed saliva sample (0.10, or 90:10), whereas her buccal swab sample had a less pronounced skew (0.33). However, when two of a patient's samples were skewed (<0.4), in all cases they were skewed in the same direction (Fig. 4B).
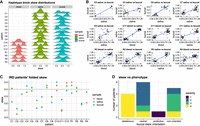
X inactivation skew in a cohort of patients with X-linked inherited retinal disorders. (A) Distribution of haplotype blocks’ skews in multiple samples from patients carrying mutations in the CHM gene (C1–C12) or RPGR gene (R1–R4). (B) Correlations of haplotype blocks’ skew between samples indicates consistency in preferential Xa choice across tissues, for each patient for whom two or more samples show skew (P < 0.4). (C) Summary of tissue skews for each patient. (D) Correlation of buccal skew with phenotypic severity. The skew is classified as either deleterious (variant allele is preferentially Xa), protective (variant allele is preferentially Xi), or neutral (no or little skew, P ≥ 0.4). For six CHM variant carriers with skewed buccal swab samples, the skew could not be oriented (nonoriented category). Retinal severity grading presented in this graph is depicted as grades 1–4 to combine retinal phenotypes for choroideremia and RPGR-associated X-linked retinitis pigmentosa (Edwards et al. 2015; Nanda et al. 2018): grade 1 is fine and normal phenotypes, grade 2 is coarse and radial pattern phenotypes, grade 3 is geographic and focal pigmentary retinopathy phenotypes, and grade 4 is male-pattern phenotypes for choroideremia and RPGR-associated X-linked retinitis pigmentosa, respectively.
To correlate skew and phenotype, we used the buccal swab skew as the most likely to reflect the retina's skew, as it is less sensitive to age-related skewing than are blood and saliva (Tukiainen et al. 2017; Mengel-From et al. 2021). Among the 10 severely affected patients (graded as 3 and 4), nine (90%) had a skew of 60:40 or greater, and five (50%) had a skew of 75:25 or greater (Fig. 4C,D). Relative to a baseline expectation of 5% of the population with skews 75:25 or greater (Tukiainen et al. 2017), we concluded that there was an overrepresentation of skew among severely affected patients (P-value = 6 × 10−5, one-sided binomial test).
We next oriented the skew relative to the allele that carries the variant. In contrast with the skew-based phasing we demonstrated previously, here we do not focus on scaffolding a consistent parental haplotype across haplotype blocks, but rather, we focus on a single haplotype block, the one that carries the disease-causing variant (e.g., on haplotype 1). Knowing the global skew for the sample and knowing the ratio of reads supporting haplotype 1 or haplotype 2 as the active X in this block, we can orient the skew relative to the variant. The skew was deemed deleterious when the predominantly active X is the one carrying the variant and as protective when the variant is instead on the predominantly inactive X (Fig. 4D). For all skewed patients with RPGR variants, the skew could be oriented, and all were deleterious (three out of three, one patient not skewed). Expressing the skew as “deleterious allele”:“healthy allele,” patients R1, R3, and R4 had ratios of 65:35, 80:20, and 64:36, respectively. For CHM variant carriers, however, skew orientation was less successful as CpG islands are sparser in this genomic region, and the length of the haplotype blocks over CHM were often limited by stretches of low complexity sequence. Out of eight skewed patients (four were not skewed), two could have their skews oriented: One was strongly deleterious (87:13), consistent with the male-pattern/grade 4 phenotype, and one was strongly protective (25:75), consistent with the mildest phenotype observed (fine phenotype/grade 1). Therefore, out of five oriented skews, all correlated with the phenotype (P-value = 0.031, one-sided binomial test). In contrast, patients with a balanced buccal swab X inactivation ratio (neutral skew) were split across phenotypic severity classification: four coarse phenotype (grade 2), one geographic phenotype (grade 3), and one male-pattern phenotype (grade 4). These data suggest that a strong deleterious skew measured in peripheral tissues could be predictive, but a neutral skew does not necessarily mean disease will not be severe.
Investigating the correlation of X inactivation skew correlation and disease severity in patient pairs
We further explored the relationship between choroideremia disease severity and X inactivation skew by focusing on a sister–sister and a mother–daughter pair, each pair sharing the same disease-causing variant as well as 50% of their genetic material (Martinez-Fernandez de la Camara et al. 2022). In addition to explaining differences in overall disease severity, skewed X inactivation may also be able to explain the geographical patterns of retinal degeneration observed in choroideremia and RPGR-linked retinitis pigmentosa (Fig. 5A,B). In particular, geographic choroideremia and focal pigmentary pattern in retinitis pigmentosa could reflect local patches of cells with elevated deleterious skews, whereas male patterns may reflect uniformly deleterious skew across the tissue.
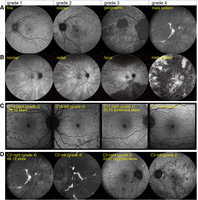
Clinical phenotypes of CHM and RPGR variant carriers and correlation with X inactivation skew. (A) Classification of retinal disease in female carriers of choroideremia based on 55° retinal fundus autofluorescence images, illustrative of the classification by Edwards et al. (2015): fine, coarse, geographic, and male-pattern degeneration. (B) Classification of retinal disease in female carriers of RPGR-associated X-linked retinitis pigmentosa based on 55° retinal fundus autofluorescence images, illustrating the classification by Nanda et al. (2018): normal, radial pattern, focal pigmentary retinopathy, and male-pattern degeneration. (C) Discordant clinical phenotypes of 34-year-old female C10 and her 59-year-old mother, C11, carrying the same pathogenic CHM variant. C10 presents with coarse (grade 2) phenotype, and C11 presents with fine (grade 1) phenotype. C11 had a pronounced buccal swab X inactivation skew (25:75) in favor of expressing the healthy allele, consistent with a milder disease phenotype. The 30° fundus autofluorescence images of right and left eyes are depicted for both carriers. (D) Discordant clinical phenotypes of 73-year-old female C2 and her 64-year-old sister, C3, carrying the same CHM variant. C2 presents with male-pattern degeneration (grade 4; picture also used to illustrate the grade 4 classification in panel A), and C3 presents with a coarse (grade 2) phenotype. C2 and C3 had skews in opposite directions, but the direction relative to the variant could not be determined. The 55° fundus autofluorescence images of right and left eyes are depicted for both carriers.
In the first pair, carrying a 4 nt deletion in CHM causing a frameshift, the nonskewed (50:50) 34-year-old female C10 presented with coarse (grade 2) phenotype, whereas her 59-year-old mother, C11, presented with a strong protective 75:25 skew had a milder fine (grade 1) phenotype (Fig. 5C,D). For a degenerative disease and given the common mutation, the mother's higher age would be expected to cause a more severe phenotype than would her daughter's; in this case, it may have been counteracted by the mother's skewed X inactivation, favoring the expression of the healthy allele, which provided her with protection against choroideremia. Although her daughter is affected, the absence of a strong deleterious skew may be beneficial in avoiding a stronger male-pattern phenotype.
In the second pair, a 1 nt deletion in the CHM coding sequence caused both of the sisters C2 (73 years old) and C3 (64 years old) to develop choroideremia, but to very different extent (Fig. 5E,F). C2 had a very strong 88:12 skew and a grade 4/male-pattern retinal degeneration, suggesting that the skew was deleterious. Unfortunately, the CHM variant was not contained within a haplotype block with informative CpG islands, so the direction of the skew could not be formally ascertained. However, we could determine that her sister, C3, had a skew in the opposite direction (33:67), and therefore protective, which matched with her much milder grade 2/coarse phenotype. These examples thus illustrate the high explanatory power of skewed X inactivation in X-linked diseases.
Discussion
In this study, we demonstrate the use of long-read genomic sequencing to measure skewed XCI. The same data also allow calling of small and large variants: We detected pathogenic intronic and exonic small variants, as well as large structural variants, in a cohort of female patients affected by X-linked retinal disorders. It confirmed previous work outlining the superiority of long reads over short reads to detect variants in the repetitive exon 15 of RPGR (Carss et al. 2017; Bonetti et al. 2023; Yahya et al. 2023; Vaché et al. 2024) or structural variants in CHM (Fadaie et al. 2021) among other genes (Nakamichi et al. 2023). Where typically multiple assays might be necessary to obtain this information (short-read exome sequencing, short-read genome sequencing, genome CNV microarray, restriction-enzyme/PCR-based tests of skew) (Caylor 2023), long-read sequencing combined with our new skew analysis method has the potential to replace all of those with one workflow, accelerating the diagnostic journey and reducing its cost. As more and more long-read population genomic projects get underway, factoring in skewed X inactivation (or epigenotype) is likely to improve the genotype–phenotype correlations in females.
Our method takes a chromosome-wide approach, drawing data from hundreds of CpG islands to derive an accurate estimate of the X inactivation skew in the sequenced sample. All samples are informative, contrary to traditional tests based on a single locus that is not sufficiently polymorphic in 20% of cases (Fahim et al. 2020; Johansson et al. 2023). By using adaptive sampling to enrich for X Chromosome reads, we obtain a threefold to fourfold reduction in sequencing cost per sample while keeping the sample preparation very straightforward (standard ligation-based library preparation protocol). Adaptive sampling is independent of methylation and therefore does not impact skew quantification. However, our analysis is equally applicable to whole-genome, unbiased sequencing as long as the X Chromosome coverage is 15 or above. This lower limit on coverage is imposed by the requirement to cluster reads overlapping CpG islands into two groups based on their methylation pattern. With lower coverage, the two groups may not be sufficiently sampled and cluster definition becomes more difficult, resulting in fewer informative CpG islands (Supplemental Table S1). When possible, sequencing longer reads (20 kb+) has multiple advantages: increases in adaptive sampling efficiency, in the overall proportion of the X Chromosome that is haplotyped, in the haplotype block lengths, in the number of haplotyped CpG islands, and in the number of CpG island–overlapping reads that are both clustered and phased. Longer reads thus provide more data for estimating the skew and also facilitate orienting the skew relative to a variant of interest.
It is important to note that skew can vary from tissue to tissue within one individual (Tukiainen et al. 2017), and certain tissues become more clonal/skewed with age (Hatakeyama et al. 2004; Juchniewicz et al. 2023). Therefore, there is a risk in extrapolating to a different, inaccessible, disease-relevant tissue. This risk can be mitigated by sampling multiple tissues. In our study, we prioritized the buccal mucosa skew to extrapolate to the retina, although the saliva and blood skews could provide additional evidence of a global, organism-wide skew and increase the confidence in the extrapolation.
Orienting the skew relative to the disease-causing variant in the absence of trio data varied in difficulty according to the locus. For a CpG island–rich, repeat-poor locus such as RPGR, in all patients (four/four) the haplotype block containing the variant spanned several informative CpG islands, and therefore, the skew could be oriented with high confidence without any trio data. In contrast, phasing of the CHM locus was often interrupted by complex regions or runs of homozygosity. With the nearest CpG island at the DACH2 promoter 300 kb away from the 3′ end of the CHM coding sequence, for six out of 12 patients the haplotype block carrying the variant did not overlap a CpG island. Two of the skews could be oriented using relatedness, but for the remaining patients, trio data would be required to definitively orient the skew. This would not require long-read sequencing but could be done simply by PCR genotyping.
Compared with transcriptome-based estimates of skewed X inactivation (Werner et al. 2022), sequencing the native genomic DNA allows orientation of the skew even for genes that are not expressed in the sampled tissue. Other benefits include a simplified sample collection and preparation, as well as the identification of all variants rather than exonic variants only.
The contribution of X inactivation skew to X-linked inherited retinal disease phenotypes in females remains understudied (Fahim and Daiger 2016; Fry and MacLaren 2023). In our cohort of 16 affected females, there was an overrepresentation of skewed X inactivation, and in all severely affected females, we found the skew orientation to be deleterious (or in one case predicted to be deleterious with high confidence). In contrast, protective skew in the female relatives of severely affected patients correlated with milder symptoms. Cumulative with previous reports of correlation between skew and RPGR-associated retinitis pigmentosa (Fahim et al. 2020), this confirmed the potential for high explanatory power of skewed X inactivation in X-linked inherited retinal disease manifestation in females and encourages investigating its use in prognostics. As gene therapy solutions for inherited retinal diseases are emerging, including for RPGR and CHM (Xue et al. 2018; Cehajic-Kapetanovic et al. 2020; Britten-Jones et al. 2022; Martinez-Fernandez de la Camara et al. 2022), early identification of candidate patients who would benefit from this treatment is crucial in order to rescue expression before irreversible retinal damage.
Besides assessing the skew for individual variants, quantifying X inactivation skew can also be used to improve phasing of the X Chromosome. For strongly skewed samples, haplotype blocks overlapping CpG islands can be linked so haplotype 1 is consistently a single parental allele. Analysis of epiallele bias may also be extended to autosomes, for instance, in pharmacogenetic panels, if there is evidence of variable expression correlating with differences in DNA methylation.
In conclusion, reporting X inactivation skew adds an important additional layer of information for all X-linked diseases or risk variants. As such, we expect our method to become part of the routine analysis pipeline for long-read population genomic data sets.
Methods
Patient sample collection
This multisite study included two cohorts of female carriers of RPGR-associated retinitis pigmentosa and choroideremia. The Australian site received ethics approval from The Royal Victorian Eye and Ear Hospital human research ethics committee (ethics ID 19-1443H). The United Kingdom site received ethics approval from the Anglia Ruskin University human research ethics committee (ethics ID ETH2223-5360). The wet laboratory component of this study received ethics approval from the Walter Eliza Hall Institute (ethics ID 20/16B). Written informed consent was obtained from each participant prior to commencement of the study, which was undertaken at the respective sites.
We used Oragene OG-600 and OCR-100 kits (DNA Genotek) for saliva and buccal mucosa collection, respectively. Patients provided saliva samples by spitting into a tube until the saliva (not foam) passed the fill line. Buccal swab involved using the cotton tip in the collection kit to rub inside the cheek (no scraping involved). The genetic variants for the participants are outlined in Table 1.
Clinical visits
The clinical component of this study was part of a larger project investigating retinal phenotypes of female carriers of X-linked inherited retinal disorders, which involved participants attending a research visit for a comprehensive ocular examination. Of interest to the current study, participants had fundus autofluorescence imaging performed to classify their retinal severity according to current grading scales for RPGR-associated retinitis pigmentosa (Nanda et al. 2018) and choroideremia (Edwards et al. 2015), and a selection of these images have been utilized in this publication.
DNA extraction
Blood, saliva, and buccal mucosa samples underwent DNA extraction at the Australian Genome Research Facility (AGRF) using a QIAgen DNeasy blood and tissue kit. Retinal DNA was extracted from 3-mm-diameter retinal discs with a QIAamp DNA micro kit.
Nanopore library preparation
Extracted genomic DNA was quantified with a Qubit, assessed for purity on a NanoDrop, and assessed for fragmentation on a TapeStation. Samples that showed excessive fragmentation were size-selected in one of two ways: when >3 µg of DNA were available, the samples were subjected to semiselective DNA precipitation (v2; https://nanoporetech.com/document/extraction-method/size-selection2) based on a method previously described (Jones et al. 2021); if <3 µg were available, we used a 0.82× high-molecular-weight bead clean-up (Promega ProNex). One microgram of DNA for each sample was then sheared to a 10–20 kb target range using Covaris g-TUBEs (two 30-sec spins at 3000g) and taken forward for library preparation following the instructions for genomic DNA sequencing with the Native Barcoding Kit 24 V14 (Oxford Nanopore Technologies SQK-NBD114.24). Three to four barcoded samples were pooled together for each library to be sequenced on one flow cell.
Nanopore sequencing
One hundred sixty nanograms of the library were loaded on R10.4.1 PromethION flow cells (FLO-PRO114M) and run to exhaustion on a PromethION P24 running MinKNOW v23.04.6, using adaptive enrichment of the X Chromosome from the T2T-CHM13v2 genome reference.
Ground-truth mouse data set with genetically skewed X inactivation
All mice were maintained and treated in accordance with approval protocols of the Walter and Eliza Hall Institute animal ethics committee (approval numbers 2018.004, 2020.048, and 2020.050). XistΔA mice (Royce-Tolland et al. 2010) were obtained from Dr. Graham Kay, Queensland Institute of Medical Research, and kept on a 129/Sv background. Triplicates of F1 129Cast neural stem cells were derived from E14.5 cortical dissections of three embryos resulting from crosses between XistΔA/+ 129 dams and Cast sires, as described previously (Jansz et al. 2018). High-molecular-weight DNA from 2 million neural stem cells was extracted with the Monarch HMW Cell kit (New England Biolab) with agitation at 300 rpm. DNA was resuspended in 750 μL EEB buffer for ultralong nanopore library preparation. Libraries were prepared with the ultralong kit (SQK-ULK001) using all the DNA from the extraction. Each sample was run on one R9.4.1 PromethION flow cell (FLO-PRO002) with two intermediate washes and reloads. Adaptive sampling was not performed for this data set. The triplicates were pooled for the skew analysis.
Adaptive sampling calibration
We start by deriving an estimate of enrichment efficiency as a factor of read lengths to inform library preparation strategy.
When enriching for a whole chromosome or in cases in which the target length is much larger than read lengths (Ltarget ≫ Lread), on-target output is maximized by having the longest read lengths possible. As reads are sampled at random, without adaptive
sampling, the proportion of the time P that a pore spends acquiring data is simply proportional to the target space (P ∝ Ltarget/Lgenome) and is independent of read lengths if the wait time between reads twait is much smaller than the time it takes to sequence a read tseq (twait ≪ tseq). However, with adaptive sampling, on average the pore has to sample noff = Lgenome/Ltarget off-target reads before finding a read that is on target. So, the proportion of time spent acquiring on-target data is reduced
to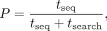 where tsearch is the time spent trying and discarding off-target reads, composed of noff repeats of treject + twait, with treject as the time it takes to sample and reject an off-target read. By denoting the sequencing speed (4∼00 b/sec) as s, we have
where tsearch is the time spent trying and discarding off-target reads, composed of noff repeats of treject + twait, with treject as the time it takes to sample and reject an off-target read. By denoting the sequencing speed (4∼00 b/sec) as s, we have 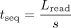 , and thus
, and thus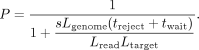 In the case of a human genome (3.1 Gb) and targeting the entire X Chromosome (154 Mb), taking treject = 2.3sec based on rejected reads of length 900 bases, and assuming twait = 0.5sec, with 1 kb reads the proportion of time spent acquiring on-target data is only P = 4.2%, whereas with 10 kb reads, it increases to 31%.
In the case of a human genome (3.1 Gb) and targeting the entire X Chromosome (154 Mb), taking treject = 2.3sec based on rejected reads of length 900 bases, and assuming twait = 0.5sec, with 1 kb reads the proportion of time spent acquiring on-target data is only P = 4.2%, whereas with 10 kb reads, it increases to 31%.
Skew analysis pipeline
Reads from POD5 files were basecalled and mapped to the T2T-CHM13v2 human genome reference using dorado v0.3.2 (https://github.com/nanoporetech/dorado) and the dna_r10.4.1_e8.2_400bps_sup@v4.2.0 model (or dna_r10.4.1_e8.2_400bps_sup@v4.0.0 for the first two flow cells that were sequenced with 4 kHz sampling). Dorado uses minimap2 (Li 2018). Modified basecalling was performed concurrently for 5mCG_5hmCG using the dorado argument ‐‐mod_bases 5mCG_5hmCG (v2 model). mosdepth v0.3.3 (Pedersen and Quinlan 2018) and NanoComp v1.21.0 (De Coster and Rademakers 2023) were used for coverage calculations and quality control. Modbam files from each patient's samples were merged to call SNVs with DeepVariant v1.5.0-gpu (Poplin et al. 2018) on the X Chromosome using the ONT_R104 model and were filtered for high-quality “PASS” calls using BCFtools (Li 2011). The variant call file (VCF) was then phased using WhatsHap v1.7 (Martin et al. 2016) using the merged modbam file. Each sample modbam (saliva, buccal, and blood and retina if available) was then processed individually by NanoMethViz v2.7.8 (Su et al. 2021) to cluster reads by their CpG island methylation pattern, using the HDBSCAN algorithm (Hahsler et al. 2019) and setting the minimum number of reads to form a cluster as five. The UCSC CHM13v2 (or mm10 for mouse) CpG island annotation was used. CpG islands for which there were exactly two clusters were used to assign Xa and Xi labels to the reads of each cluster (Xa for the low methylation cluster, Xi for the high methylation cluster, reversing these labels in the case of the XIST/Xist promoter CpG island). Within each haplotype block, the number of haplotype 1/Xa, haplotype 2/Xi, haplotype 1/Xi, and haplotype 2/Xa reads were counted to estimate the block-wise skew and to estimate the sample skew as the value that maximizes the joint probability of observing those counts over all blocks.
Given this sample skew, the log-odds of haplotype 1 being the predominantly active versus inactive X for a given haplotype block is given by logodds = (2 × successes − trials) × log10(P) + (trials − 2 × successes) × log10(1 − P).
We used R packages plyranges v1.20.0 (Lee et al. 2019) for genomic interval manipulation and cowplot v1.1.1 for visualization.
The pipeline is made available as a nextflow pipeline (https://github.com/QGouil/SkewX) and is archived as Supplemental Code.
Data access
The mouse neural stem cells ultralong nanopore gDNA sequencing data have been submitted to the EBI ArrayExpress database (https://www.ebi.ac.uk/biostudies/arrayexpress/) under accession number E-MTAB-13706. The patients’ genomic nanopore sequencing data generated in this study have been submitted to the European Genome-Phenome Archive (EGA; https://ega-archive.org/) under accession number EGAS50000000440.
Competing interest statement
Q.G. received travel support from Oxford Nanopore Technologies to attend a conference.
Acknowledgments
We thank Kelsey Breslin for the production of mouse neural stem cells; Sarah MacRaild, Stephen Wilcox, and Marek Cmero for their assistance running the PromethION; Jafar Jabbari for sharing his custom bead clean-up protocol; and Kathleen Zeglinski, Edward Yang, and the WEHI Research Computing team for their help with the Nextflow pipeline. The illustrations were created with BioRender. This study was supported by a Choroideremia Research Foundation (USA) grant to S.A.G., L.N.A., T.L.E., J.K.J., and M.E.B. Q.G., L.N.A., M.E.R., and M.E.B. are supported by Australian National Health and Medical Research Council (NHMRC) investigator grants (GNT2007996, GNT1195713, GNT2017257, and GNT1194345, respectively), a University of Melbourne Driving Research Momentum Fellowship awarded to L.N.A., and an Australian Government Research Training Program Scholarship to S.A.G. The Walter and Eliza Hall Institute and the Centre for Eye Research Australia receive support from the Victorian State Government through its Operational Infrastructure Support Program. Additional support was provided by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program and an Australian National Health and Medical Research Council IRIISS grant (9000719).
Author contributions: Study conception and design were by S.A.G., J.L., T.L.E., D.G.H., M.E.B., L.N.A., and Q.G. Data collection was by S.A.G., J.L., and J.K.J. Analysis and interpretation of results were by S.A.G., J.L., S.S., M.E.R., M.E.B., L.N.A., and Q.G. Draft manuscript preparation was by S.A.G. and Q.G. Intellectual contribution and text revisions were by S.A.G., J.L., J.K.J., T.L.E., D.G.H., M.E.R., M.E.B., L.N.A., and Q.G. All authors reviewed the results and approved the final version of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279396.124.
- Received March 22, 2024.
- Accepted September 10, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








