
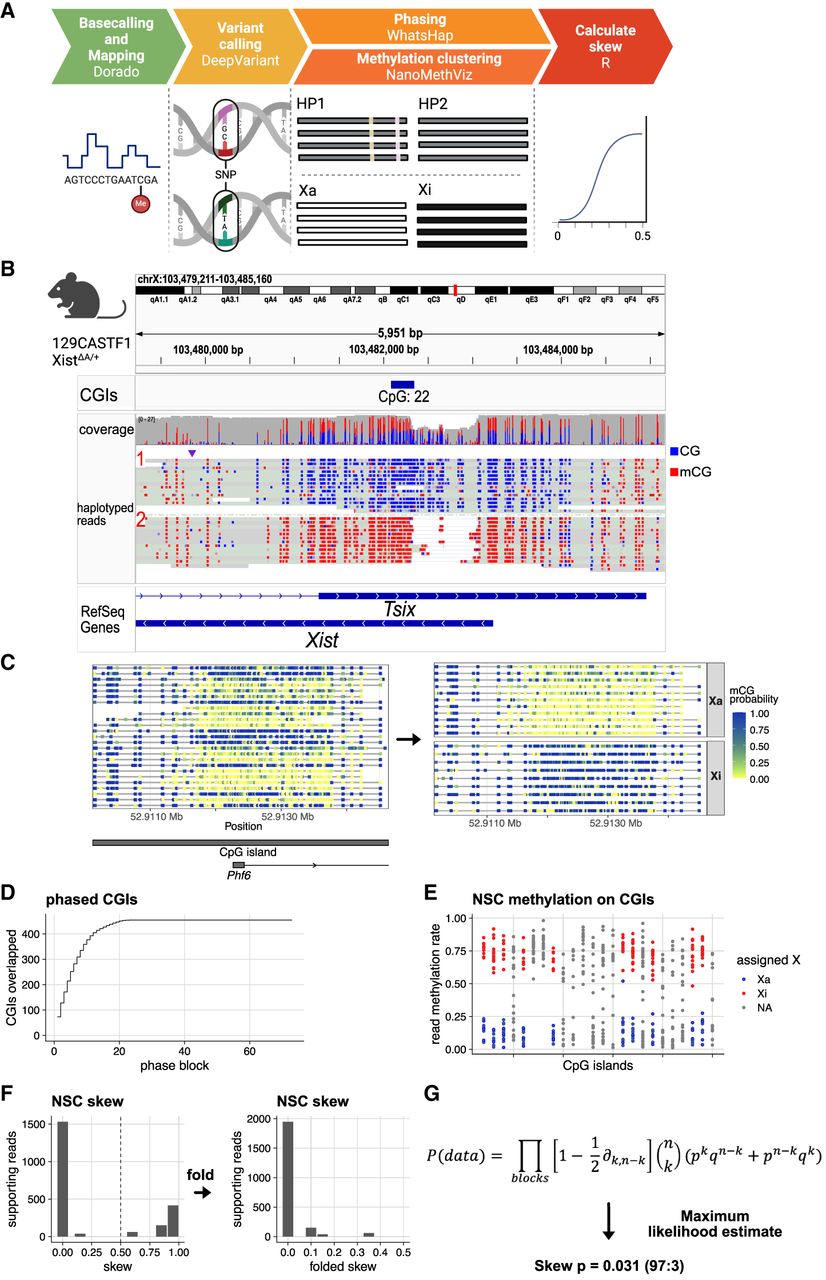
Bioinformatic workflow to measure X inactivation skew with long-read genomics, demonstrated on ground-truth data. (A) Five-step bioinformatic pipeline: basecalling and mapping reads including modified bases, SNV calling, phasing alleles and reads based on SNVs, clustering reads based on their methylation profile, and tabulating the allele/epiallele combinations to calculate the skew. (B) Validation on mouse data with genetically engineered skewed X inactivation: A deletion of the Xist A-repeat (haplotype 2) triggers its constitutive choice as the Xi, as confirmed by the DNA methylation profile of the Xist promoter (red indicates methylated CpG; blue, unmethylated CpG). (C) Example of read clustering by methylation profile for the Phf6 CpG island: reads split into two clusters, one with low methylation (CpG sites with low probability of methylation in yellow; cluster assigned as Xa) and one with high methylation (CpG sites with high probability of being methylated in blue; cluster assigned as Xi). (D) Cumulative sum of CpG islands overlapped by haplotype blocks. Large haplotype blocks overlap many CpG islands, providing additional power to estimate the X inactivation skew in those haplotype blocks. (E) CpG island methylation clustering results for one haplotype block. Each point represents a read. CpG islands with gray reads failed to split into Xi/Xa groups. (F) Nonfolded and folded histograms of the haplotype block-wise skews, binned in 10% bins and scaled by the underlying number of reads. Skew is measured as the proportion of reads supporting haplotype 1 as Xa. Because skewed X inactivation is genetically engineered in this experiment, haplotype blocks are expected to yield skews close to either zero or one. In the case of perfectly balanced X inactivation, haplotype blocks should return skews of 0.5. Folding is done around the x = 0.5 axis, to account for the random assignment of haplotype 1/2 labels to the two alleles in each block. Folding would not be necessary if we could assign haplotype 1 to a single parental haplotype consistently. (G) From the block skews, the sample skew P is calculated by maximizing the joint probability of observing the data. q = 1 − P; for each haplotype block, k is the number of haplotype 1 reads that are Xa and haplotype 2 reads that are Xi (“successes”), and n is the total number of haplotyped and clustered reads (“trials”).








