
Gapless assembly of complete human and plant chromosomes using only nanopore sequencing
- Sergey Koren1,
- Zhigui Bao2,3,
- Andrea Guarracino4,
- Shujun Ou5,
- Sara Goodwin6,
- Katharine M. Jenike7,
- Julian Lucas8,
- Brandy McNulty8,
- Jimin Park8,
- Mikko Rautiainen1,
- Arang Rhie1,
- Dick Roelofs9,
- Harrie Schneiders9,
- Ilse Vrijenhoek9,
- Koen Nijbroek9,
- Olle Nordesjo10,
- Sergey Nurk10,
- Mike Vella10,
- Katherine R. Lawrence10,
- Doreen Ware6,11,
- Michael C. Schatz7,
- Erik Garrison4,
- Sanwen Huang3,12,
- William Richard McCombie6,
- Karen H. Miga8,
- Alexander H.J. Wittenberg9 and
- Adam M. Phillippy1
- 1Genome Informatics Section, Center for Genomics and Data Science Research, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892, USA;
- 2Department of Molecular Biology, Max Planck Institute for Biology Tübingen, 72076 Tübingen, Baden-Württemberg, Germany;
- 3Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518120, China;
- 4Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, Tennessee 38163, USA;
- 5Department of Molecular Genetics, Ohio State University, Columbus, Ohio 43210, USA;
- 6Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA;
- 7Department of Computer Science, Johns Hopkins University, Baltimore, Maryland 21218, USA;
- 8Genomics Institute, University of California Santa Cruz, Santa Cruz, California 95060, USA;
- 9KeyGene, 6708 PW Wageningen, Netherlands;
- 10Oxford Nanopore Technologies, Oxford OX4 4DQ, United Kingdom;
- 11USDA ARS NEA Plant, Soil and Nutrition Laboratory Research Unit, Ithaca, New York 14853, USA;
- 12State Key Laboratory of Tropical Crop Breeding, Chinese Academy of Tropical Agricultural Sciences, Haikou, Hainan 571101, China
Abstract
The combination of ultra-long (UL) Oxford Nanopore Technologies (ONT) sequencing reads with long, accurate Pacific Bioscience (PacBio) High Fidelity (HiFi) reads has enabled the completion of a human genome and spurred similar efforts to complete the genomes of many other species. However, this approach for complete, “telomere-to-telomere” genome assembly relies on multiple sequencing platforms, limiting its accessibility. ONT “Duplex” sequencing reads, where both strands of the DNA are read to improve quality, promise high per-base accuracy. To evaluate this new data type, we generated ONT Duplex data for three widely studied genomes: human HG002, Solanum lycopersicum Heinz 1706 (tomato), and Zea mays B73 (maize). For the diploid, heterozygous HG002 genome, we also used “Pore-C” chromatin contact mapping to completely phase the haplotypes. We found the accuracy of Duplex data to be similar to HiFi sequencing, but with read lengths tens of kilobases longer, and the Pore-C data to be compatible with existing diploid assembly algorithms. This combination of read length and accuracy enables the construction of a high-quality initial assembly, which can then be further resolved using the UL reads, and finally phased into chromosome-scale haplotypes with Pore-C. The resulting assemblies have a base accuracy exceeding 99.999% (Q50) and near-perfect continuity, with most chromosomes assembled as single contigs. We conclude that ONT sequencing is a viable alternative to HiFi sequencing for de novo genome assembly, and provides a multirun single-instrument solution for the reconstruction of complete genomes.
Recently, long-read sequencing has revolutionized genome assembly, and the combination of long and accurate circular consensus sequencing (Wenger et al. 2019) with ultra-long (UL) nanopore sequencing (Jain et al. 2018b) has revealed the first truly complete sequence of a human genome (Nurk et al. 2022). In addition, trio sequencing (Koren et al. 2018; Cheng et al. 2021), Strand-seq (Porubsky et al. 2021), and Hi-C (Garg et al. 2021; Garg 2023; Lorig-Roach et al. 2024) approaches can be used to assemble phased haplotypes directly from heterozygous diploid genomes (Rautiainen et al. 2023) and are enabling comparative genomics studies of complete chromosomes (Hallast et al. 2023; Rhie et al. 2023; Makova et al. 2024). However, these approaches require input from multiple sequencing platforms: Pacific Bioscience (PacBio) for the long and accurate High Fidelity (HiFi) data (15–25 kb at 99.5% accuracy), Oxford Nanopore Technologies (ONT) for the UL data (>100 kb at 95% accuracy), and Illumina short-read sequencing for the trio, Strand-seq, or Hi-C phasing data. While this combination of data types has proven effective, it complicates data generation and limits accessibility, especially in developing countries where instrument placement is expensive and limited (Helmy et al. 2016).
Nanopore-based DNA sequencing relies on single-stranded molecules passing through a pore embedded in a membrane (Branton et al. 2008; Deamer et al. 2016). Typically, the pore has a motor protein (helicase) which serves to control the transit speed of the DNA as well as separate the double-stranded DNA into single strands. As a DNA strand passes through the pore, it creates deviations in electrical current related to its nucleotide composition, and changes in this current over time are subsequently decoded through a basecalling algorithm. This process is challenged by noise in the electrical signal and different sequence contexts that share similar current profiles (Kovaka et al. 2024).
Since the commercial release of ONT sequencing, several techniques have been proposed to combine information from both strands of a DNA molecule to increase sequencing accuracy (Jain et al. 2016). By reading both strands of a single molecule, ambiguous, or noisy signal measurements on one strand can be resolved by comparing them to the corresponding measurements on the second strand. The initial data generated for a closed Escherichia coli genome was named 2D (Loman et al. 2015), where both strands were read through the use of a hairpin adapter linking the two strands. ONT later transitioned to 1D2 which eliminated the adapter and relied instead on the physical proximity of the complementary strand to initiate sequencing. Early instances of this technology required a custom pore and library preparation and had low success rates (Wang et al. 2021), but ONT has continued to refine this process, leading to the current method of Duplex sequencing and basecalling via the Stereo Duplex algorithm (Fig. 1A). Duplex sequencing has the potential to produce highly accurate, double-stranded measurements with improved throughput and efficiency compared to the prior chemistries. With these developments, ONT now provides all three of these modes of sequencing on a single instrument: a high-accuracy protocol, named Duplex; a length-optimized protocol, named UL Simplex (Jain et al. 2018b); and a chromatin contact mapping protocol named Pore-C (Deshpande et al. 2022). This is particularly promising on the recently released Oxford Nanopore “P2” instrument, which uses the same high-throughput flow cells as the larger PromethION sequencer, although the instrument is substantially less expensive (https://store.nanoporetech.com/). Using this combination of protocols, we assess the potential of ONT sequencing alone to generate complete, telomere-to-telomere (T2T) genome assemblies.
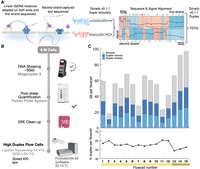
Duplex data generation. (A) Sequences with an adapter on both strands are sequenced sequentially. Once sequenced, the reads are processed using the stereo basecaller. First, each strand of the sequence is converted to basecalls using the super high-accuracy mode of the basecallers. The segmented signals and the bases of the strands are then aligned to each other and a “stereo” basecalling model is run which combines this information to give a final sequence for the double-stranded molecule. Note that the basecaller in this study was run both on the instrument to detect and call reads where both strands were sequenced as well as on the output reads marked as single-stranded to identify missed double-strand junctions. (B) The process for library preparation before sequencing. DNA is sheared to 50 kb followed by clean-up before sequencing on the PromethION. (C) The throughput and yields from the cells used for HG002 in this study. The yield in terms of total bases is indicated by the bars. After conversion to Duplex, the forward and reverse strands are combined, yielding a single read. While variable, the Duplex yield stabilized at ∼20 Gb per flow cell in the later sequencing runs with the newest flow cells (Supplemental Table 1, mustard yellow).
Results
Duplex reads
The Stereo Duplex algorithm first basecalls all DNA reads as in Simplex sequencing, producing nucleotide sequences for each read as well as a partitioning of the signal to each nucleotide, known as the move table. Second, pairs of reads passing sequentially in time through the same pore have their sequences aligned to each other. If the reads align well, they are considered a Duplex pair and proceed through the pipeline. Trimming of the reads is then performed where edges of the sequence do not align well (e.g., sequencing adapters on opposite ends). Third, the sequence alignment is used along with the move tables, which represent the approximate correspondence between signal coordinate and base-called bases, to align the first read signal to the time-reversed second read signal, inserting padding as necessary. Finally, the resulting aligned sequences, signals, and per-base quality scores are encoded as a matrix of values for input to the Stereo Duplex basecalling model (Fig. 1A). This model is very similar in architecture and implementation to models used for Simplex basecalling (https://nanoporetech.com/document/data-analysis). The primary difference is that it is trained with Duplex reads on the augmented signal/sequence input matrices. Importantly, the Stereo Duplex basecalling model functions as a de novo basecaller producing a new sequence for the Duplex molecule. It is not constrained to agree with or choose between the input Simplex sequences.
Using early access Duplex chemistry, we generated 15 PromethION flow cells of data for the well-characterized human reference genome HG002 (Fig. 1B; Zook et al. 2016; Jarvis et al. 2022; Liao et al. 2023), totaling 227 Gb or approximately 70× coverage (Supplemental Table 1). The Duplex efficiency, defined as the fraction of sequenced bases successfully converted to Duplex reads, was relatively stable with a median of 55% (Fig. 1C; Supplemental Table 1). Throughput increased over time as chemistry and library preparation improved. The last three Duplex runs using “high-yield” flow cells on this sample averaged 22 Gb (Fig. 1C). The instrument-reported Phred quality scores varied between Q10 and Q40 with a median of approximately Q30 (error rate of 0.1%), as expected (https://nanoporetech.com/about-us/news/oxford-nanopore-tech-update-new-duplex-method-q30-nanopore-single-molecule-reads-0). In contrast, the single-stranded ONT Simplex data currently averages an instrument-reported accuracy below Q20 (error rate of 1%) (Supplemental Fig. 1).
We evaluated the accuracy of Duplex sequencing using the recently released Chromosome X of HG002 (Rhie et al. 2023) and compared it to publicly available PacBio HG002 HiFi Revio sequencing data, basecalled with DeepConsensus (https://downloads.pacbcloud.com/public/revio/2022Q4/) (Fig. 2). The true read quality, as measured by alignment to the HG002 Chromosome X (Nurk et al. 2020; Rhie et al. 2023), is similar to the instrument-reported quality, with most reads falling around Q30 for both platforms (Supplemental Fig. 2). In the case of ONT Duplex, there is a broad read length distribution at this quality value, indicating no drop in quality with increasing read length. In contrast, the HiFi length distribution does not exceed ∼25 kbp and there is a negative correlation between read length and quality. The quality of the HiFi read is determined by the number of passes (Wenger et al. 2019; Baid et al. 2022), that is the number of times the polymerase can read the same molecule, with a plateau between 10 and 14 passes. Since the maximum number of bases a polymerase can read is capped by the experiment runtime, longer sequences will have fewer passes and lower consensus accuracy
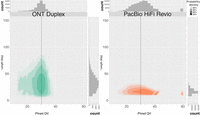
Duplex data from ONT is accurate and long. A comparison of human HG002 sequencing read length and quality for ONT Duplex (this paper) and PacBio HiFi (https://downloads.pacbcloud.com/public/revio/2022Q4/) Phred QV was measured as in Nurk et al. (2020), using the finished X Chromosome from HG002 as a ground truth (Nurk et al. 2020; Rhie et al. 2023). Plotted using R (R Core Team 2024) and ggplot2 (Wickham 2016).
Diploid human genome assembly
To evaluate assembly quality using the Duplex data, we ran the Verkko (Rautiainen et al. 2023) assembler titrating Duplex coverage from 20× to 70× in combination with 30× and 70× of UL Simplex data and trio information. Any Simplex data generated as a byproduct of the Duplex sequencing was combined with the UL Simplex data (Supplemental Table 2). We measured assembly continuity using NG50, the shortest contig such that half of the diploid genome is present in contigs of this size or greater. We also identified T2T contigs and scaffolds, that is sequences containing canonical vertebrate telomere sequences (TTAGGG) within 10 kbp of both ends, following the Vertebrate Genome Project (VGP) (Rhie et al. 2021) methodology. The assembly continuity saturated at 50× Duplex coverage, similar to HiFi data (Rautiainen et al. 2023), with the T2T contig count improving with 70× versus 30× of UL coverage (Supplemental Table 2). The Duplex assemblies exceed the T2T counts of recently published Sequel II HiFi + UL assemblies (Rautiainen et al. 2023; Cheng et al. 2024) at equivalent coverage, resolving over 50% more T2T contigs and 30% more T2T scaffolds with similar gene completeness statistics. However, the QV was five points lower (99.9997% vs. 99.9999%) and the hamming error rate for haplotype switches was approximately fourfold higher (Table 1; Supplemental Table 2).
Assemblies of the reference human genome HG002
We additionally processed the 50× assembly with Hi-C and Pore-C data using GFAse (Lorig-Roach et al. 2024) integration within Verkko (Table 1; Supplemental Table 2). The assemblies generated using either trio, Hi-C, or Pore-C information for phasing haplotypes had similar scaffold, QV, gene completeness, and T2T statistics. The assemblies achieved nearly 30 T2T scaffolds in both cases, with approximately half as many gapless T2T contigs (Supplemental Table 2).
We next investigated the chromosomes which were not completely assembled. Current tools cannot yet assemble or scaffold across the large and repetitive rDNA arrays on the human acrocentric chromosomes (13, 14, 15, 21, and 22), leaving the distal satellite region of these chromosomes unassigned and typically resulting in at least 10 incomplete chromosomes (5 per haplotype). However, in HG002, paternal Chromosome 13 has a short rDNA array with only six copies (https://github.com/marbl/HG002/blob/main/README.md) and the trio assembly was able to resolve it with a single scaffold. No previous automated HiFi + ONT assembly was able to resolve this chromosome, despite the short rDNA array and higher coverage (Jarvis et al. 2022; Rautiainen et al. 2023; Cheng et al. 2024). Excluding isolated scaffolds of the distal satellite, spanning from the short-arm telomere into the rDNA array, all nine remaining acrocentric chromosomes were resolved in the trio assembly (five as gapless contigs). In comparison, a total of 6 out of 10 distal satellites were resolved as scaffolds (four as gapless contigs) by the Pore-C assembly. The remaining nonacrocentric chromosomes had coverage gaps that were resolved by higher Duplex coverage, with the exception of Chromosome 9, which was fragmented into multiple components in all assemblies. We found that Duplex coverage dropped in the HSat3 array located on this chromosome, which has a unique inverted arrangement of repeat blocks (Altemose et al. 2022; Hoyt et al. 2022; Nurk et al. 2022) and matched a pattern of coverage dropouts at the inversion breakpoints (interestingly, only at half of the breakpoints, e.g., from rev-to-fwd but not fwd-to-rev transitions) (Supplemental Fig. 3).
Since ONT UL data require high-molecular-weight (HMW) DNA, which can be difficult to extract for certain sample types (Jain et al. 2018b), we also generated assemblies of only Duplex data. The scaffold statistics, hamming error, and QV are similar between the Duplex-only and Duplex + UL assemblies. As expected, without the UL data, the longest repeats cannot be resolved and the T2T contig count drops. Nevertheless, Duplex-only assembly improves on published HiFi-only results (Nurk et al. 2020; Cheng et al. 2021; Rautiainen et al. 2023) and provides an alternate approach for the generation of highly continuous, haplotype-resolved assemblies.
Lastly, we identified and validated centromeric arrays in these assemblies and evaluated their methylation patterns in comparison to the HG002 v1.1 assembly. Over 10 centromeric arrays were resolved without gaps in all assemblies (Chromosomes 1, 2, 7, 9, 11–13, 15, 16, 19, 21, 22, X, and Y) (Supplemental Figs. 4–17). As an example, Figure 3 and Supplemental Figure 15 show the methylation, self-similarity (Vollger et al. 2022), and NucFreq (Vollger et al. 2019; Mc Cartney et al. 2022) plots for the Chromosome 22 centromeric array. NucFreq supports the correctness of these arrays, with the exception of local increases of second-most frequent variants, likely due to the lower QV and higher hamming error rate of the ONT-only assemblies. However, the assembled haplotypes and methylation patterns are consistent in all assemblies with the reference HG002 assembly.
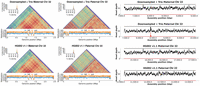
ONT-only assemblies accurately resolve centromeric arrays for both haplotypes. The figure shows StainedGlass (Vollger et al. 2022) and methylation plots for Chromosome 22 of HG002 on the left and NucFreq (Vollger et al. 2019; Mc Cartney et al. 2022) validation using HiFi sequencing (Jarvis et al. 2022; Liao et al. 2023) on the right. The top row shows the 50× Duplex + 30× UL + trio assembly while the bottom is the HG002 v1.1 reference assembly (https://github.com/marbl/HG002/blob/main/README.md). The alpha satellite repeat pattern is consistent between both assemblies for both haplotypes. The methylation pattern, including the location of the centromeric dip region (CDR) (Logsdon et al. 2021; Altemose et al. 2022; Gershman et al. 2022), is also consistent between assemblies. Lastly, NucFreq shows the assembly is overall accurate, with a few local quality issues indicated by an increase in secondary allele frequency (red), likely due to a missing centromeric repeat unit caused by the lower read accuracy.
Near T2T agricultural genomes
To demonstrate the utility of Duplex sequencing beyond human genomes, we selected two important agricultural genomes, Solanum lycopersicum Heinz 1706 (tomato) and Zea mays B73 (maize), and sequenced them to approximately 40× Duplex coverage each. In addition, we generated 30× and 16× of 100 kbp or longer UL data for tomato and maize, respectively (Supplemental Fig. 18; Supplemental Tables 3–5). Since both of these strains are inbred and almost fully homozygous, there was no need for Pore-C or trio data.
Both tomato and maize assemblies were highly continuous with N50s of 63.8 Mbp and 152.5 Mbp, respectively, exceeding their current reference assembly N50s of 41.7 Mbp (SL5, based on PacBio HiFi data) (Zhou et al. 2022) and 47.0 Mbp (Zm-B73-NAM-5.0, based on PacBio CLR long reads and BioNano optical maps) (Hufford et al. 2021). The tomato assembly resolved 5 of 12 chromosomes as T2T contigs while maize resolved 2 of 10 chromosomes. As with HG002, we investigated the source of the remaining gaps. In the tomato, Chromosome 2 harbored a complex unresolved repeat, corresponding to the 45S rDNA array, which has been estimated at 2300 (Ganal et al. 1988) copies or over 20 Mbp in size (Fig. 4). Chromosomes 11 and 12 shared high similarity in a peri-telomeric region that could not be resolved, and Chromosome 3 had a gap in an AT-rich region that was only spanned by a single ONT UL read. Chromosomes 8, 9, 10, 11, and 12 had seven unresolved regions which could not be resolved using Duplex and ONT UL data alone. Manual inspection indicated three were due to retained heterozygosity while the remaining four were due to low-coverage Duplex sequencing, retaining error in the assembly graph (Fig. 4).
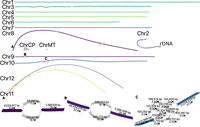
Duplex + UL assembly graph for S. lycopersicum before manual resolution. In the tomato assembly graph, most chromosomes are linear and fully resolved, except for regions of remaining heterozygosity (highlighted in red boxes): the shared sequence between Chromosomes 11 and 12 (red box bottom left), a gap on Chromosome 3, and the 45S rDNA array on Chromosome 2. ChrCP denotes the chloroplast and ChrMT denotes the mitochondria genomes, respectively. The callouts (A–C) show some unresolved structures in detail. The simple bubble on Chr 8 (A) and a simple bubble on Chr 9 (B) were resolved by picking a random haplotype. The region on Chr 10 (C) corresponds to a low-coverage Duplex region, indicated by low coverage on the nodes. These regions were gap-filled using ONT UL sequences, generating additional noise in the graph. This prevents automated resolution which requires support from at least twice as many ONT UL reads as the next best. A path consistent with the largest number of ONT UL sequences was selected.
In the maize assembly, three chromosomes (Chr 1, 8, and 9) had four unresolved regions of heterozygosity (Fig. 5). Chromosome 6 had a complex repeat, again corresponding to the rDNA array. Unlike the tomato, there were multiple coverage gaps in several chromosomes (Chr 1, 2, and 4) (Fig. 5). These regions intersect current gaps in the Zm-B73-NAM-5.0 reference assembly (Supplemental Fig. 19) and the sequence surrounding these gaps is AT-rich. We compared these regions to the recently published T2T assembly of a different maize line (Mo17) sequenced using HiFi and ONT UL data (Chen et al. 2023) and found that these locations corresponded to gaps and a low-coverage region in the initial UL ONT assembly. The resolved sequence was high in AT-repeats and neither the Duplex nor the UL data covered the regions in question. While we cannot be sure that the lack of coverage is due to a difference between maize lines, given the coincidence of gaps in our assembly, the initial ONT-based Mo17 assembly, and the Zm-B73-NAM-5.0 reference, it is likely that sequencing bias is causing coverage dropouts and the resulting gaps.
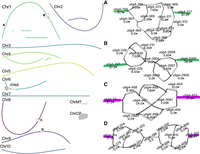
Duplex + UL assembly graph for Z. mays before manual resolution. In the maize assembly graph, most chromosomes are linear and resolved, except for regions of unresolved repeats (highlighted in red boxes), gaps on Chromosomes 1, 2, and 4, and the 45S rDNA array on Chromosome 6. ChrCP denotes the chloroplast and ChrMT denotes the mitochondria genomes, respectively. The callouts (A,B,C,D) correspond to low-coverage Duplex region, indicated by low coverage on the nodes. These regions were gap-filled using ONT UL sequences, generating additional noise in the graph. In each case, the path agreeing with the majority of ONT UL read alignments was selected for resolving the tangle. One end of Chromosome 3 was missing a telomere which was incorporated using ONT UL read consensus.
Starting with the above assemblies, we performed manual curation of the assembly graphs to resolve the remaining heterozygosity, resolved any cross-chromosome homology via ONT UL alignments, and performed targeted assembly of the chloroplast, mitochondria, and rDNA sequences (Rautiainen 2024). As a final step, we used DeepVariant (Poplin et al. 2018) with Duplex data to polish the consensus sequence. The resulting assemblies are nearly T2T with only 20 and 26 contigs for tomato and maize, respectively. Consensus sequence accuracy exceeds 99.999% (Table 2). The relatively lower QV for tomato is due to errors at the ends of Chromosomes 11 and 12 (Fig. 4) where Duplex coverage was low and the consensus relied solely on ONT UL reads. The last 250 kbp of these two chromosomes accounts for 78% of their error and 45% of the total assembly errors. Excluding these two regions, the QV increases from 51.81 to 54.41. The assemblies were colinear with previous references (Supplemental Figs. 20, 21) while adding missing sequences (Supplemental Figs. 19, 22). We also evaluated the structural accuracy of the assemblies using polishing scripts from the T2T-CHM13 project (Mc Cartney et al. 2022) and VerityMap (Mikheenko et al. 2020), and identified <1% of the assembled bases as potential issues. The majority of flagged regions were localized near gaps or rDNA, as expected.
Duplex + ultra-long curated assembly statistics for S. lycopersicum and Z. mays compared to existing reference genomes
Discussion
Here, we have demonstrated the complete assembly of human and plant chromosomes using a single sequencing platform. The high accuracy of ONT Duplex data (exceeding 99.9%) makes it a suitable alternative to PacBio HiFi data for the construction of genome assembly graphs that can then be untangled with the integration of ONT UL reads and, if needed, haplotype phased using ONT Pore-C reads. The ability to generate all three of these data types, originating from diverse species, on a single sequencing instrument greatly simplifies the overall workflow and has the potential to democratize access to the construction of high-quality reference genomes. Sarashetti et al. (2024) independently evaluated ONT Duplex data on a different human sample with a different assembler, hifiasm (Cheng et al. 2024) instead of Verkko (Rautiainen et al. 2023). However, they mirror our conclusions that ONT Duplex read quality is similar to HiFi and it can produce more continuous assemblies than HiFi. Applying this sequencing recipe to human, tomato, and maize genomes, we show that the resulting assemblies exceed the continuity of reference genomes and state-of-the-art approaches, albeit with a modestly lower final assembly consensus quality. While we observed higher hamming rates for ONT Duplex assemblies, this appears to be an issue with assembly base accuracy in homopolymer stretches. When evaluated on homopolymer-compressed assemblies using homopolymer-compressed parental markers, the hamming error rate for HiFi and Duplex assemblies are equivalent. We expect continuing improvements in read quality and improved models for postassembly polishing will close this gap in the future.
It is important to recognize that our study used a prerelease version of Duplex sequencing, with a large variability in Duplex throughput (6 ± 3 Gbp) and conversion rate (26% ± 18%). The production high-yield flow cells were more stable with a Duplex throughput of 18 ± 5 Gbp and conversion rate of 51% ± 12%. We also observed sequencing biases, most notably on HSat3 on human Chromosome 9 (Supplemental Fig. 3) but in other regions as well (Supplemental Fig. 23). Similar, context-specific biases are a common issue for other sequencing technologies as well, e.g., GC bias for Illumina sequencing (Ross et al. 2013), GA bias for HiFi sequencing (Supplemental Fig. 23A; Nurk et al. 2020), and AT bias in older HiFi sequencing kits (Rhie et al. 2023). Some of the ONT biases we identified were successfully addressed by updated versions of the sequencing and basecalling methods. Additionally, the accuracy of ONT Simplex sequencing is also rapidly improving, with Simplex quality scores now reaching Q28 (https://labs.epi2me.io/gm24385_ncm23_preview/). A recent preprint (Stanojević et al. 2024) demonstrated that Simplex data can be corrected to a similar quality as PacBio HiFi. This enables de Bruijn-style assembly graph construction using Verkko or LJA (Bankevich et al. 2022) directly from Simplex data with similar T2T counts and QV to Duplex data (Stanojević et al. 2024). This can obviate the need for ONT Duplex sequencing. Regardless, we expect continued improvements in long-read quality and throughput to further reduce the barriers to complete genome assembly. When combined with the affordable yet high-throughput Oxford Nanopore P2 sequencer, the single-instrument, T2T assembly recipes presented here open the exciting possibility of personalized human genomes and complete genomes for any other species, in any country and potentially any institution in the world.
Methods
Sequencing and basecalling
HG002
HG002 cell line was purchased from Coriell Institute (GM24385) and cultured in RPMI-1640 media with 2 mM l-glutamine and 15% FBS at 37°C, 5% CO2. HMW DNA was extracted from cells using NEB Monarch HMW DNA Extraction Kit for Tissue (NEB T3060). Isolated DNA was then sheared using the Diagenode Megaruptor 3, DNAFluid+ Kit (E07020001). The size of sheared DNA fragments was analyzed on an Agilent Femto Pulse System using the Genomic DNA 165 kb Kit (FP-1002-0275). The fragment size distribution of postsheared DNA had a peak at ∼50 kbp. Small DNA fragments were removed from the sample using the PacBio SRE Kit (SKU 102-208-300). Library preparation was carried out using Oxford Nanopore Technologies’ Ligation Sequencing Kit V14 (SQK-LSK114). PromethION high duplex flow cells were provided by ONT for sequencing on the PromethION 48 sequencer. Three libraries were prepared per flow cell. Flow cells were washed using ONT's Flow Cell Wash Kit (EXP-WSH004) and reloaded with a fresh library every 24 h for a total sequencing runtime of 72 h. HG002 data were basecalled using Duplex Tools (v0.2.20) and Dorado v0.1.1 (https://github.com/nanoporetech/dorado) with the following commands:
-
# Simplex calling
-
## FAST5 files were converted to POD5 and then grouped by channel with:
-
pod5 convert fast5 ‐‐force-overwrite ‐‐threads 90 ${FAST5}/*.fast5 ${POD5}/output.pod5
-
pod5 subset ‐‐force_overwrite ‐‐output ${POD5_GROUPED} ‐‐summary $SEQSUMMARY ‐‐columns $POD5_GROUPING -M ${POD5}/output.pod5
-
## Call Simplex data with Dorado:
-
MODEL_PATH=“dorado_v4_duplex_beta_models/dna_r10.4.1_e8.2_400bps_sup@v4.0.0”
-
dorado basecaller -x “cuda:all” $MODEL_PATH $POD5_GROUPED > ${OUTPUT}/${output_name}_Dorado_v0.1.1_400bps_sup.sam
-
# Duplex calling:
-
duplex_tools pair ${OUTPUT}/${output_name}_Dorado_v0.1.1_400bps_sup.bam
-
dorado duplex ${MODEL_PATH} $POD5_GROUPED ‐‐pairs ${OUTPUT}/pairs_from_bam/pair_ids_filtered.txt > ${OUTPUT}/${output_name}_Dorado_v0.1.1_400bps_sup_stereo_duplex.sam
-
## Read rescue and duplex calling on rescued reads:
-
## For extra duplex, first fast-call (with ‐‐emit-moves)
-
FAST_MODEL_PATH=“dorado_v4_duplex_beta_models/dna_r10.4.1_e8.2_400bps_fast@v4.0.0”
-
dorado basecaller ${FAST_MODEL_PATH} ${POD5} ‐‐emit-moves > ${OUTPUT}/${output_name}_unmapped_reads_with_moves.sam
-
## Second, use duplex tools split pairs to recover non-split duplex reads
-
duplex_tools split_pairs ${OUTPUT}/${output_name}_unmapped_reads_with_moves.sam ${POD5} pod5s_splitduplex/
-
## Finally, duplex-call with sup
-
dorado duplex ${MODEL_PATH} pod5s_splitduplex/ ‐‐pairs split_duplex_pair_ids.txt > ${OUTPUT}/${output_name}_duplex_splitduplex.sam
-
## SAM files were converted to BAM and filtered using SAMtools
More recent versions of Dorado have incorporated read rescue and allow basecalling with a single command.
Tomato
For tomato Heinz1706 (2n = 2x = 24 [The Tomato Genome Consortium 2012] also available as CGN15437) young seedlings were grown and young leaves were bulk harvested. HMW DNA was extracted by KeyGene using nuclei isolated from frozen leaves ground under liquid nitrogen, as previously reported (Zhang et al. 2012; Datema et al. 2016).
Library preparation was carried out using the ligation sequencing kits (Oxford Nanopore Technologies) SQK-LSK112 for two R10.4 (translocation speed 260 bps) PromethION flow cells. Constructed libraries were loaded on R10.4 FLO-PRO112 flow cells and sequenced on PromethION P24 sequencer using the super accuracy model (Supplemental Table 3). See also the data release at https://www.keygene.com/newsitem/fast-contiguous-and-accurate-arabidopsis-col-0and-tomato-heinz-1706-genome-assembly-thanks-to-new-chemistry-nano-pores-and-plant-trained-basecaller.
In addition, seven R10.4.1 FLO-PRO114 PromethION flow cells were run in which the library preparation was carried out using the ligation sequencing kit (Oxford Nanopore Technologies) SQK-LSK114 (Supplemental Table 3). Finally, three high duplex PromethION flow cells were run in which the library preparation was carried out using the ligation sequencing kit (Oxford Nanopore Technologies) SQK-LSK114 (Supplemental Table 3). One HMW DNA sample was fragmented and SRE (circulomics) treated, other two samples were unfragmented and not SRE treated.
The data were basecalled using Duplex Tools (v0.2.20) and Dorado v0.1.1 following the same steps as HG002.
To generate ONT UL data, HMW DNA was extracted by the SDS method without purification step to sustain the length of DNA; 8–10 μg of gDNA was size selected (>50 kb) with SageHLS HMW library system and processed using the Ligation Sequencing 1D kit (SQK-LSK109) and sequenced on the PromethION P48 at the Genome Center of GrandOmics. The data from five PromethION cells were basecalled using Guppy 6.5.7 with SUP mode (Supplemental Table 4).
Maize
For maize B73 (2n = 2x = 20, PI550473) young seedlings were grown and young leaves were bulk harvested. HMW DNA was extracted by KeyGene using nuclei isolated from frozen leaves ground under liquid nitrogen, as previously reported (Zhang et al. 2012; Datema et al. 2016). Library preparation was carried out using the ligation sequencing kits (Oxford Nanopore Technologies) SQK-LSK112 for a total of 22 R10.4 (translocation speed ∼260 bps) PromethION flow cells. Constructed libraries were loaded on R10.4 FLO-PRO112 flow cells (Supplemental Table 5). These data were basecalled into FASTQ reads, using Guppy v.6.0.1 with the “sup” accurate models, “dna_r10.4_e8.1_sup.cfg” for R10.4 reads. Duplex calling was performed using Duplex Tools v0.2.7 followed by Guppy v6.0.0. See also the data release at https://www.keygene.com/newsitem/maize-b73-oxford-nanopore-duplex-sequence-data-release.
In addition, five R10.4.1 FLO-PRO114 PromethION flow cells were run in which the library preparation was carried out using the ligation sequencing kit (Oxford Nanopore Technologies) SQK-LSK114 (Supplemental Table 5). Finally, one high duplex PromethION flow cell was run in which the library preparation was carried out using the ligation sequencing kit (Oxford Nanopore Technologies) SQK-LSK114 (Supplemental Table 5). The data were basecalled using Duplex Tools (v0.2.20) and Dorado v0.1.1 following the same steps as HG002.
To generate ONT UL data, HMW DNA was extracted according to Bionano Prep Plant Tissue DNA Isolation Base Protocol (Bionano Genomics doc#30068) utilizing a gel-plug based extraction. Library preparation was performed using the Ultra-Long DNA Sequencing Kit (SQK-ULK001) compatible with the R9.4.1 flow cells and the Ultra-Long DNA Sequencing Kit V14 (SQK-ULK114) compatible with the R10.4.1 flow cells. Data from a total of 14 PromethION cells were generated (Supplemental Table 4). The data were basecalled using Dorado (version 0.2.1 + c70423e) in super accuracy mode.
Statistics for yield and Duplex conversion rate were calculated for all samples in pre-high-yield cells using R (R Core Team 2024) summary and sd functions. High-yield flow cell statistics excluded two tomato cells which were run without DNA shearing or the use of the SRE kit.
Assembly
Assemblies were generated with Verkko v1.3.1 (Rautiainen et al. 2023). Duplex data were provided using the ‐‐hifi parameter. We observed chimeras in Simplex sequences from the Duplex runs. Similar to the chimera in CLR where a SMRTbell adapter is not found (Eid et al. 2009; Koren et al. 2017), a read combining both strands corresponds to a missed read end signal. Rather than joining the two strands and calling a single Duplex read, a chimera Simplex read is output. The chimera for HG002 was not random, with consistent chimera at telomeric ends. To avoid introducing these systematic errors into the assembly, all Simplex data generated from Duplex cells was filtered for a telomere signal in the middle of the sequence using the VGP pipeline (Rhie et al. 2021). These reads, along with the ONT UL, were then input using the ‐‐ont option to Verkko with the command:
-
verkko ‐‐hifi {duplex reads} ‐‐nano {ont reads} -d asm ‐‐screen human ‐‐unitig-abundance {minimum coverage, see below} ‐‐hap-kmers maternal.k30.hapmer.meryl paternal.k30.hapmer.meryl trio
For HG002, the Minimizer based sparse de Bruijn Graph constructor (MBG) (Rautiainen and Marschall 2021) parameter ‐‐unitig-abundance was changed from the default of 2 based on the Duplex coverage, using 2 for 20×, 30×, 3 for 40×, and 4 for >40×. We ran GFAse (Lorig-Roach et al. 2024) using the Verkko wrapper (https://github.com/skoren/verkkohic). First, we reran the assembly without trio information to generate consensus, reusing steps 0-correction through 5-untip from the trio run with the commands:
-
mkdir asm_notrio
-
cd asm_notrio
-
ln -s ../asm/1-buildGraph
-
ln -s ../asm/2-ProcessGraph
-
ln -s ../asm/3-align
-
ln -s ../asm/4-processONT
-
ln -s ../asm/5-untip
-
cd ..
-
<path to verkko>/verkko ‐‐hifi {duplex reads} ‐‐nano {ont reads} -d asm_notrio ‐‐screen human ‐‐unitig-abundance {abundance value}
Followed by GFAse git tag f19f969cfe5da51b841c3222faec32bdf6c95e6c
-
export VERKKO=<path to verkko>/verkko-v1.3.1/
-
export GFASE=<path to GFAse>/GFAsebuild/
-
bash $GFASE/gfase_wrapper.sh/gfase_wrapper.sh asm_notrio asm_gfase `pwd`
For both Hi-C and Pore-C data. For maize and tomato, we removed the ‐‐screen human option and used ‐‐unitig-abundance 4 for both. Maize included the ‐‐copycount-filter-heuristic option to MBG.
Manual curation was based on inspecting the assembly graph output by Verkko. Retained heterozygosity was assumed when node coverage indicated an approximately 50/50 split of average genome-wide coverage. Low-coverage nodes compared to expectations were assumed to indicate sequencing error and/or sequencing dropout. To resolve such regions, we generated multiple candidate paths and evaluated their support using ONT UL sequences as described in Nurk et al. (2022). A similar strategy was used to resolve the ends of Chromosomes 11 and 12 in tomato, extending the consensus with ONT UL reads to include the canonical telomere repeat. No nodes produced by the assembler were split during curation.
Data and assemblies are mirrored at https://obj.umiacs.umd.edu/marbl_publications/duplex/index.html.
Previously generated HG002 HiFi + ONT assemblies were downloaded from https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=submissions/53FEE631-4264-4627-8FB6-09D7364F4D3B‐‐ASM-COMP/HG002/assemblies/, hifiasm*0.19.5 and verkko*1.3.1. HG002 Pore-C data downloaded from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under acccession number SRR27664048 (87×), HG002 Hi-C data downloaded from https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=NHGRI_UCSC_panel/HG002/hpp_HG002_NA24385_son_v1/hic/downsampled/*NovaSeq* (71×). Tomato HiFi data were downloaded from SRA accession number SRR15243707. Maize HiFi data were downloaded from https://downloads.pacbcloud.com/public/revio/2023Q1/maize-B73-rep1/.
Validation
Switch and hamming errors were measured using yak (Liao et al. 2023). HG002 QV were measured using Merqury with a k = 21 Illumina k-mer database. For maize and tomato, we built databases from both HiFi and Illumina data, removed any k-mers occurring only once in either, and merged them to create a hybrid database.
Missing and duplicated gene stats were computed using compleasm git tag dbf13d032cd6790c7d992f993abc3b604acc5cea (Huang and Li 2023) with the primate ODBv10 lineage (Zdobnov et al. 2021).
-
miniprot -u ‐‐outs=0.95 -t8 ‐‐gff assembly.haplotype[12].fasta primates_odb10/refseq_db.faa.gz>assembly.haplotype[12].aln.gff
-
python3 AnalysisMiniprot.py -g assembly.haplotype[12].aln.gff ‐‐full_table_file assembly.haplotype[12].full_table.tsv ‐‐complete_file assembly.haplotype[12].summary.txt
The missing column combines the total genes reported and either missing or fragmented. T2T contigs were identified using VGP telomere scripts (Rhie et al. 2021) with the telomere sequence of TTAGGG for human and TTTAGGG for tomato/maize within 10 kb of the chromosome ends and longer than 1000 bp. To confirm the telomere arrays, we identified are true telomere ends, we checked the reference assemblies for human (HG002 v1.1), tomato (SL5) (Zhou et al. 2022), or maize (Zm5.0, Mo17) (Hufford et al. 2021; Chen et al. 2023). With the exception of Chr 2 in maize, which was not considered T2T in our assembly, we did not identify any telomeric signal longer than 1000 bp outside of the chromosome ends. Any scaffolds with gaps were counted toward T2T scaffolds while those without gaps were counted as T2T contigs. rDNA was identified by mapping a canonical unit (KY962518.1) (Kim et al. 2018) with mashmap v2.0 (Jain et al. 2018a) and retaining any match with >95% identity and 10 kbp length. We required that an assembly extended from the q-arm of the acrocentric chromosome to include at least 10 kbp of an rDNA repeat unit to consider a chromosome resolved. However, the assembly could include gaps and thus not have base-level representation of all regions before the rDNA, such as the centromere or proximal junction (PJ).
NGA50 was computed with the Q100 toolkit (https://github.com/nhansen/q100bench; Supplemental Code) git commit 20787e46dd2cab2679e682017cd6213d4e308e99. Assemblies were split at gaps of at least three Ns and assessed with the commands:
-
seqtk cutN -n 3 assembly.haplotype[12].fasta |sed ‘s/:/_/g’ |sed ‘s/-/_/g’ > assembly.haplotype[12].ctgs.fasta
-
minimap2 -t $cores -I 8G -ax asm5 v1.0.1.fasta.gz assembly.haplotype[12].ctgs.fasta | samtools sort -O bam>assembly.haplotype[12].bam
-
samtools index assembly.haplotype[12].bam
-
q100bench -b assembly.haplotype[12].bam -r v1.0.1.fasta.gz -q assembly.haplotype[12].ctgs.fasta -p assembly.haplotype$i -A “v1.0.1” -B “$prefix.haplotype[12]”
And the NGA50 statistics were calculated by taking the alignment block lengths from both haplotype's test*covered.v1.0.1.merged.bed file, sorting by length, and finding the NGA50 using G set to 6.08 Gbp.
Two reference-free validation methods were run on the tomato and maize assemblies. T2T-Polish (https://github.com/arangrhie/T2T-Polish) (Mc Cartney et al. 2022) was used to align both ONT Duplex and HiFi reads to the assembly with the commands:
-
T2T-Polish/pattern/microsatellites.sh asm.fasta
-
T2T-Polish/winnowmap/_submit.sh asm.fasta hifi|duplex map-pb
-
T2T-Polish/coverage/issues.sh hifi|duplex.pri.paf t2t_asm asm HiFi
The microsatellites.sh script creates IGV-compatible tracks (Robinson et al. 2011) of different dinucleotide frequencies in the reference shown in Supplemental Figure 23. The issues were merged if they overlapped by at least 50% in both intervals using the BEDTools merge intersect (Quinlan and Hall 2010). Errors in alternate/unassigned sequences, mitochondria, chloroplast, and a canonical rDNA unit were excluded. Total bases in issues were summed to report the fraction of bases with potential issues.
Second, VerityMap (Mikheenko et al. 2020) git commit d24aa797be9c977dbcb9164ecfe18b3af6e4a026 was run using HiFi data available for each data set with the command:
-
veritymap ‐‐reads hifi.reads.fastq -d hifi -haploid-complete -t 32 -o output_asm
We reported errors by counting entries in the <asm>_kmers_dist_diff.bed when the allele frequency was at least 25 and the length of error was at least 2 kbp. We attempted to run VerityMap on HG002 with reads partitioned by haplotype but the program did not complete after running for more than 2 weeks on 32 cores.
We also validated our assemblies against the existing reference to test for large-scale rearrangements. We aligned the published genomes (Zm-B73-v5 and SL5) to our assemblies with minimap2 (Li 2018, 2021) v2.26 with the options—eqx -ax asm5 and called variants by SyRI v1.6.3 (Goel et al. 2019).
Methylation processing and visualization
HiFi BAM and ONT FASTQ files with 5mC methylation calls as MM and ML tags were aligned against the generated assemblies and HG002 v1.1 using pbmm2 v.1.13.0 (for HiFi reads) and Winnomap v2.03 (Jain et al. 2022) (for ONT reads). The alignments were then converted to sorted BAM files containing only primary mappings:
-
# HiFi reads
-
pbmm2 align {genome}.mmi {bam_with_meth_calls} -j 42>{output.bam}
-
samtools view -@ 24 -Sb -F 2048 {output.bam} |samtools sort -@ 24 -T {temporary_directory} ->{output.bam}
-
samtools index {output.bam}
-
# ONT reads
-
winnowmap -t 48 -W {genome}_repetitive_k15.txt -ax map-ont -y {assembly_fasta} {fastq_with_meth_calls}>{output.sam}
-
samtools view -@ 24 -Sb -F 2048 {output.sam} |samtools sort -@ 24 -T {temporary_directory} ->{output.bam}
-
samtools index {output.bam}
Aggregated methylation percentages at all CpGs were obtained using modbam2bed v0.10.0 (https://github.com/epi2me-labs/modbam2bed) with bases with >0.8 probability called “methylated” and bases with <0.2 probability called “unmethylated”:
-
modbam2bed -t 48 -e -m 5mC ‐‐cpg -a 0.20 -b 0.80 {assembly_fasta} {output.bam}>{output.bed}
Finally, we used StainedGlass (Vollger et al. 2022) to generate and visualize centromere similarity heatmaps with the aggregated methylation profiles on the bottom.
Annotation
Transposable elements were annotated using EDTA v2.1.5 (Ou et al. 2019) with curated TE libraries from maize and tomato, respectively. LTR assembly index (LAI) was calculated using LAI beta3.2 (Ou et al. 2018) and standardized using parameters of -iden 94.70 -totLTR 73.63 -genome_size 2200000000 for maize genomes and parameters of -iden 92 -totLTR 32.2 -genome_size 850000000 for tomato genomes.
Data access
The ONT Duplex and UL data generated in this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra under accession number SRP320775 (HG002 Duplex), the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB49840 (tomato and maize Duplex, maize UL), and the CNCB BioProject database (https://ngdc.cncb.ac.cn/bioproject/) under accession number PRJCA028625 (tomato UL).
Competing interest statement
S.K. has received travel funds to speak at events hosted by Oxford Nanopore Technologies. A.H.J.W. has received free-of-charge flow cells and kits for nanopore sequencing for this and other studies, and travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences. W.R.M. is a founder, shareholder, and board member of Orion Genomics, which focuses on plant genomics. S.N., O.N., M.V., and K.R.L. are employees of Oxford Nanopore Technologies. The remaining authors declare no competing interests.
Acknowledgments
This work was supported, in part, by the Intramural Research Program of the National Human Genome Research Institute, the National Institutes of Health (S.K., A.R., M.R., and A.M.P.) as well as the National Science Foundation awards IOS-2216612 and IOS-1758800 (to M.C.S.) and the Human Frontier Science Program award RGP0025/2021 (to M.C.S.) and NIH/NHGRI RO1:R01HG011274-01 and U01HG010971 (to K.H.M.). S.H. was supported by the National Key Research and Development Program of China 2019YFA0906200, the National Natural Science Foundation of China 31991180, and the Shenzhen Outstanding Talent Training Fund. Z.B. is supported by Max Planck Society funds to Detlef Weigel. W.R.M. is the Davis Family Professor of Human Genetics. S.O. is supported by the OSU Global Gateways Initiative Grant. W.R.M. would further like to acknowledge funding support from the CSHL/Northwell Health Affiliation for the purchase of an ONT PromethION sequencer used in this study and the NIH 5P30CA045508 Cancer Center support grant. S.G. was supported by the National Institutes of Health (5R50CA243890). This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov). We thank Willem van Rengs who curated and uploaded the Duplex data to the European Nucleotide Archive (ENA) for tomato and maize.
Author contributions: H.S., B.M., and S.G. performed HMW extractions, library preparation, and sequencing. I.V., K.N., J.L., and J.P. performed primary data analysis/Duplex calling. S.K., Z.B., A.G., S.O., K.M.J., M.R., and A.R. performed assembly experiments and secondary data analysis. O.N. and S.N. analyzed/improved Duplex sequencing data. M.V. and K.R.L. developed and implemented the stereo basecalling algorithm. D.R., D.W., E.G., S.H., and W.R.M. conceived and planned the experiments. A.H.J.W., K.H.M., M.C.S., and A.M.P. conceived and planned the experiments and supervised the project. S.K., Z.B., A.G., S.O., A.H.J.W., M.C.S., and A.M.P. drafted the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279334.124.
- Received March 15, 2024.
- Accepted October 8, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








