
GenomeMUSter mouse genetic variation service enables multitrait, multipopulation data integration and analysis
- Robyn L. Ball1,
- Molly A. Bogue1,
- Hongping Liang1,
- Anuj Srivastava2,
- David G. Ashbrook3,
- Anna Lamoureux1,
- Matthew W. Gerring1,
- Alexander S. Hatoum4,5,
- Matthew J. Kim6,
- Hao He1,
- Jake Emerson1,
- Alexander K. Berger1,
- David O. Walton1,
- Keith Sheppard1,
- Baha El Kassaby1,
- Francisco Castellanos2,
- Govindarajan Kunde-Ramamoorthy1,
- Lu Lu3,
- John Bluis1,
- Sejal Desai1,
- Beth A. Sundberg1,
- Gary Peltz7,
- Zhuoqing Fang7,
- Gary A. Churchill1,
- Robert W. Williams3,
- Arpana Agrawal8,
- Carol J. Bult1,
- Vivek M. Philip1 and
- Elissa J. Chesler1
- 1The Jackson Laboratory, Bar Harbor, Maine 04609, USA;
- 2The Jackson Laboratory for Genomic Medicine, Farmington, Connecticut 06032, USA;
- 3University of Tennessee Health Science Center, Memphis, Tennessee 38163, USA;
- 4Psychological and Brain Sciences, Washington University in St. Louis, St. Louis, Missouri 63130, USA;
- 5Artificial Intelligence and the Internet of Things Institute, Washington University School of Medicine, St. Louis, Missouri 63110, USA;
- 6University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada;
- 7Department of Anesthesia, Pain and Perioperative Medicine, Stanford University School of Medicine, Stanford, California 94305, USA;
- 8Department of Psychiatry, Washington University School of Medicine, St. Louis, Missouri 63110, USA
Abstract
Hundreds of inbred mouse strains and intercross populations have been used to characterize the function of genetic variants that contribute to disease. Thousands of disease-relevant traits have been characterized in mice and made publicly available. New strains and populations including consomics, the collaborative cross, expanded BXD, and inbred wild-derived strains add to existing complex disease mouse models, mapping populations, and sensitized backgrounds for engineered mutations. The genome sequences of inbred strains, along with dense genotypes from others, enable integrated analysis of trait–variant associations across populations, but these analyses are hampered by the sparsity of genotypes available. Moreover, the data are not readily interoperable with other resources. To address these limitations, we created a uniformly dense variant resource by harmonizing multiple data sets. Missing genotypes were imputed using the Viterbi algorithm with a data-driven technique that incorporates local phylogenetic information, an approach that is extendable to other model organisms. The result is a web- and programmatically accessible data service called GenomeMUSter, comprising single-nucleotide variants covering 657 strains at 106.8 million segregating sites. Interoperation with phenotype databases, analytic tools, and other resources enable a wealth of applications, including multitrait, multipopulation meta-analysis. We show this in cross-species comparisons of type 2 diabetes and substance use disorder meta-analyses, leveraging mouse data to characterize the likely role of human variant effects in disease. Other applications include refinement of mapped loci and prioritization of strain backgrounds for disease modeling to further unlock extant mouse diversity for genetic and genomic studies in health and disease.
Hundreds of inbred mouse strains (NCBI:txid10090) and thousands of intercross progeny have been characterized for disease-related traits, developmental characteristics, age-related phenotypes, microbiota, response to infections, and other environmental factors, as well as molecularly characterized endpoints, such as transcriptomes, proteomes, and metabolomes across many tissues. Advances in genetics, including transcriptome-wide and phenome-wide association analysis methods (Gamazon et al. 2015; Bastarache et al. 2022; Dai et al. 2023) and new methods for integration of data obtained from multiple species (Reynolds et al. 2021) create compelling new opportunities for using fully-reproducible and widely studied inbred mouse strains to characterize the polygenetic basis for individual differences in disease-related traits and to more precisely associate phenotypic variation with genetic variation. These applications require uniformly dense information on genotypic variation across populations. However, the genomes of most mouse inbred strains and intercross progeny have been very sparsely and non-uniformly characterized through studies performed using a variety of genotyping arrays over the past few decades. In contrast, extensive sequencing efforts for a small number of the most widely used inbred strains have generated a tremendous amount of information about polymorphisms, structural variants, and other types of genetic diversity. These data have been presented in various interfaces to support evaluation of the state of region-specific, known variants across inbred strains (Keane et al. 2011; Mulligan et al. 2017; Blake et al. 2021), but often, cannot be accessed programmatically to support genome-wide calculation within or across populations. The problems associated with genotyped data sets, the technical difficulty of merging disparate data sets, the scalability and interoperability of the resources and other challenges thus make it difficult to use these data collectively to perform robust and integrative genetic analyses, such as meta-analysis of genome-wide association studies (GWAS), with adequate power and density. Therefore, an integrated data resource is needed.
Prior efforts to harmonize variant data sets and address the sparsity of data incorporated known or inferred similarity of sequence across well-characterized strains into genotype imputation algorithms (Szatkiewicz et al. 2008; Kirby et al. 2010; Wang et al. 2012). Most prior studies included a small number of strains, represented a relatively narrow subset of mouse diversity, and evaluated a limited set of genotypes. Furthermore, the algorithms these studies used require computationally expensive parameter estimation methods and often rely heavily on close or known ancestral relations among the strains. Szatkiewicz et al. (2008) and Kirby et al. (2010) trained hidden Markov models (HMMs) with various numbers of hidden states, estimating the HMM parameters with expectation-maximization (EM) algorithms (Churchill 1989; Frazer et al. 2007). Szatkiewicz et al. (2008) imputed genotypes for 51 strains using the trained HMM and the Viterbi algorithm (Viterbi 1967), which converges on the most probable path given the observed data. Kirby et al. (2010) imputed genotypes with the trained HMM and an EM algorithm, EMINEM (Kang et al. 2010), assembling known and imputed genotypes for 94 strains at 8.27 million or more locations. Wang et al. (2012) incorporated phylogenetic information, imputing genotypes for 100 classical inbred strains by using the four-gamete rule (Hudson and Kaplan 1985) to define haplotype blocks and infer locally perfect phylogeny. This requirement for close phylogeny and the need to estimate large numbers of free parameters for the HMMs, an O(n) computation that grows linearly with the number of SNPs (Szatkiewicz et al. 2008), limits the application of these algorithms and makes them unsuitable to a comprehensive merged variant set, with sparse genotypes encompassing the extensive diversity of extant mouse strains and their intercross derivatives. Data from prior efforts are also somewhat limited in their analytic applications and presentation in authoritative data resources because the methods used do not provide a locally precise estimate of the accuracy of imputed results, as would be needed for calculations involving weighted variant states.
Therefore, we present an imputation approach and implemented data service to provide a broad and more comprehensive mouse variant resource. The approach is not limited to classical inbred strains, allowing data integration across diverse mouse populations. It can be applied in a computationally efficient manner without the need to estimate large numbers of free parameters, incorporates local phylogenetic information over larger regions without prior knowledge of strain relatedness, is sensitive to co-occurring missingness while leveraging aggregate genetic similarity measures to predict the most likely allelic state, and provides a local accuracy estimate that can be used in downstream analyses. We evaluated the strain-specific imputation accuracy on a “held-out” test set that was not used in the imputation process. We deliver these data in a harmonized, scalable, and programmatically accessible annotated variant resource, GenomeMUSter, which will remain essential even when the majority of strains are sequenced. We present its application to multipopulation and multispecies analyses of complex trait variation in type 2 diabetes and substance use disorders (SUDs) and compare these results to human genetics studies.
Results
GenomeMUSter brings together the known sequence variation among Mus musculus strains. The data resource includes merged and harmonized allelic state data from 16 data sets that include two Sanger whole-genome sequencing data sets (REL2004 and REL1505) (Keane et al. 2011), whole-genome sequencing of the collaborative cross (CC) recombinant inbred strains (Srivastava et al. 2017), BXD recombinant inbred strains (Ashbrook et al. 2022; Sasani et al. 2022), C57BL/6J Eve (B6Eve) (Sarsani et al. 2019), long-read whole-genome sequencing of 42 inbred strains (Arslan et al. 2023), and 10 SNP genotype data sets previously curated on the Mouse Phenome Database (MPD; https://phenome.jax.org) (Frazer et al. 2007; Kirby et al. 2010; Yang et al. 2011; Morgan et al. 2016; Srivastava et al. 2017). We merged data from these 16 variant data sets in each 10-Mb region within each chromosome, and when there were disagreements in the genotype call among data sets for a strain and location, we harmonized the merged variant data set by setting the genotype to the consensus vote (i.e., the majority allelic state) across data sets. Genotypes were removed and later imputed if no consensus was reached. Genotype imputation was performed within each 10-Mb region with a data-driven approach that incorporates local phylogenetic information and the Viterbi algorithm, as implemented in HaploQA (https://haploqa.jax.org). For each target strain in each region, a strain-specific “held-out” test set was randomly selected and not used in the imputation process. Next, optimal predictor strains were chosen for each target strain based on phylogenetic similarity and the degree of co-occurring missingness with the target strain in the region. The Viterbi algorithm inferred allelic states for the target strain in the region based on shared information among the target strain and predictor strains. The local strain-specific accuracy was estimated by comparing the predicted allelic states to the observed allelic states in the “held-out” test set, producing a single strain- and region-specific estimated accuracy level for imputed allelic states in the region.
After merging and harmonizing these 16 variant data sets, the resulting resource contains segregating allelic data at 106,811,517 genomic locations across 657 inbred strains and includes genotype data for Chromosomes Y and MT across 654 and 483 strains, respectively. Although some of the variant sets have no data across Chromosomes Y and MT, many of the strains overlap among the 16 data sets, providing a more complete genome for most strains (Fig. 1) than any single data set alone. The merging process resulted in a comprehensive set of sites across these data sets but sparsely observed allelic state information for most strains. The largest whole-genome sequencing data set (Sanger REL2004) contains allelic state data for 53 strains at 79.8 million sites, yet this represents <10% of the 657 strains in GenomeMUSter, and these sites account for only 74.8% of the 106.8 million sites in GenomeMUSter. Array-based genotype data sets contained allelic states for <1% of the sites in the merged data set (Fig. 1). The relative density of observed allelic state data across variant sets varies greatly overall (Supplemental Table S1) and by chromosome (Supplemental Table S2).
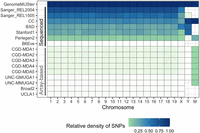
Relative density of information in each data set to GenomeMUSter. The intensity of the color represents the density of SNPs for each data set and chromosome, with white, light green, and dark blue representing a density near 0, less than 0.25, and 1, respectively. Whole-genome sequencing data sets (labeled “sequenced”) include two Sanger variant sets, collaborative cross (CC), BXD recombinant inbred strains, long-read whole-genome sequencing data set of classical inbred strains (Arslan et al. 2023), Perlegen2, and B6Eve; that is, C57BL/6J Eve. The relative density of the B6Eve data set is low because it only includes allelic states that differ from the reference, C57BL/6J. All other variant sets are array-based.
After merging and harmonizing across the 16 variant data sets but before imputation, we evaluated the completeness of observed allelic state data for each strain across the genome and found that 90% of strains were missing allelic state information at >74% of the sites in the merged data set (Fig. 1), revealing the need for an accurate imputation algorithm. Within the chromosome and within each 10-Mb region, missing genotypes for each strain were imputed with a data-driven approach that incorporates local phylogenetic information and the Viterbi algorithm, as implemented in HaploQA (https://haploqa.jax.org). HaploQA was designed to perform quality assurance analyses on genotype array data to resolve haplotypes. Here, we take advantage of the computationally efficient implementation of the Viterbi algorithm in the HaploQA software platform along with local phylogenetic information from the merged and harmonized variant data set to impute genotypes. After imputation, every strain had a known or imputed genotype for at least 84,606,399 sites (>80% complete allele calls) with the median of complete allele calls at 97,442,189 sites (91.6% complete) (Supplemental Tables S3, S4). Across the genome, the median number of imputed genotypes per strain was 92,940,001 (87.0% of 106.8 million) with interquartile range (IQR) [74,979,703 (70.2%), 95,706,897 (89.6%)].
To evaluate the accuracy of the imputation approach for each strain, a strain-specific “held-out” test set was randomly selected in each 10-Mb region. This test set was not used in the imputation process. Based on aggregated held-out test sets in each 10-Mb region, the median strain-specific imputation accuracy on the test set across 657 strains was 0.944 with IQR [0.923, 0.991] and achieved accuracies greater than 0.80 for 636 of 657 (96.8%) of inbred strains (Supplemental Tables S3, S4). The median strain-specific imputation accuracy across 30 wild-derived inbred strains was 0.769 with IQR [0.629, 0.845], yet imputation remained low for a few wild-derived inbred strains such as PAHARI/EiJ, CAROLI/EiJ, and SPRET/EiJ, with estimated strain-specific accuracies and 95% confidence intervals of 0.331 (0.326, 336), 0.485 (0.480, 0.490), and 0.532 (0.529, 0.535), respectively (Fig. 2A; Supplemental Table S3). These strains, of increasing interest to mammalian geneticists (Chang et al. 2017, Poltorak et al. 2018), represent different Mus species; namely, Mus pahari, Mus caroli, and Mus spretus. They are distinct from the most deeply characterized M. musculus domesticus strains, which include classical inbred strains, many of which have been sequenced, yielding higher accuracy across the genome (Fig. 2A). To gain a deeper insight as to how imputation accuracy varies across the genome for each strain, we provide a ridgeline plot of the distribution of estimated accuracies across 10-Mb regions for every strain in GenomeMUSter (Supplemental Fig. S1; Supplemental Table S5). It is evident from these distributions (Fig. 2B; Supplemental Fig. S1) that outside of wild-derived strains, CC strains, which are a multiparent population bred from five common and three wild-derived inbred strains, have more variable imputation accuracies across their genomes than some of the other inbred strains. The distribution of imputation accuracies for CC strains reflects the admixture of these common and wild-derived parental strains in that it appears to be a multimodal mixture distribution with one mode centered around the common strain imputation accuracy and a long tail with a second mode centered around the phylogenetically distant wild-derived founders’ imputation accuracy (Fig. 2B). This is unlike the fully wild-derived strains; for example, PAHARI/EiJ and CAROLI/EiJ, which have more symmetrical distributions of imputation accuracy centered around a value that reflects their distance from the majority of sequenced strains (Fig. 2B).

Imputation accuracy. (A) Histogram of estimated strain-specific imputation accuracies across Chromosomes 1–19 and X for 657 strains. Wild-derived strain accuracies are colored purple. Strains with accuracies below 0.70 are annotated. (B) Ridgeline plots of the strain-specific distribution of imputation accuracies across Chromosomes 1–19 and X (all 657 strains in Supplemental Fig. S1). Shown are the common inbred CC founders (C57BL/6J, 129S1/SvImJ, A/J, NOD/ShiLtJ, NZO/HILtJ), wild-derived CC founders (CAST/EiJ, PWK/PhJ, WSB/EiJ), two CC strains (CC003/Unc, and CC051/TauUnc), and two more-distant wild-derived strains (CAROLI/EiJ, PAHARI/EiJ).
We investigated the role of phylogenetic similarity, co-occurring missingness, and the number of predictor strains on imputation accuracy and found that to achieve higher imputation accuracies, phylogenetic similarity and the amount of shared data in the region are more important to consider than the number of predictor strains included in the algorithm. We calculated the median phylogenetic distance of the predictor strains to the target strain, the median percentage of co-occurring missingness among predictor strains and the target strain (i.e., the amount of shared data), and the number of predictor strains used in the algorithm for every strain in each 10-Mb region and compared them with the strain-specific accuracy in the region (Supplemental Table S5). Across Chromosomes 1–19 and X, the median phylogenetic distance of the predictor strains to the target strain was highly negatively correlated to the strain's imputation accuracy with Spearman's ρ = −0.738 (P < 0.001), indicating that imputation accuracies were lower for strains in regions where predictor strains were more dissimilar to the target strain (Supplemental Fig. S2). This finding helps explain the lower imputation accuracies of wild-derived strains and underscores the need for local accuracy estimates. The median percentage of co-occurring missingness among predictor strains and the target strain was also negatively correlated to the strain's imputation accuracy (Spearman's ρ = −0.288, P < 0.001), underscoring the importance of overlapping genotype observations among strains. The number of predictor strains used in the algorithm was also negatively correlated to the target strain's imputation accuracy (Spearman's ρ = −0.106, P < 0.001); however, it was unclear whether this was owing to lower phylogenetic similarity in the region, which influenced the number of predictor strains included in the algorithm. After accounting for phylogenetic similarity among predictor strains and the target strains, we found that co-occurring missingness was associated with strain-specific imputation accuracy (likelihood ratio test, X1 = 2680.2, P < 0.001), yet after accounting for both phylogenetic similarity and co-occurring missingness, the number of predictor strains used in the algorithm was not significantly associated with imputation accuracy (likelihood ratio test, X1 = 1.8, P = 0.180). These results indicate that both phylogenetic similarity and the amount of shared data in the region are the most important factors to consider in the imputation algorithm. Over Chromosomes 1–19 and X, the median phylogenetic distance among predictor strains and the target strain was 0.05 with IQR [0.00, 0.197]; the median percentage of co-occurring missingness among predictor strains and the target strain was 14.5% with IQR [5.9%, 23.1%]; and the median number of predictor strains used in the algorithm was four with IQR [4, 5].
To gain a deeper understanding of the genetic mechanisms influencing imputation accuracy, we calculated the observed minor allele frequency (MAF). For individual variants with very low MAF, it is difficult to accurately predict the allele state and to estimate the uncertainty of the prediction owing to the insufficient representation of strains with the observed minor allele state. Indeed, it is even impossible to accurately estimate the MAF in such cases. Therefore, we instead compared the regional mean observed MAF to the mean imputation accuracy across strains in each 10-Mb region across the autosomes (249 regions) (Supplemental Fig. S3; Supplemental Table S6). Across autosomes, the mean MAF ranged from 0.063 to 0.131, and the mean imputation accuracy ranged from 0.880 to 0.963. As suspected, the mean MAF is negatively correlated with mean imputation accuracy (Spearman's ρ = −0.363; P < 0.001); however, it is important to note that the lowest mean accuracy across the autosomes was 0.880. Including X, Y, and MT (268 regions), mean MAF is negatively correlated with mean accuracy, with an estimated Spearman's ρ = −0.338 (P < 0.001). The mean MAF across all chromosomes ranged from 0.019 to 0.156, and the mean accuracy ranged from 0.744 to 0.963 (Supplemental Table S6).
GenomeMUSter enables multipopulation, multitrait meta-analysis
A major utility of GenomeMUSter is that it enables genetic discovery across the wealth of recent and historical studies of mouse population variation in a deep and extensible collection of complex disease-relevant characteristics. We illustrate the utility of GenomeMUSter for meta-analyses across mouse populations in two complex diseases, SUDs and type 2 diabetes, using expertly curated trait measures in the MPD (RRID:SCR_003212). Briefly, we first performed GWAS on each project-specific MPD measure (separated by sex, diet, treatment, dose, as appropriate) with a mixed-effect model that accounts for population structure. These GWAS summary statistics served as inputs to a mixed-effect meta-analysis (Han and Eskin 2011). To focus our efforts on variants with the strongest evidence across meta-analyses, downstream analyses included loci with top peaks in the 99.5th percentile, as well as variants linked to the peak locus whose GWAS summary statistics were highly correlated with the peak locus. By including variants highly correlated with peak variants that exceed a somewhat stringent threshold, we guard against false negatives and ensure that we include other relevant, perhaps causal, variants that may otherwise be excluded.
We evaluated the meta-analysis in the realm of diabetes and obesity research by analyzing a heterogenous set of relevant phenotypic endpoints and comparing the genetic associations to known gene–disease annotations. We analyzed 279 phenotypic endpoints obtained from seven studies in the MPD that measured the response to a high-fat diet, as well as another set of 122 phenotypic endpoints containing the term “glucose” in their metacontent (Supplemental Table S7). Studies measured response to a high-fat diet through various methods that include histopathology, lipid profiles and metabolic panels, hormone and metabolite quantification, bone density, and organ and body weights and dimensions. Studies that measured phenotypic endpoints related to glucose used glucose- and insulin-tolerance methods, metabolic and metabolite panels, and hormone quantification methods. These cross-population analyses on the response to a high-fat diet and phenotypic endpoints related to glucose were computed on about 500 Diversity Outbred (J:DO) mice, C57BL/6 consomic strains, CC, CC Pre, the Hybrid Mouse Diversity Panel, and other classical and wild-derived inbred strains (Supplemental Table S7; Paigen et al. 2000; Lake et al. 2005; Lin et al. 2005; Jackson Laboratory 2006; Svenson et al. 2007; Champy et al. 2008; Li et al. 2008; Center for Genome Dynamics 2009; Shockley et al. 2009; Philip et al. 2011; Tsuchiya et al. 2012; Ghazalpour et al. 2014; Morgan et al. 2014; Keller et al. 2018; Kollmus et al. 2020; Bachmann et al. 2022). Studies in these analyses that measured inbred strains included six to 44 strains (median of 14 strains) in the high-fat diet response analysis and two to 190 (median of eight strains) in the analysis across phenotypic endpoints related to glucose.
We performed a GWAS meta-analysis across this heterogeneous set of measurements to detect variants that affect the high-fat diet response (Fig. 3A) and glucose-related endpoints (Fig. 3B). We identified 1169 variants with effects on high-fat diet response and 1284 variants with effects on glucose-related endpoints across this heterogeneous set of measurements; 597 variants with effects in the 99.5th percentile (top peaks) for each analysis, and, to account for linkage, an additional 572 variants (high-fat diet response) and 687 variants (glucose) highly correlated with those variants with effects in the 99.5th percentile. These variants are associated with 224 (high-fat diet response) and 287 (glucose) mouse genes and their human orthologs through evidence from PLAC-seq, ChIA-PET, EPD/CAGE, and eQTL results from Diversity Outbred (DO) mouse populations (Reynolds et al. 2021). Of the human ortholog genes associated with a high-fat diet response, 93 were associated with 284 Disease Ontology terms (Supplemental Table S8). Of the human ortholog genes associated with glucose-related endpoints, 128 were associated with 384 Disease Ontology terms (Supplemental Table S9). When we evaluated the 284 Disease Ontology annotations associated with high-fat diet response in our analysis, Alzheimer's disease was the most frequent, followed by end-stage renal disease, asthma, hepatocellular carcinoma, hypertension and obesity, and other inflammatory diseases that include type 2 diabetes (Fig. 3C; Supplemental Table S8). When we evaluated the 384 Disease Ontology annotations for genes associated with glucose-related endpoints in our analysis, type 2 diabetes was the most frequent annotation to the mouse-derived ortholog set, followed by Alzheimer's disease, obesity, and other inflammatory diseases (Fig. 3D; Supplemental Table S9). Examining these results together, we find that 148 variants have effects on both the high-fat diet response and glucose-related endpoints, and these are associated with 43 human ortholog genes. These results indicate that mouse multitrait meta-analysis produces disease-relevant information and further, by examining the precise traits and genes, can facilitate characterization of their role in disease.
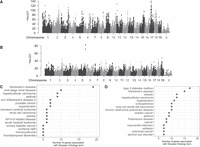
Mouse cross-population multitrait meta-analysis of physiological measurements related to the response to high-fat diet (A) and glucose-related endpoints (B), identifying gene orthologs annotated to Alzheimer's disease (C), type 2 diabetes (D), and other inflammatory diseases.
Characterizing human GWAS with mouse genetic traits
We applied this multispecies approach in the opposite direction, going from human to mouse, to assess the utility of GenomeMUSter to interpret and characterize human GWAS discoveries with mouse genetic traits. Hatoum et al. (2023) performed a multiancestry GWAS meta-analytic study to identify variants and genes broadly associated with the use of multiple addictive substances in humans and identified 42 significantly associated genes. However, it is not clear what specific processes are impacted by these variants because the use of multiple substances simultaneously is a highly complex phenotype that results from many aspects of response to environment, patterns of substance use, and effects of substance use. In contrast, mouse studies typically focus on more granular phenotypes meant to model specific aspects of the disease. To interpret the role of human genetic variants in this process in light of the many hundreds of mouse traits that have been characterized in studies of phenomena associated with SUDs, we performed a meta-analysis of 366 mouse traits in the MPD annotated to the Vertebrate Trait ontology term, VT:0010488, response to addictive substance, and separate meta-analyses of each of the following child terms: response to alcohol, cocaine, and nicotine and withdrawal response to addictive substance (VT:0010489, VT:0010718, VT:0010719, VT:0010721, and VT:0010722, respectively) (Fig. 4). These phenotypic endpoints were obtained from 15 studies in the MPD that measured the response to addictive substances (Supplemental Table S7). These cross-population analyses on the response to addictive substances were computed on about 300 Diversity Outbred (J:DO) mice, CC, BXD recombinant inbred strains, and other classical and wild-derived inbred strains. Studies in these analyses that measured inbred strains included six to 58 strains (median of 14 strains).
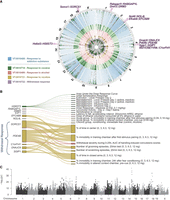
Cross-species integration of human multisubstance GWAS with mouse cross-population multitrait meta-analyses of 360 or more traits grouped by vertebrate trait (VT) ontology term. (A) The circular plot provides a bird's eye view of these five mouse meta-analyses, variant effects, and associated pleiotropic genes detected in both mouse and human GWASs across multiple substances (Hatoum et al. 2023). Genomic regions of variants associated to these genes are highlighted, with associated mouse genes and their human orthologs annotated along the outer ring. The colors of each ring correspond to separate meta-analyses for response to addictive substance (blue), cocaine (green), alcohol (red), nicotine (orange), and withdrawal response to addictive substance (purple). Dots, colored by substance, indicate measures in which the variant effect exists (m-value > 0.9) (Han and Eskin 2011) to enable discernment between substance-specific effects and polysubstance effects. A full resolution version with annotations of all associated mouse genes is available (Supplemental Fig. S4). (B) The Sankey plot shows the number of variants affecting the mouse traits and the associated orthologous genes. The width of the line represents the number of variants, and the color of the line represents the substance (green indicates cocaine; red, alcohol; orange, nicotine). (C) The Manhattan plot of the meta-analysis of the parent term, response to addictive substance (VT: 0010488) on 366 mouse traits.
Across these five meta-analyses, we detected a total of 5705 variants with effects in the 99.5th percentile or highly correlated with the variants in the 99.5th percentile. We associated variants to mouse genes and their human orthologs as illustrated in work by Reynolds et al. (2021). These 5705 variants were associated with 1784 mouse genes and 1850 of their human orthologs, annotated on the outer ring (Supplemental Fig. S4), that were identified through additional variant to gene information including intergenic coding variation and eQTLs from GTEx (The GTEx Consortium 2013), mouse striatum (Philip et al. 2023), and hippocampus (Skelly et al. 2019). Of these 5705 variants, 1153 were detected in the meta-analysis of the parent term, response to addictive substance (VT:0010488), and are associated with 525 human ortholog genes. Across the substance-specific meta-analytical results, 99 variants, associated with 65 human ortholog genes, were shared among multiple substances, indicating a possible role in general addiction processes. Fifty-one of these 99 variants, associated with 41 human ortholog genes, were also detected both in the meta-analysis of the parent term, response to addictive substance, and in the meta-analysis of withdrawal response to addictive substance, indicating that these variants have a possible role in regulating a general mechanism of withdrawal across substances (Fig. 4, Supplemental Fig. S4).
Gene products identified through the mouse meta-analyses were compared with two sets of human ortholog genes that were found
to be significantly associated to multiple SUDs (Hatoum et al. 2023). Specifically, we used MAGMA (de Leeuw et al. 2015) to conduct whole-gene burden tests using summary statistics and also considered “mapped genes” that were within 10 kb of
a genome-wide significant SNP as statistically significant. Compared with the results from Hatoum et al. (2023), our multisubstance meta-analyses in the mouse revealed HS6ST3, NOL4L, and PDE4B from the MAGMA-identified gene set and TNN, RABGAP1L, DNM3, EFCAB8, BPIFA2, E2F1, DNAJC6, PDE4B, SGIP1, C1orf141, and SORCS1 from the mapped gene set, annotated on the outer ring (Fig. 4A). One of the top hits, PDE4B, is thought to affect the dopaminergic pathway (Snyder and Vanover 2017) and is one of the 15 genes that is associated with SNP effects observed across two human populations (Hatoum et al. 2023). We evaluated the GWAS summary statistics for individual measures for the variants that mapped to Pde4b, and across measures, the maximum measure-level t-statistic 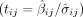 was 2.13, well below the significance threshold (Supplemental Fig. S5) needed to detect effects at the individual measure GWAS level. Yet, by pooling power across studies and by including variants
near top peaks that are highly correlated, the meta-analysis not only identified Pde4b but also, by examining the meta-content of the mouse measures associated with Pde4b-related variation, found that these were traits that measured the withdrawal response from addictive substances (Fig. 4B), including withdrawal severity scores from alcohol based on handling-induced convulsions, the number of grooming and scratching
episodes at different nicotine doses, and the percentage of time in closed-arms and time in the center of an open field assay
at different nicotine doses. Although Hatoum et al. (2023) identified PDE4B as a gene associated with multisubstance use in humans, the association does not reveal what biobehavioral process is affected
by PDE4B variation. The mouse meta-analyses and GenomeMUSter support the specific hypothesis that Pde4b variation mediates withdrawal response, supporting the potential utility of therapeutic interventions in this process.
was 2.13, well below the significance threshold (Supplemental Fig. S5) needed to detect effects at the individual measure GWAS level. Yet, by pooling power across studies and by including variants
near top peaks that are highly correlated, the meta-analysis not only identified Pde4b but also, by examining the meta-content of the mouse measures associated with Pde4b-related variation, found that these were traits that measured the withdrawal response from addictive substances (Fig. 4B), including withdrawal severity scores from alcohol based on handling-induced convulsions, the number of grooming and scratching
episodes at different nicotine doses, and the percentage of time in closed-arms and time in the center of an open field assay
at different nicotine doses. Although Hatoum et al. (2023) identified PDE4B as a gene associated with multisubstance use in humans, the association does not reveal what biobehavioral process is affected
by PDE4B variation. The mouse meta-analyses and GenomeMUSter support the specific hypothesis that Pde4b variation mediates withdrawal response, supporting the potential utility of therapeutic interventions in this process.
We examined the P-values for both the individual measures and meta-analysis for the response to addictive substance study, which included the largest number of measures (n = 366). In the individual analyses, we identified only two variants, which were next to each other on Chr 6, for which two studies had a measure level of −log10(P) ≥ 7, typically considered genome-wide significant, but these variants were not detected in the meta-analysis. In the meta-analysis at a stringent −log10(P) ≥ 12.8, we detected 658 variants. Thus, at these thresholds the meta-analysis is far more sensitive, as expected. However, we suspect the individual threshold may be so stringent that it may lead to false negatives so we also examined individual results at a measure level of −log10(P) ≥ 5, typically considered an unpublishable suggestive significance level. This revealed an additional 515 variants associated with at most one or two measures. Seven of the 515 variants detected at suggestive levels were detected at the stringent threshold in the meta-analysis, presumably amid a large number of false positives. Although most individual studies lack power to detect variant effects, the meta-analysis pools power across them and, once detected across studies, allows us to make inferences about variant effects at the measurement-level through the METASOFT software. METASOFT calculates the posterior probability that the variant affects the phenotypic endpoint (m-value) for each variant and measure included in the meta-analysis.
Discussion
Here, we present GenomeMUSter, intended to be the most comprehensive and interoperable mouse genetic variation data resource service to date, containing more than 106.8 million genotyped, sequenced loci, or imputed variant states for 657 inbred mouse strains (~70 billion allelic states) representing common, wild-derived, recombinant inbred strains, and their intercross derivatives. GenomeMUSter provides a comprehensive analytical SNP resource that can be used for a variety of research applications, which include characterizing the effect of genetic variants and their influence on transcripts and traits and refinement and resolution of genetic mapping experiments. It is important to note, however, that GenomeMUSter is not a static resource; rather, it is a dynamic and versioned resource that will continually be updated as new mouse sequence data are acquired. Because the sequenced and typed data used to build GenomeMUSter are on GRCm38 coordinates, we chose to keep GRCm38 coordinates for this version and provide access to the recently released Sanger (REL2021) data set (Keane et al. 2011) in the GRCm39 assembly, as well as an option to use liftOver to map GRCm38 coordinates onto GRCm39, while more rigorous annotation of the GRCm39 is in progress. Notably, assemblies will be continuously updated as more regions are sequenced and as we learn more of the structural variation present in the mouse with newer long-read sequencing-based assemblies. For example, long-read whole-genome sequencing, as used by Arslan et al. (2023), included in GenomeMUSter, and Ferraj et al. (2023), who sequenced a set of 20 diverse inbred strains from three subspecies of M. musculus, provides rich information about structural variation in regions previously unexamined. Additionally, alignment to an appropriate reference genome or pseudogenome has been shown to greatly improve the accuracy of RNA-seq and eQTL analyses (Munger et al. 2014). GenomeMUSter can be used for alignment to a pseudoreference or used for genotyping by RNA-seq (Choi et al. 2020) to more precisely address the needs of functional genomics. Importantly, the analytical approach we present here not only can be applied to new mouse assemblies and new variation data but also can be applied to any other widely studied genetic model organisms, including Drosophila, rat, Caenorhabditis elegans, yeast, and many agricultural species for which incomplete reference genotypes are available (Sulston et al. 1992; Güldener et al. 2005; Hillier et al. 2008; Mackay et al. 2012; Shimoyama et al. 2015). We note that the laboratory mouse is a somewhat unique model organism owing to its well-developed collection of typed strains and populations and that some of our specific analysis implementation may require adaptation to the data formats used in research on other species. However, extensive developments in rat genomics, including a well-developed panel of inbred strains and an increasingly used outbred population, provide similar opportunities for data integration. C. elegans, Drosophila, and other organisms also have similar resource collections, including inbred panels, heterogenous stocks, and others. The applicability of imputation to derive full sequence from genotype information is well established. The state of genotype, sequence and archival phenotype resources varies across these organisms, but as they are expanded, the integrative analyses we show are certainly adaptable.
Imputation will ultimately be replaced by full sequence data for many strains, but for legacy populations, heterogeneous stocks, and inter-crosses, augmentation of sparse genotype data will still be required. As more strains are sequenced, particularly covering a broader range of mouse genetic variation, performance of the imputation will improve for a greater number of strains. Genealogical analysis of the extant strain population reveals that most widely used laboratory strains are derived from closely related stocks, and several large subpopulations and collections of unrelated strains provide great variety in phylogenetic distance among strains (Beck et al. 2000). Prior imputation efforts provided high-confidence imputations for only the well-characterized set of classical inbred strains, likely owing both to the extreme sparseness of the data for some strains, which were genotyped at as few as 1638 loci, and to the tremendous genetic diversity of the population, which limits the utility of known or inferred ancestry information owing to a lack of similar cases that have sufficiently dense data in any given region. For example, although Wang et al. (2012) incorporated phylogenetic information, they excluded wild-derived strains because of the abundance of private alleles, which make it difficult or impossible to construct local haplotype blocks using the four-gamete rule in regions <5 kb. Moreover, some approaches that use HMMs require estimation of many parameters, and importantly, the number of parameters to estimate in these methods increases linearly with the number of SNPs (Szatkiewicz et al. 2008). Given the lack of dense genotypes for the majority of strains and the number of private alleles among wild-derived strains, GenomeMUSter makes use of a larger genomic region on which to calculate phylogenetic similarity, does not require free parameter estimation, and uses the computationally efficient Viterbi algorithm, as implemented in HaploQA.
Imputation accuracy is influenced by phylogeny. The phylogenetic trees support a data-driven approach in which imputed genotypes are based on known genotypes from phylogenetically similar predictor strains with limited co-occurring missingness, and both phylogenetic distance and the proportion of co-occurring missingness were shown to be associated to imputation accuracy. Thus, if the most phylogenetically similar strains differ in important ways (i.e., are a different species, as is the case for the strains with the lowest imputation accuracies) or the amount of observed allelic state data in the region are exceptionally sparse, we expect lower imputation accuracies. We found that for most strains the imputation algorithm produced high strain-specific imputation accuracies with a median imputation accuracy of 0.944 on the held-out test set. Lower imputation accuracies (<0.70) in some wild-derived strains, such as PAHARI/EiJ, CAROLI/EiJ, and SPRET/EiJ (Fig. 2A), were expected, as these strains are a different species (M. pahari, M. caroli, and M. spretus, respectively) from classical inbred strains (M. musculus), and the data for these strains were sparse in the data sets we included in GenomeMUSter. New full sequence data are being added for wild-derived strains (Thybert et al. 2018). As more wild-derived inbred mouse strains are sequenced and included in GenomeMUSter, imputation accuracy for all strains will improve (Chang et al. 2017; Poltarak et al. 2018; Gambogi et al. 2023).
The phylogenetic trees also provide a rich genomic landscape to explore possible regions of cryptic genetic variation among strains; namely, variation that may have little effect on phenotypic variation within populations but may contribute to trait heritability across populations (Paaby and Rockman 2014). It may be possible to evaluate multiple populations in a single model for some easily standardized phenotypes, in which case one can evaluate the effects of cryptic variation, particularly that which is fixed in different states and in different populations. Admixed populations also allow for detection of these effects.
A challenge with imputing genotypes for all missing allele states is that we inadvertently impute SNPs for insertions and deletions, thus known structural variation needs to be masked. Pangenome representations are a compact and even more scalable graphical means of presenting individual variation in the context of population diversity (Wang et al. 2022; Liao et al. 2023) that address this challenge and will be used in future releases of GenomeMUSter.
GenomeMUSter serves as an analytical resource that is available programmatically for a wealth of applications in the study of genetic variation and phenotypic diversity in health and disease. For example, GenomeMUSter can be used for GWAS and genetic meta-analysis, used to refine genetic loci underlying trait variation across mouse strain panels (Raza et al. 2023), and used for a host of other genome-scale operations. The extant inbred mouse strains have been broadly characterized over decades of mouse phenomic research. Examination of the large set of mouse traits that share orthologous regulatory variant targets provides a means to interpret and characterize GWAS variants. Our meta-analysis of genetic associations used many legacy strain surveys in a new way. For many of these data sets, no genetic mapping accompanied the publication, and in many cases, there is insufficient power or genetic precision to perform individual mapping studies. Although most individual studies lack power to detect variant effects, the meta-analysis pools power across them and, once detected across studies, allows us to make inferences about variant effects at the measurement level. Therefore, a major novel outcome of the approach is that these data sets can now be combined and reused in a new way for genetic association analyses that is sufficiently powered.
Many GWAS studies are followed with mouse models of causal variants, preclinical in vivo studies, and other experimental research, but the phenotypic endpoints to pursue are not always obvious. The comparison of human GWAS results to variation in the mouse genome through tools like GenomeMUSter enables investigators an initial path for characterizing when, where, and how human genetic variants influence the disease. We illustrated the utility of using GenomeMUSter for characterization of variants and orthologous genes for two complex diseases: addiction and type 2 diabetes. For example, our analysis of genes pleiotropic across human studies of multisubstance use reveals that the GWAS candidate Pde4b most likely plays a role in withdrawal-mediated response to a drug. It is speculated that this is indeed a broad mechanism of SUD across all classes of drugs (Hatoum et al. 2023), as people often use substances to ameliorate the consequences of withdrawal; namely, negative reinforcement is a general addiction process regardless of substance class. This is consistent with neurobiological mechanisms of SUD and provides evidence in support of the GWAS finding that PDE4B-related variation is associated with addiction but not specific to any one class of substance. Notably, the interpretation of multipopulation, multitrait meta-analyses in mice indicates future research directions in the functional characterization of human GWAS variants pharmacologically. Indeed, early clinical trials have already shown promise for PDE4B drug targets in treating alcohol and opioid withdrawal (Cooper et al. 2016; Burnette et al. 2021).
In our evaluation of the use of GenomeMUSter in GWAS meta-analysis to discover genes and variants related to type 2 diabetes and obesity, we found that the physiological response to high-fat diet was associated with genes that had a general involvement in metabolic traits, based on their annotation to terms in the Disease Ontology, and that meta-analysis of mouse measures associated with glucose-related endpoints netted terms that were more specific to type 2 diabetes. In the former, terms retrieved are related to conditions such as Alzheimer's disease, renal disease, liver cancer, hypertension, and obesity, each known to be associated with type 2 diabetes (Ritz et al. 1999; Okosun et al. 2001; Dyson et al. 2014; Chatterjee and Mudher 2018). In the latter analysis on glucose-related endpoints, a more specific diabetes-related endophenotype, netted terms were associated directly with type 2 diabetes in addition to Alzheimer's disease and obesity. This reveals that the multitrait meta-analysis could identify specific disease-associated genes when a disease feature was used, and it was capable of detecting more general indirect environmental drivers of disease etiology such as high-fat diet, when this type of term was used.
It should be noted that in meta-analysis, the quality of the result is driven by the study inclusion criteria. Broad search terms for disease-related traits, although rapidly computationally reproducible and readily applicable to whole vocabularies and databases, may result in false-positive search results, and manual filtering, although labor intensive, may improve the precision of global associations of variants to disease. Use of specific mappings of traits to disease developed in efforts such as the Ontology of Biological Attributes (Stefancsik et al. 2023) will further refine the precision of disease-relevant queries of mouse trait data. Overall, we observed a somewhat low overlap of genes implicated in both human and mice, particularly for multiple substance use. Human GWASs are often underpowered, particularly in SUDs. The human study identified only 19 significant loci, whereas the mouse meta-analyses identified 5705 significant loci. The overlap is potentially also limited by the specific conservation of variant effects, and pathway-based approaches are showing promise in expanding beyond the observed similarity of genetic associations across species (Wright et al. 2023).
Human GWASs often connect variants to disease diagnoses using a very limited set of clinical assays. Mouse phenotyping studies combined with genomic harmonization through GenomeMUSter can add extensive breadth and depth, allowing an understanding of the specific processes and temporal involvement of genetic variants in disease risk, manifestation, and progression. GenomeMUSter unlocks the utility of all genotyped inbred mouse populations for integrative, predictive genetic, and genomic analyses by providing insight into previously unavailable genetic variation, enhancing the effective use of inbred mouse strains, and establishing their versatility as disease models. Parallel efforts to refine the alignment of disease and trait vocabularies across species and identify variants with orthologous targets will greatly expand the potential for analytic operations using integrated genomics and phenomics in the laboratory mouse. GenomeMUSter and its accompanying analytical approach increase the scope, scale, and power to find relations among genetic variants and individual phenotypic variation and use this variation to better model, map, and characterize the variation associated with complex human disease.
Methods
Preprocessing of whole-genome sequence data
Whole-genome sequence data of the CC (Srivastava et al. 2017) were downloaded from the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB14673; Merged_69_flagged.tab.vcf.gz. Insertions and deletions (indels) and multiple-nucleotide polymorphism (MNP) alleles were excluded by filtering out loci with either INDEL or COMPLEX in the “INFO” column. Single-nucleotide polymorphism (SNP) alleles with read depths of five or more were included. Heterozygous genotypes were called “H” if both alleles met the allele depth requirement and accounted for at least 20% of the total read depth. This 20% threshold for the heterozygous genotype percentage of total read depth was determined based on agreement with University of North Carolina (UNC) GMUGA genotyped SNP calls (Morgan et al. 2016) across Chromosome 1.
We adopted the same filtering approach for read depth and calling heterozygous genotypes for the other whole-genome sequencing variant sets. For the C57BL/6J Eve (B6Eve) (Sarsani et al. 2019) whole-genome sequencing set, we included only genomic loci that had “PASS” in the filter column. For the BXD recombinant inbred strains variant set, we included the three lane C57BL/6J (∼80×) and two-lane DBA/2J (∼100×) genotypes. The single-lane genotype calls for the parental strains were not used. The long-read whole-genome sequencing variant set (Stanford1) (Arslan et al. 2023) was processed similarly to the other whole-genome sequencing data sets.
Merging and harmonizing variant sets
Allelic state calls were harmonized across 16 disparate data sets that include two Sanger releases (REL2004, REL1505) (Keane et al. 2011), whole-genome sequencing of the CC (Srivastava et al. 2017), BXD recombinant inbred strains (Ashbrook et al. 2022; Sasani et al. 2022), C57BL/6J Eve (B6Eve) (Sarsani et al. 2019), long-read whole-genome sequencing of 42 inbred strains (Stanford1) (Arslan et al. 2023), and sparse genotypes from 10 legacy data sets available from MPD (e.g., Frazer et al. 2007; Kirby et al. 2010; Yang et al. 2011; Morgan et al. 2016; Srivastava et al. 2017). Raw and processed whole-genome sequencing data for the CC and the BXD data sets are available at the ENA: PRJEB14673; Merged_69_flagged.tab.vcf.gz (CC) and PRJEB45429 (BXD). Raw whole-genome sequencing data for B6Eve (Sarsani et al. 2019) and long-read whole-genome sequencing data for the Arslan et al. (2023) data set are available from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession numbers PRJNA318985 (B6Eve) and PRJNA788143. Strain and allelic state data were merged by chromosome and within chromosome in 10-Mb segmented regions. When there were disagreements among data sets regarding the genotype call for a strain and position, the consensus vote across all data sets was used. Genotypes were removed and imputed if no consensus was reached unless the disagreement was complementary; for example, A and T. Across Chromosomes 1–19 and X (about 70 billion strain-site allelic states), there were about 14.7 million of 70B (0.021%) total strain-site disagreements among the 16 data sets. Of these, consensus was reached for 5.3 million strain-site allelic state disagreements among data sets. The remaining 9.4 million (0.013%) strain-site allelic state disagreements were resolved by imputation.
Imputation approach
Missing genotypes were imputed for each strain in each 10-Mb region spanning the genome using the Viterbi algorithm as implemented in HaploQA (https://haploqa.jax.org). Unlike other HMMs in which many free parameters must be estimated (Szatkiewicz et al. 2008; Kirby et al. 2010), the implementation of the Viterbi algorithm in HaploQA relies on given transition probability parameters, resulting in a computationally efficient estimate of the most probable path (Supplemental Table S10). We incorporate local phylogenetic information without prior knowledge of relatedness in an approach that accounts for the degree of information in the region, namely, co-occurring missingness. Our choice for using a 10-Mb region in the imputation algorithm and not a smaller region was twofold: (1) We wanted to provide a local strain-specific estimated accuracy, and (2) to achieve reasonable accuracies, we needed to have enough shared information in the region about both the target strain and predictor strains. We calculated the number sites with known allelic states across all strains in each region and evaluated the completeness across Chromosomes 1–19 and X. Genotyped or sequenced allelic state data were sparse in the merged and harmonized data set (Fig. 1). For 10-Mb regions, the minimum completeness across the genome for all strains ranged from three to 508 sites with a median minimum completeness of 262 sites. For 10-Mb regions, the median completeness across the genome for all strains ranged from 55 to 2842 sites, with a median of median completeness of 1830 sites (Supplemental Table S11). We chose a 10-Mb region and not a smaller region to balance the need for a local imputation accuracy with the ability to accurately impute genotypes in each region based on shared information between the target and predictor strains.
To impute genotypes on a target strain, we identified the optimal predictor strains, considered “haplotypes” in the algorithm, by selecting strains that, first, had the fewest co-occurring missing genotypes with the target strain in the region and, second, had the smallest phylogenetic distance and thus were the most phylogenetically similar to the target strain in the region. The local phylogenetic distance between each strain and all other strains was calculated per 10-Mb region using the merged variant set and the function “dist.dna” in the R package “ape” (Paradis and Schlep 2019) with model = “F84” (Felsenstein and Churchill 1996). For each target strain in each 10-Mb region, potential predictor strains were ranked by the number of positions missing genotype calls that coincided with the positions missing genotype calls in the target strain. An alternative approach is to partition the genome based on local phylogeny (White et al. 2009); however, the sparsity of genotype-based analyses may yield limited benefits. We selected the 20 potential predictor strains with the most genotype information in the region that coincided with missing data in the target strain, namely, had the lowest co-occurring missingness in the region. Next, we selected predictor strains most phylogenetically similar to the target strain in the region, lower than the 10th percentile of phylogenetic distances between the target strain and all potential predictor strains. We required at least two predictor strains, and thus if no predictor strains could be chosen based on this threshold, we increased the phylogenetic distance threshold in increments of 10th percentiles until we found at least two predictor strains with the lowest co-occurring missingness that were phylogenetically closest to the target strain. Although we required at least two predictor strains for each target strain, we aimed for at least four predictor strains for each target strain, including a maximum of 20 predictor strains.
Imputation algorithm
For each target strain within a 10-Mb region, the major (A) and minor (B) alleles were determined across the optimal predictor strains for each locus. Using these classifications, genotypes for the target and predictor strains were translated to one for A if the genotype matched the major allele, two for B if the genotypes matched the minor allele, three if the genotype matched a third allele or H, and zero for N if the genotype was unknown. The transition probability parameters in the HMM used by the Viterbi algorithm implemented in HaploQA are given, not estimated, and are based on expected genotyping or sequencing errors. Except for missing genotypes, the transition probabilities were set by assuming there were minimal errors in the merged and harmonized variant set (Supplemental Table S10). The percentage of discordant observations among the merged data set was 0.021%, but for the vast majority of loci, there is insufficient replicate genotyping or sequencing with which to observe such errors. We therefore chose a conservative estimate of the error rates of 1% (haplohmm.py in https://github.com/TheJacksonLaboratory/GenomeMUSter derived from https://github.com/TheJacksonLaboratory/haploqa).
Validation of the imputation accuracy
To evaluate the accuracy of the imputation approach, a “held-out” test set for each strain was randomly selected in each 10-Mb region for 1000 locations or 10% (whichever was smaller) of known genotypes. This test set was not used in the imputation process. We calculated the strain-specific accuracy on the held-out test set and set the confidence level in each 10-Mb region to the estimated accuracy on the held-out test set. If the imputation accuracy equaled one, we set it to 0.999 to distinguish it from known genotype data. Because most strains had sparse data for the Y Chromosome and mitochondria, we assessed the overall accuracy for each strain over Chromosomes 1–19 and X and provide a 95% Clopper–Pearson confidence interval. We calculated accuracy assuming that nucleotide complements were equivalent but provide both this and a traditional accuracy measure for completeness. After evaluating accuracy, allelic state data in the “held out” test sets were merged back into the full data set. All analyses were completed in the R environment for statistical computing (R Core Team 2020), except the Viterbi algorithm, which was in HaploQA.
We collected data during the imputation process to assess which factors (if any) impacted imputation accuracy. These included the median observed MAF in each 10-Mb region, the median imputation accuracy across all strains in each 10-Mb region, and strain-specific data in each 10-Mb region: the number of predictor strains used to impute genotypes on the target strain, the median proportion of co-occurring missingness among the predictor strains and the target strains, and the median phylogenetic distance between the predictor strains and the target strain. We assessed whether these measures were correlated to imputation accuracy in the region with the Spearman's rank order correlation. Next, we used the likelihood ratio test to evaluate whether, after accounting for one or more variables (restricted model), adding an additional variable (full model) was predictive of imputation accuracy. Test statistics and associated P-values are reported.
Validation of the utility of GenomeMUSter for multispecies integrative analyses
To evaluate the utility of this dense SNP resource for integrative genetic analysis, we performed multiple GWAS meta-analyses in two complex disease areas. First, we performed two cross-population meta-analyses on mouse traits that measured the physiological response to a high-fat diet and glucose-related endpoints (Supplemental Table S7). Second, we performed five meta-analyses on addiction-related mouse traits (Supplemental Table S7) annotated to Vertebrate Trait ontology terms in the MPD. We identified traits measuring the response to high-fat diet by searching “high-fat” in the “intervention” column of trait measures in the MPD, and similarly, for endpoints related to glucose, we searched the measure description column for “glucose” (“measurements.csv” at https://phenome.jax.org/downloads). We identified addiction-related traits as those mouse traits annotated to the Vertebrate Trait ontology term VT:0010488, response to addictive substance, and the following child terms: response to alcohol, cocaine, and nicotine and withdrawal response to addictive substance (VT:0010489, VT:0010718, VT:0010719, VT:0010721, and VT:0010722, respectively). The substances studied included morphine, cocaine, methamphetamine, nicotine, and ethanol (alcohol) and measured various aspects of addiction-related endpoints, including sensitivity and tolerance; intravenous self-administration [IVSA] endpoints; observations related to balance, strength, cognition, and dexterity; bottle or lever choice; fear conditioning; as well as histopathology, and measures of lipids and metabolites (Supplemental Table S7; Crabbe et al. 2003a,b; Rustay et al. 2003; Metten et al. 2004; Kamens et al. 2005; Metten and Crabbe 2005; Yoneyama et al. 2008; Thomsen and Caine 2011; Portugal et al. 2012; Tsuchiya et al. 2012; Kutlu et al. 2015; Wiltshire et al. 2015; Dickson et al. 2016; Bubier et al. 2020; Chesler et al. 2021; Bagley et al. 2022; Schoenrock et al. 2022). Before performing GWAS, we used the qtl2 R package (Broman et al. 2019) to impute genotypes of Diversity Outbred (DO) animals from the “Attie2” study (Keller et al. 2018) and the “CSNA03” study (Chesler et al. 2021) onto a common SNP set so the GWAS summary statistics could be combined across inbred and outbred studies (Bogue et al. 2023). The strains in GenomeMUSter are inbred and largely homozygous, and the DO mice have a high rate of heterozygosity from a constrained set of eight founders.
For all mouse meta-analyses, we first ran a GWAS mixed-effect model that accounts for population structure, namely, kinship (pyLMM; https://github.com/nickFurlotte/pylmm), on each project-specific measure, so that within a project, a separate GWAS was performed for each measure, sex, and treatment (e.g., substance, diet, dose). Importantly, parental strains were not included in the same GWAS analysis; namely, C57BL/6J and DBA/2J were not included in GWAS analyses across BXD strains. Next, we performed a mixed-effect meta-analysis (METASOFT) (Han and Eskin 2011) on the summary statistics, as described by Bogue et al. (2023). METASOFT estimates variant effects in two parts: the fixed effect (i.e., average effect) and the random effect, which accounts for the heterogeneity in the effect across measures (Han and Eskin 2011). It is important to note that if performing meta-analyses only across the BXDs or Diversity Outbred mice, a linkage-based approach is generally recommended (Parker et al. 2017); however, in this case, we performed meta-analyses across a wide range of mouse populations and thus controlled for the population structure through robust mixed-effect models. Although it is possible to incorporate sex into the model as an additive or, more often, interacting cofactor, it would require post hoc modeling of sex effects and other factor effects. However, combining GWAS summary statistics in a mixed-effect meta-analysis such as METASOFT enables the discovery of both main and interaction effects (Kang et al. 2014). Currently, we only use phylogenetic information in the imputation, not in the genetic association analyses. As stated above, we model effects within population and integrate across the populations in large part because of the sparsity of phenotype data. Although phylogenetic information is not presently incorporated into mapping models, within-population structure is absorbed in the mixed-effect models. The data resource, in combination with phenotype data in the MPD, provides the users of GenomeMUSter with the ability to develop additional genetic and phenotypic analyses with integration of local phylogenetic structure.
For each meta-analysis, we included variants in downstream analyses if they had effects in the 99.5th percentile (top peaks),
and then, to account for any remaining linkage among variants, we also included variants near each peak that were strongly
correlated with the peak locus. We chose a 99.5th percentile to focus our efforts on variants with the strongest evidence
while accounting for linkage by also including variants near top peaks that were highly correlated with the top peak. For
each variant i and trait measure j in the meta-analysis, we calculated the t-statistic 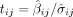 for each measure's GWAS summary statistic: GWAS variant effect
for each measure's GWAS summary statistic: GWAS variant effect 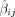 divided by the estimated standard deviation of the GWAS variant effect
divided by the estimated standard deviation of the GWAS variant effect 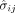 ; so, for example, the meta-analysis on response to addictive substance was computed on 484 GWAS summary statistics (estimated
variant effects and standard errors) so each variant included in the meta-analysis had 484 t-statistics. A variant within 500,000 bp of a variant in the 99.5th percentile (top peaks) was included if the Pearson's r correlation coefficient between the variant's t-statistics and the top peak variant's t-statistics was ≥0.80. Variants detected in the meta-analyses were then passed through gene regulatory data resources, such
as GTEx (The GTEx Consortium 2013), PLAC-seq, ChIA-PET, and eQTL from Diversity Outbred mouse tissues, including striatum and hippocampus, to identify targeted
mouse genes and their human orthologs (Reynolds et al. 2021). To enable others to access these data and use these analytical tools, we integrated the meta-analysis service with expertly
curated mouse data in the MPD (https://phenome.jax.org/, RRID:SCR_003212). After selecting trait data and running the GWAS meta-analysis tool, users can adjust thresholds, examine
variant effects, and map variants to mouse genes and their human orthologs with the “Fuji plot” tool within GWAS meta-analysis
(Bogue et al. 2023). Additionally, within the GWAS meta-analysis tool on MPD, users can view the variant effect on each measure with the P-M
plot and Forest plot (Han and Eskin 2012) to explore whether the effect is broad or specific to a measure or factor (e.g., sex, treatment).
; so, for example, the meta-analysis on response to addictive substance was computed on 484 GWAS summary statistics (estimated
variant effects and standard errors) so each variant included in the meta-analysis had 484 t-statistics. A variant within 500,000 bp of a variant in the 99.5th percentile (top peaks) was included if the Pearson's r correlation coefficient between the variant's t-statistics and the top peak variant's t-statistics was ≥0.80. Variants detected in the meta-analyses were then passed through gene regulatory data resources, such
as GTEx (The GTEx Consortium 2013), PLAC-seq, ChIA-PET, and eQTL from Diversity Outbred mouse tissues, including striatum and hippocampus, to identify targeted
mouse genes and their human orthologs (Reynolds et al. 2021). To enable others to access these data and use these analytical tools, we integrated the meta-analysis service with expertly
curated mouse data in the MPD (https://phenome.jax.org/, RRID:SCR_003212). After selecting trait data and running the GWAS meta-analysis tool, users can adjust thresholds, examine
variant effects, and map variants to mouse genes and their human orthologs with the “Fuji plot” tool within GWAS meta-analysis
(Bogue et al. 2023). Additionally, within the GWAS meta-analysis tool on MPD, users can view the variant effect on each measure with the P-M
plot and Forest plot (Han and Eskin 2012) to explore whether the effect is broad or specific to a measure or factor (e.g., sex, treatment).
We took two different approaches to assess the utility of using GenomeMUSter for GWAS meta-analyses for the discovery of variants and gene targets related to complex disease. For the meta-analyses on measures of the physiological response to exposure to a high-fat diet and glucose-related endpoints, we evaluated the human ortholog genes identified from these meta-analyses against the set of human diseases and conditions annotated to the human ortholog genes by the Alliance of Genome Resources (Kishore et al. 2020; for disease Homo sapiens associations, see https://www.alliancegenome.org/downloads). For the addiction-related meta-analyses, we took the opposite approach, going from human to mouse. We compared the set of human genes associated with poly-SUD through a cross-ancestry meta-analysis of observations from 1,025,550 individuals of European ancestry and 92,630 individuals of African ancestry (Hatoum et al. 2023) with the human ortholog genes identified through five meta-analyses of mouse measures annotated to Vertebrate Trait ontology terms in the MPD. Two methods were used to assign significant genes from the human GWAS. First, in the European sample, we used MAGMA gene-based testing at the level of whole genes (de Leeuw et al. 2015), controlling for linage disequilibrium across genes and gene size. Second, we took genome-wide SNPs that were significant in the European GWAS and the cross-ancestry GWAS and mapped them to any gene within 10 kb of the significant gene for Europeans and directly on the gene for cross-ancestry (Hatoum et al. 2023).
Accessing and using GenomeMUSter
GenomeMUSter and all individual data sets that went into building GenomeMUSter are stored in a specially constructed 8.1T instance of Google BigQuery, with a database schema consistent with many human data sets in BigQuery, for example, 1000 Genomes Project (Siva 2008), ClinVar (Landrum et al. 2016), gnomAD (Koch 2020). The database was designed to enable the efficient query of about 70 billion genotypes across these strains and to be easily joined with elements from other data sets in BigQuery. This database is integrated with the publicly accessible MPD user interface (UI) and application programming interfaces (APIs), providing the research community with ease of access to these data. Variant annotations to authoritative nomenclature, coordinates, and disease attributions are provided by the Mouse Variant Registry (MVAR; https://mvar.jax.org). MVAR is an annotation service that evaluates submitted variant data to determine whether any of the variants are equivalent to ones already in a repository (i.e., canonicalization). MVAR determines the type and consequence of variants using Jannovar (Jäger et al. 2014). Associations of variants to phenotypes using terms in the Mammalian Phenotype Ontology (MPO) (Smith and Eppig 2009) are provided through expertly curated annotations from the Mouse Genome Database (MGD) (Blake et al. 2021).
GenomeMUSter and each of the individual data sets are publicly available via UI and API at https://muster.jax.org (RRID:SCR_024214). Search inputs include gene symbols, reference SNP identifiers (rsIDs), coordinates and/or coordinate ranges, and flanking regions. These inputs are used to search GenomeMUSter or, optionally, the individual variant sets across the selected strains or strain panels. All data in GenomeMUSter are in Genome Reference Consortium Mouse Build 38 (GRCm38) coordinates; however, we also provide access to Sanger SNP data in GRCm39 (REL2021), as well as an optional liftOver from GRCm38 to GRCm39 coordinates via pyliftover v0.4 (https://pypi.org/project/pyliftover/). The search result table (Supplemental Fig. S6) contains chromosome, location (bp), observed alleles, functional annotations from MVAR, gene symbol (MVAR), and reference strain genotype data (classical C57BL/6J), followed by the remaining strains (default in alphabetical order). The confidence level, shown as a heat map (Supplemental Fig. S6), equals one for known genotypes and is between zero and 0.999 for imputed genotypes. It represents the strain-specific estimated accuracy in each 10-Mb region, and the slider can be used to adjust the confidence level threshold to return only genotypes with confidence levels above a user-selected threshold. Results may also be filtered to only show rows with strain polymorphisms by selecting the radio button in the search query. Thresholds persist when downloading the table or the full query via API. GenomeMUSter can be accessed directly at https://muster.jax.org and through variant association tools in MPD (Bogue et al. 2023; https://phenome.jax.org).
GenomeMUSter is a dynamic and versioned resource, which is updated as new data sets become available. Additional genotype or sequence data submissions may be made to this public data resource by contacting phenome@jax.org.
Data access
The data set generated for this paper and individual variant sets used to construct the merged and harmonized data set are available at https://muster.jax.org. Code used to harmonize these data and impute missing allelic states is available at GitHub (https://github.com/TheJacksonLaboratory/GenomeMUSter) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
Funding was provided (NIH) by National Institutes of Health DA028420, Center for Systems Neurogenetics of Addiction P50 DA039841 and by The Jackson Laboratory, The Cube Initiative Program Fund. We thank Drs. Laura Reinholdt and Beth Dumont for editorial review of this manuscript, and Dr. Reinholdt's assistance with HaploQA. HaploQA was partially funded by the Mutant Mouse Resource and Research Center and Special Mouse Strain Resource at The Jackson Laboratory (U42 OD10921, P40 OD011102). A.S.H. was supported by NIH K01 AA030083. D.G.A., L.L., R.W.W., and the BXD sequencing effort were supported by the UT Center for Integrative and Translational Genomics and funds from the UT-ORNL Governor's Chair. G.P. and F.Z. were supported by NIH/National Institute on Drug Abuse (1 U01DA044399-01), NIH/Office of Research Infrastructure Programs (1R24OD03540801), and NIH/National Institute on Deafness and Other Communication Disorders (1R01DC021133) awarded to G.P. We thank Lisa Tarantino, J. David Jentsch, Jane S. Adams, Stephen Grubb, Matthew Dunn, Benjamin L. Walton, Beena Kadakkuzha, and members of the Computational Sciences Service at The Jackson Laboratory, supported by the JAX Cancer Center support grant (P30 CA034196), for expert assistance with the work described in this publication.
Author contributions: All authors reviewed the final manuscript. G.A.C., Z.F., G.P., L.L., R.W.W., and D.G.A. provided variation data and assisted in their incorporation into GenomeMUSter. A.L., J.B., M.K., A.K.B., S.D., B.A.S., and D.O.W. contributed to the user interface design and development. B.E.K., F.C., G.K.-R., and C.J.B. provided the variant accessioning and annotations. R.L.B., H.L., A.S., M.W.G., H.H., K.S., and J.E. performed the analysis and built the data platform. A.S.H. and A.A. provided scientific expertise and data for the polysubstance use meta-analysis. R.L.B., M.A.B., V.M.P., and E.J.C. contributed the overall design and scientific leadership for this work.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278157.123.
-
Freely available online through the Genome Research Open Access option.
- Received August 8, 2023.
- Accepted January 10, 2024.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








