
Dissecting and improving gene regulatory network inference using single-cell transcriptome data
- 1Center for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, China, 100871;
- 2The MOE Key Laboratory of Cell Proliferation and Differentiation, School of Life Sciences, Peking University, Beijing, China, 100871;
- 3Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, China, 100871
Abstract
Single-cell transcriptome data has been widely used to reconstruct gene regulatory networks (GRNs) controlling critical biological processes such as development and differentiation. Although a growing list of algorithms has been developed to infer GRNs using such data, achieving an inference accuracy consistently higher than random guessing has remained challenging. To address this, it is essential to delineate how the accuracy of regulatory inference is limited. Here, we systematically characterized factors limiting the accuracy of inferred GRNs and demonstrated that using pre-mRNA information can help improve regulatory inference compared to the typically used information (i.e., mature mRNA). Using kinetic modeling and simulated single-cell data sets, we showed that target genes’ mature mRNA levels often fail to accurately report upstream regulatory activities because of gene-level and network-level factors, which can be improved by using pre-mRNA levels. We tested this finding on public single-cell RNA-seq data sets using intronic reads as proxies of pre-mRNA levels and can indeed achieve a higher inference accuracy compared to using exonic reads (corresponding to mature mRNAs). Using experimental data sets, we further validated findings from the simulated data sets and identified factors such as transcription factor activity dynamics influencing the accuracy of pre-mRNA-based inference. This work delineates the fundamental limitations of gene regulatory inference and helps improve GRN inference using single-cell RNA-seq data.
An overarching goal of biology is understanding the regulatory principles of cell states and fates (Waddington 1957; Huang et al. 2009; MacArthur et al. 2009; Ferrell 2012). A critical step toward this goal is to reconstruct gene regulatory networks (GRNs) (Tavazoie et al. 1999; D'haeseleer et al. 2000; Lee et al. 2002; Yeung et al. 2002; Segal et al. 2003; Friedman 2004; Levine and Davidson 2005; Alon 2006; Davidson 2010; Gerstein et al. 2012; Thompson et al. 2015), with which one could delineate the global transcriptional regulatory architecture, and identify key players in the network that control cell states and transitions between them. The reconstruction of GRNs involves inferring edges linking between regulator genes and target genes, and has been traditionally based on population-level microarray or RNA-seq data (D'haeseleer et al. 2000; Segal et al. 2003; Friedman 2004; Margolin et al. 2006; Hecker et al. 2009; Huynh-Thu et al. 2010; Marbach et al. 2012; Sonawane et al. 2017). With the advent of single-cell RNA-seq (scRNA-seq) techniques, it is becoming possible to reconstruct GRNs using scarce and precious tissue samples, yielding unprecedented details into the regulatory underpinnings of key biological processes, including animal development, tumor progression, and immune response (Bendall et al. 2014; Trapnell et al. 2014; Aibar et al. 2017; Chan et al. 2017; Matsumoto et al. 2017; Fiers et al. 2018; Iacono et al. 2019; Pratapa et al. 2020; Qiu et al. 2020, 2022; Van de Sande et al. 2020; Deshpande et al. 2022). However, despite numerous efforts, reconstructing GRNs from scRNA-seq data has remained challenging (Fiers et al. 2018; Pratapa et al. 2020; Argelaguet et al. 2021).
Despite the growing list of methods and algorithms to infer regulatory links from scRNA-seq data (Nguyen et al. 2021), benchmarking results have shown that methods that purely rely on gene expression data often cannot consistently outperform a random predictor (Pratapa et al. 2020). Alternative methods have been developed to overcome this by integrating gene expression data with additional knowledge (Aibar et al. 2017; Yuan and Bar-Joseph 2019; Qiu et al. 2020; Van de Sande et al. 2020). For example, the widely used method, SCENIC (Aibar et al. 2017), leverages regulator binding motif information derived from ChIP-seq data sets to purge links learned from the gene expression data. Nevertheless, despite some success, these alternative methods do not address the fundamental challenge of GRN inference using scRNA-seq data, that is, the challenge to accurately infer gene regulatory relationships from gene expression data only.
To better understand this challenge, it would be helpful to closely examine gene regulation, and revisit the rationale underlying GRN inference using scRNA-seq data. During gene regulation, the transcription factor (TF)'s activity level influences the target gene's expression level (Fig. 1A). The rationale to infer such a relationship using scRNA-seq data is conventionally built upon the assumption that the target's mRNA level can report upstream regulatory activity, and the latter can be approximated by the TF's mRNA level (Fig. 1B). However, a series of factors, including the inherent stochasticity of biochemical reactions and expression levels or dynamics of genes in the network, could undermine this assumption, affecting the accuracy of relationship inference. Although the effect of these factors in GRN inference has been previously suggested or analyzed (Cao et al. 2020; Qiu et al. 2020; Gupta et al. 2022), we nevertheless lack a systematic understanding of how network inference is limited by various factors, and addressing this issue could help improve GRN inference.
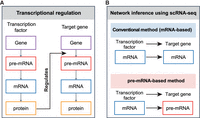
Schematics of gene regulation and regulatory inference methods. (A) Schematic diagram of molecular species involved in transcriptional regulation. (B) Schematics comparing conventional and proposed methods for gene regulatory network (GRN) inference. Conventionally, mRNA levels of transcription factors and target genes from scRNA-seq data are used to infer GRN (i.e., the mRNA-based method). In contrast, we propose to use mRNA levels of transcription factors and pre-mRNA levels of target genes for inferring GRN (i.e., the pre-mRNA-based method).
Here, we quantitatively delineated constraints on GRN inference using chemical kinetic modeling, simulated data sets, and public experimental data sets, and showed that incorporating intronic reads in raw scRNA-seq data can improve GRN inference. Our systematic dissections illustrated fundamental constraints for network inference and helped pave new avenues for improving GRN inference using scRNA-seq data.
Results
A bottom-up dissection of gene regulatory inference using kinetic modeling
To delineate constraints for gene regulatory inference, we first took a bottom-up approach by focusing on the chemical reactions involved in gene regulation, and asked how parameters in these reactions could affect the accuracy of network inference. The rationale for a bottom-up dissection is that network inference involves deducing the relationship between regulator activity and target expression, yet regulator activity level may not be accurately reported by target expression level during gene regulation, placing fundamental limits for network inference. In other words, when the target gene expression level barely reports regulator activity level, it would be very challenging to infer their relationships with any algorithms.
To quantitatively describe gene regulation, we included the reaction of transcription, which produces pre-mRNAs of the target gene, the reaction for splicing, which produces mature mRNAs from pre-mRNA molecules, and the reaction for mRNA degradation (Fig. 2A; Supplemental Fig. S1A; Methods). Based on these reactions, we obtained differential equations describing the rates of change for pre-mRNA and mRNA levels (Supplemental Fig. S1A; Methods). To make the model more amenable to systematic parameter analysis, we used a Boolean description of the gene activity, with its initial state being the OFF state and its subsequent state being controlled by pulse-like regulator activity dynamics (Methods). Using this model, we could quantify how accurate target gene expression level can be used to infer upstream regulator activity level, and how this accuracy is affected by a range of factors, including stochasticity in the reactions, regulator dynamics, and rates of reactions for transcription, splicing, and degradation (Fig. 2A). These analyses would help elucidate how factors intrinsic to gene regulatory reactions could affect the accuracy of gene regulatory inference.

Using kinetic modeling to compare pre-mRNA-based and mRNA-based GRN inference methods. (A) The inference of regulatory activity, g(t), using target gene expression level (either pre-mRNA level or mRNA level) can be affected by various factors. These factors include the dynamics of the regulatory activity (i.e., Ton), the transcription rate of the target gene (α), the splicing rate of the pre-mRNA (β), and the degradation rate of the mRNA (γ). (B) Heatmaps showing the relative accuracy between pre-mRNA-based and mRNA-based inference of the regulatory activity in the absence of stochasticity. The model (Supplemental Fig. S1A) was analytically solved using indicated parameter combinations and the relative inference accuracies between pre-mRNA-based and mRNA-based methods were calculated (Methods). The redder the color, the more accurate the pre-mRNA-based method is. (C) Heatmap showing the relative accuracy between pre-mRNA-based and mRNA-based inference of the regulatory activity in the presence of stochasticity. The model (Supplemental Fig. S1A) was stochastically solved using indicated parameter combinations, and the relative inference accuracies between pre-mRNA-based and mRNA-based methods were calculated. Note that under low gene expression levels and slow regulatory dynamics, the pre-mRNA-based method underperforms compared to the mRNA-based method.
We quantified inference accuracy by measuring how accurately the target gene expression level can capture upstream regulator activity level, defined as the fraction of time that gene expression level matches with regulator activity level (Supplemental Fig. S1B). We then modeled how a typical gene (e.g., with experimentally determined parameters averaged across genes; see Methods) responds to a pulse of regulator activity (Supplemental Fig. S1C), and found that using the mRNA level of the target gene to infer the regulator activity level resulted in a much lower accuracy compared to the pre-mRNA level. The difference in inference accuracy was observed under different types of regulator activity dynamics (Supplemental Fig. S1D). Mechanistically, such a difference comes from the different half-lives of pre-mRNA and mRNA, as the time scale of splicing is ∼10 min and the time scale of mRNA degradation is typically several hours, allowing the pre-mRNA level to reach steady state faster than the mRNA level. As a result, the degree of improvement in inference accuracy using pre-mRNA depends on the time scales of splicing and mRNA degradation (Fig. 2B, left). It also depends on the time scale of regulator dynamics (i.e., fast or slow regulation, Fig. 2B, right; Supplemental Fig. S1E). Notably, for most parameters, pre-mRNA can provide a more accurate inference of regulatory activity (Fig. 2B).
Although the preceding results indicated that pre-mRNA could better capture upstream regulator activity, we speculated that network inference using pre-mRNA level would be more sensitive to noise than using mRNA level, as the steady-state level of pre-mRNA is much lower than that of mRNA. Indeed, when introducing stochasticity to the model (Supplemental Fig. S1F; Methods), the advantage of pre-mRNA was reduced for most parameter sets, and could even reverse when the transcription rate is very low and the regulator dynamics are very slow (Fig. 2C; Supplemental Fig. S1G).
In summary, using a chemical kinetic model, we demonstrated that factors intrinsic to the gene regulatory system can individually or combinatorially affect the accuracy of network inference, resulting in a (theoretical) upper limit of inference accuracy (which depends on gene-specific and regulator-specific kinetic parameters). Importantly, we found that the upper limit of the inference accuracy is generally higher when using the pre-mRNA level of the target gene than using the mRNA level, except for genes whose transcription rates are very low and are under the regulation of very slow regulatory dynamics. It should be noted that whereas parameters in our model simulations were estimated from experimental data, these simulations are only for deriving general principles of how various factors affect inference accuracy and likely cannot capture many of the biologically relevant scenarios (please refer to Supplemental Note S1 for further discussions on model simulations).
Comparing mRNA-based and pre-mRNA-based inference methods using simulated data sets
Because we only considered the regulation of one gene in the chemical kinetic model, we asked whether the conclusions from this model would apply to complex systems containing networks of genes. To systematically address this, we resorted to synthetic single-cell data sets generated by a state-of-the-art single-cell simulation engine, dyngen (Cannoodt et al. 2021), which could simulate stochastic pre-mRNA and mRNA dynamics for gene regulatory networks. With this tool, we could assess the effects of network-level and gene-level factors on GRN inference using either pre-mRNA or mRNA levels of target genes (Fig. 1B), which may provide a systematic picture of factor dependency of inference accuracy (Fig. 3A; Supplemental Fig. S2; Methods).
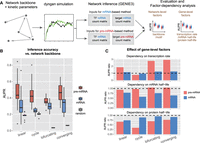
Using simulated single-cell data sets to compare pre-mRNA-based and mRNA-based methods. (A) Schematic illustration of pipelines used for the generation of simulated data sets, the inference of the GRN, the evaluation of inferred GRN, and factor-dependency analysis. Simulated data sets were generated using the dyngen package, whereby different network backbones and kinetic parameters were used for performing dynamic simulations. The output count matrices were then used for network inference using GENIE3. The accuracies of the inferred networks were then calculated and used for factor-dependency analysis. See Methods for details. (B) Boxplots comparing the performance of pre-mRNA-based and mRNA-based methods in four different network backbones. The performance was determined by the accuracy of the inferred network, measured by AUPR (area under precision-recall curve). Meanwhile, AUPR for a random predictor is included for comparison. N = 20. (C) Factor-dependency analysis for pre-mRNA-based and mRNA-based methods. Simulations were performed under parameter ranges of three gene-level factors (transcription rate, mRNA half-life, and protein half-life). The effect of each factor on the inference accuracies of different network backbones was evaluated using AUPR ratio, calculated as the ratio of the AUPR under the largest parameter value to the AUPR under the lowest parameter value (see also Supplemental Fig. S4A–C). Black dashed lines indicate AUPR ratio of 1 (i.e., no effect of the parameter choice on inference accuracy).
We first asked whether leveraging pre-mRNA levels of target genes could improve GRN inference compared to using mRNA levels. To address this, we simulated networks containing different backbones (Supplemental Fig. S3A), including linear, cycle, bifurcating, and converging backbones. In these networks, we observed four general patterns of regulator activity dynamics, with each network displaying a distinct combination of dynamic patterns (Supplemental Fig. S3B,C). We then used a random forest-based algorithm, GENIE3 (Huynh-Thu et al. 2010), to reconstruct GRNs from synthetic single-cell data sets, and quantified the inference accuracy using AUPR (area under the precision-recall curve) (Fig. 3A; Methods). We found that the pre-mRNA-based method confers a significantly higher accuracy of the reconstructed GRN compared to using mRNA levels for all four network backbones (Fig. 3B). In addition to GENIE3, we tested five other methods of GRN inference (including Pearson's correlation, propr [Quinn et al. 2017], ARACNE [Margolin et al. 2006], PIDC [Chan et al. 2017], and TIGRESS [Haury et al. 2012]) and found that the pre-mRNA-based method consistently outperforms the mRNA-based method, and that combining pre-mRNA and mRNA counts did not provide an advantage compared to the pre-mRNA-based method (Supplemental Fig. S3D).
When comparing the relative performance between pre-mRNA-based and mRNA-based methods, we found that the pre-mRNA-based method achieved the highest relative performance for the cycle backbone, whereas for the bifurcating backbone, the relative performance is the lowest (Supplemental Fig. S3E). This result indicated that network topology affects the extent of improvement when using the pre-mRNA-based method, and in networks where regulator activities exhibit more dynamics (e.g., the cycle backbone), the pre-mRNA-based method appears to confer a relatively higher advantage compared to the mRNA-based method. It should be noted that we simulated relatively small networks because of limited computational resources and it would be helpful to further investigate the above findings by simulating larger networks.
Analyzing the effect of gene-level factors using simulated data sets
Although GRN inference could be improved by using pre-mRNA, the inference accuracy (as quantified by AUPR) is far below 1 (Fig. 3B). We speculated that the relatively low accuracy of inferred GRNs could arise from at least two different categories of factors. The first category is gene-level factors, such as kinetic parameters of genes, as suggested by the results from the kinetic model. The other category is related to network topology, such as the abundance of motifs. For example, the two leaf nodes of a bi-fan motif could be falsely connected during inference (Marbach et al. 2010). Both types of factors would lower the theoretical upper bound of the inference accuracy and pose challenges for GRN inference. Quantifying how the accuracy of inferred GRNs depends on gene-level and motif-level factors may provide guidance for improving GRN inference.
To dissect gene-level factors, we explored how the inference accuracy of all four types of networks is affected by kinetic parameters of genes in the network, such as transcription rate, mRNA degradation rate, and protein degradation rate (which affects regulator activity dynamics). To do so, we measured the parameter dependency of AUPR using simulated data sets generated by scanning individual kinetic parameters across a wide range (Supplemental Fig. S4). We found that the pre-mRNA-based method is generally more susceptible to changes in transcription rate compared to the mRNA-based method, and when the transcription rate is very low, the performance of the pre-mRNA-based method drops below that of the mRNA-based method (Supplemental Fig. S4A). This result is consistent with the finding from the kinetic model (Supplemental Fig. S1G). Notably, the cycle backbone behaved differently from other backbones (Supplemental Fig. S4A), which we will dissect later.
For the second kinetic parameter, mRNA half-life, we found that the mRNA-based method is much more susceptible to it than the pre-mRNA-based method for all four backbones (Supplemental Fig. S4B). More specifically, with a shorter mRNA half-life, the mRNA-based method can achieve higher precision. Such dependency is consistent with the finding from the kinetic model (Fig. 2B), that is, when mRNA half-life is shorter, the time delay between mRNA level and upstream regulator activity is shorter, and thus the mRNA level can allow a more accurate inference. For the third kinetic parameter, protein half-life, we found that the pre-mRNA-based method is much more susceptible to it compared to the mRNA-based method for all four backbones (Supplemental Fig. S4C). More specifically, with a shorter protein half-life, the pre-mRNA-based method can achieve higher precision, as the regulator dynamics would be more rapid and thus could be better distinguished from other regulators. This dependency was similarly observed in the kinetic model (Supplemental Fig. S1E). Notably, these parameters can combinatorially affect the inference accuracy (Supplemental Fig. S4D,E). For completeness, we also analyzed the effect of splicing time (Supplemental Fig. S4F) and found that it affects pre-mRNA-based method but not mRNA-based method, and that if the splicing time is longer at a given transcription rate, the count number of pre-mRNA will be larger, and thus the inference will be less affected by stochastic noise.
Together, except for the dependency of cycle backbone on transcription rate, other networks display parameter dependencies that are overall consistent with findings from the kinetic model. These results suggest that whereas the pre-mRNA-based method generally outperforms the mRNA-based method, gene-level factors greatly affect the inference accuracy of either method (Fig. 3C).
Analyzing network-level factors using simulated data sets
Because genes can assemble into network motifs, gene pairs in the same motif may have correlated expression levels without having regulatory relationships, giving rise to motif-related errors that affect inference accuracy. It was previously shown that four types of motif-related errors are common in network inference, namely fan-out error, fan-in error, cascade error, and feed-forward loop (FFL) error (Marbach et al. 2010). Out of the four motifs, we focused on fan-out and cascade motifs (Supplemental Fig. S5A,B), as the other two motifs are not enriched in the four simulated network backbones. By analyzing pre-mRNA-based inferred GRNs from the simulated data sets, we found that fan-out error pervades all four backbones (Supplemental Fig. S5C; Methods), whereas cascade error is only significant in bifurcating and converging backbones (Supplemental Fig. S5D). These results demonstrated that motif-related errors (Supplemental Fig. S5E) affect the accuracy of inferred GRNs in a network backbone-dependent manner.
To further dissect network backbone-specific motif errors, we focused on the cycle backbone, as the preceding gene-level analysis showed that the inference accuracy of the cycle network exhibits unexplained dependency on the gene transcription rate (Supplemental Fig. S4A). We thus sought to address whether such unexplained dependency could be accounted for by motif-related errors.
As a notable feature in the motif analysis result, the fan-out error in the cycle network has prediction confidence even higher than true links in the network (Supplemental Fig. S5C), that is, the two genes at the leaf nodes of the fan-out motif were inferred as having regulatory relationships much more often than gene pairs that have true regulatory relationships. By examining the temporal dynamic traces of all three genes in a fan-out motif inside a cycle network, we found that indeed the two leaf-node genes are temporally correlated (Supplemental Fig. S5F). Thus, the relatively high fan-out error appeared to depend on the cycling dynamics of the upstream regulator. We observed that the dependency of network inference accuracy on transcription rate is dependent on protein half-life (e.g., the degree of regulator dynamics) (Supplemental Fig. S5G), and thus regulator dynamics-dependent motif errors (e.g., fan-out errors) could potentially account for the abnormal dependency of inference accuracy on gene transcription rate for the cycle backbone (Supplemental Fig. S4A). To test this, we calculated fan-out and cascade errors under varying kinetic parameters, and found that the fan-out error becomes more significant as the protein half-life increases to 5 h or above (Supplemental Fig. S5H). In contrast, when the protein half-life is short (1 or 2 h), the fan-out error is not as significant (Supplemental Fig. S5H), and the abnormal dependency of inference accuracy on gene transcription rate disappears (compare Supplemental Fig. S4A cycle backbone with Supplemental Fig. S5G).
Together, these results explained the abnormal dependency of inference accuracy on transcription rate for the cycle network when using the pre-mRNA-based method, and more generally, highlighted that gene-level and network-level factors can combinatorially constrain GRN inference accuracy in a network backbone-dependent manner.
Evaluating GRN inference using public scRNA-seq data sets
Thus far, we have systematically dissected factors constraining GRN inference accuracy, and have demonstrated that the pre-mRNA-based method outperforms the mRNA-based method under most scenarios. Yet, the comparison between the two methods was based on in silico data, and it is unclear whether the pre-mRNA-based method would still perform as well in real data sets.
To compare the two methods using real data sets, we needed to address two challenges. First, unlike simulated data sets that contain measurements of mRNA and pre-mRNA levels, typical scRNA-seq data is acquired by isolating and sequencing poly(A)-containing mRNA molecules, and thus in principle does not measure pre-mRNA molecules directly. However, several recent studies have shown that many scRNA-seq data contains reads that can be mapped to intronic regions of genes (Gaidatzis et al. 2015; La Manno et al. 2018; Wu et al. 2022), most likely arising from the binding of Oligo(dT) primers to the internal adenosine-rich regions of pre-mRNA molecules (La Manno et al. 2018), allowing one to estimate the expression levels of pre-mRNAs using intronic reads. Second, compared to simulated data sets that have ground-truth GRNs, real data sets acquired with natural systems do not have ground-truth GRNs, posing a critical challenge to the evaluation of inference accuracy. To alleviate the challenge, we relied on two types of “ground-truth” GRNs for evaluation. The first ground-truth GRN is a network with edges having A or B confidence scores in the DoRothEA database (Garcia-Alonso et al. 2019; Methods), a carefully curated database integrating multiple data sources. DoRothEA network has a relatively low number of nodes and edges, but high confidence levels. The second ground-truth GRN is from TF binding motifs (curated using ChIP-seq data, designated as Motif GRN) (Aibar et al. 2017), which has many more nodes and edges compared to DoRothEA network, but with relatively low confidence levels (Methods).
Next, by using intronic reads to approximate pre-mRNA levels and using two complementary ground-truth GRNs, we sought to infer GRNs and evaluate inference accuracy using 30 public scRNA-seq human and mouse data sets (Supplemental Table S1; Methods). The rationale for choosing these data sets is twofold: the underlying biological processes are representative of key mammalian processes, and these data were acquired using 10x Genomics platform and are thus comparable in terms of sequencing library construction. To infer GRNs, we created pre-mRNA (i.e., based on intronic reads) and mRNA (i.e., based on exonic reads) expression matrices for each data set, which were fed into the GENIE3 algorithm (Huynh-Thu et al. 2010; Methods). Because of the large size and the sparse nature of GRNs, we devised a metric, average early precision (AEP), to evaluate the inferred GRNs, which measures the accuracy of the top network (Methods).
Using DoRothEA network as the ground truth, we found that above a threshold level of precision (i.e., ∼0.15), the pre-mRNA-based GRNs were more accurate than mRNA-based GRNs (Fig. 4A,C). Yet for GRNs with generally low precision (below 0.15), the two methods were comparable (Fig. 4A,C), which could be due to the relatively small network size of the DoRothEA GRN. We then asked whether the enhanced inference accuracy of pre-mRNA-based GRNs would be susceptible to dropouts in reads. To address this, we subsampled both intronic and exonic reads to ∼30% and performed GRN inference using subsampled reads. We found that whereas the inference accuracies of both methods were reduced, pre-mRNA-based GRNs were generally still more accurate than mRNA-based GRNs (Supplemental Fig. S6A), suggesting that the performance of the pre-mRNA-based method was not affected by dropouts more than that of the mRNA-based method. We next evaluated the results with the Motif GRN, which has a much larger network size. The pre-mRNA-based method outperformed the mRNA-based method for most networks (Fig. 4B,D). We further analyzed the performance of our inferred networks using AUPR as a metric, and our evaluation results were found to be similar to those obtained using the AEP (Fig. 4E,F). However, we observed that AUPR is generally less sensitive than AEP, particularly when evaluating the results using the Motif network as ground-truth (Fig. 4F). For some data sets, the mRNA-based method outperformed the pre-mRNA-based method, suggesting the presence of factors contributing to data set–specific performance difference (see Supplemental Note S2 for further discussions). Moreover, because different data sets exhibited varying AUPRs, we asked whether the observed difference in inference accuracy could be attributed to difference in data sparsity in the count matrix. We found that AUPR ratio (i.e., relative to random) from either method is not positively correlated with the numbers of exon or intron UMI per cell (Supplemental Fig. S6B), implicating that inference accuracy is more affected by factors other than data sparsity.
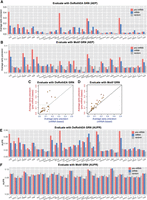
Using experimental single-cell data sets to compare pre-mRNA-based and mRNA-based methods. (A) Comparison between pre-mRNA-based and mRNA-based methods for 30 scRNA-seq data sets of human and mouse cells using DoRothEA GRN as the ground truth. The performance was measured by average early precision (Methods). Precision for a random predictor is shown for comparison. (B) Analogus to A, except that Motif GRN was used as the ground truth. (C) Scatter plots showing the comparison between pre-mRNA-based and mRNA-based methods for 30 scRNA-seq data sets of human and mouse cells using DoRothEA GRN as the ground truth. Diagonal line denotes equal performance. Dashed lines denote precision value at 0.15. (D) Scatter plots analogous to C, except that Motif GRN was used as the ground truth. (E,F) Analogous to A and B, except that AUPR was used as the metric.
Furthermore, we compared inferred GRNs using both methods from data sets in which exogenously induced TFs were used to program cell fates, such that we should be able to recover the TFs being induced in the inferred network without needing to rely on GRN databases as the ground truth. By analyzing public data sets taken at two different time points postinduction (Hersbach et al. 2022), we found that whereas both methods identified exogenously induced TFs as hub nodes in the GRN, only the pre-mRNA-based method appeared to capture the temporal behaviors of the TFs (Supplemental Fig. S6C,D; Supplemental Note S3). Additionally, we compared the two methods using a single-nucleus RNA-seq data set of mouse skeletal myofibers (Petrany et al. 2020), and found that even with an increased intronic read fraction (an average of ∼74%), the performance of the pre-mRNA-based method was not much enhanced when compared to the mRNA-based method and the evaluation of inferred GRNs is limited by the ground-truth database (i.e., the DoRothEA network may lack regulatory interactions specific to this particular cell type) (Supplemental Fig. S6E). Therefore, it is necessary to use multiple evaluation methods to accurately assess the performance of different approaches.
We next focused on the human forebrain data set (hFB) (La Manno et al. 2018), as the pre-mRNA-based method consistently and greatly outperformed the mRNA-based method when evaluated using two ground-truth GRNs. For the top links in the inferred network, pre-mRNA-based GRN achieved a precision of ∼50% when recall is ∼4%, whereas mRNA-based GRN could only achieve a precision of ∼20% when the recall is ∼2% (Fig. 5A).
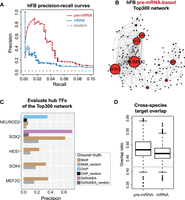
Using the forebrain data sets to demonstrate the improved performance of the pre-mRNA-based method. (A) Precision-recall curves for GRNs inferred by pre-mRNA-based and mRNA-based methods for the human forebrain data set. The ground-truth network is DoRothEA GRN. (B) Inferred GRN for the human forebrain data set using the pre-mRNA-based method. In this network, the edges represent the inferred transcriptional regulation from one TF (transcription factor) to one target gene. The size of the node represents the number of inferred target genes for the TF. 300 interactions (edges) of the highest confidence were shown (i.e., Top300 network). (C) Evaluation of the hub TFs in the pre-mRNA-based Top300 network using different types of ground-truth network. Note that because the DoRothEA ground-truth GRN contains much fewer nodes and edges compared to the Motif ground-truth GRN, only one of the hub TFs could be evaluated using DoRothEA GRN whereas all of them could be evaluated with the Motif GRN. (D) Ground-truth-free comparison between GRNs inferred by pre-mRNA-based and mRNA-based methods using cross-species target overlap. In particular, GRNs from the human forebrain data set (hFB) and the mouse forebrain (mFB) were evaluated, whereby the overlap ratio of top 500 targets in two networks for each TF was calculated. Such overlap ratio represents the cross-species similarity between the two networks, and was compared between pre-mRNA-based and mRNA-based GRNs. N = 372 TFs and P-value = 5 × 10−9 from Wilcoxon test. Gray horizontal line indicates random overlap ratio.
To analyze the biological relevance of the inferred GRN from the human forebrain data set, we extracted the high-precision top network of the pre-mRNA-based GRN, and analyzed the hub TFs in the top network (Fig. 5B). We found that the hub TFs are all related to human neural development, consistent with the biological process during which the data was acquired (which contrasts with mRNA-based GRN (Supplemental Fig. S6F)). We further evaluated the precision of each hub TF using the Motif GRN as the ground truth, and found that all these hub TFs, except the largest hub TF NEUROD2, have much higher precision than random, with a median precision of ∼35% (Fig. 5C). To account for the small increase in precision relative to random control for NEUROD2, we speculated that it was because of the lack of reliable motif data for this TF, and thus resorted to cell type–specific ChIP data (Bayam et al. 2015) as the ground truth to further evaluate the inferred targets of NEUROD2. With the cell type–specific ChIP data, NEUROD2 has a precision of ∼35% (Fig. 5C), illustrating the importance of using relevant ground truth for evaluation. Furthermore, using cross-species target overlap as a ground-truth-free evaluation method, we found that between mouse and human forebrain data sets (mFB and hFB), pre-mRNA-based GRNs contain more overlapping target genes compared to mRNA-based GRNs (Fig. 5D).
Besides the mouse and human forebrain data sets, we also analyzed the biological relevance of top five hub TFs from mRNA-based or pre-mRNA-based GRN for four other data sets, and found that hub TFs inferred by the pre-mRNA-based method were more related to the corresponding biological processes (Supplemental Table S2). Additionally, we noted that ribosomal proteins were more frequently inferred as TFs in the mRNA-based method compared to the pre-mRNA-based method (Supplemental Table S2).
Taken together, we demonstrated that the pre-mRNA-based method generally outperforms the mRNA-based method for experimental scRNA-seq data sets, and that the ground-truth-free method of evaluation using forebrain data sets provides consistent support.
Factor-dependency analysis of inferred GRNs from experimental data sets
Although we have established the overall advantage of the pre-mRNA-based method over the mRNA-based method with experimental data sets, it remained unclear what affects the relative performance between the two methods and whether such an advantage arises from the same factors as illustrated in the simulated data sets.
We focused on the factor-dependency analysis of specific GRNs and explored whether the inference accuracy of links within GRNs depends on factors related to the target gene or the TF (Fig. 6A). We chose four experimental data sets, whereby for all data sets, the pre-mRNA-based method outperformed the mRNA-based method using Motif ground-truth GRN, but with varying degrees of improvements (Fig. 4B). We first focused on factors related to target genes, including expression level and mRNA half-life, which relate to kinetic parameters of genes and have been shown to affect inference accuracy in simulated data sets. To analyze the effect of the target gene's expression level on inference accuracy, we sorted target genes into two bins by their expression levels (separately for pre-mRNA and mRNA), and averaged the top-10-precision of each target (i.e., mean inference precision of the top 10 inferred TFs of each target evaluated using Motif GRN) within each bin (Supplemental Fig. S7A). We found that for all four data sets, the mean top precision of the GRN inferred with the pre-mRNA-based method exhibits a much stronger dependency on the target's pre-mRNA level compared to the dependency of the mRNA-based method on the target's mRNA level (Fig. 6B). These results are consistent with the findings from the kinetic model (Supplemental Fig. S1G) and simulated data sets (Fig. 3C), indicating that target gene expression levels affect inference accuracy in real data sets as well.
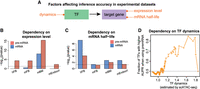
Factor-dependency analysis for GRNs inferred from experimental data sets. (A) Schematic diagram of the factor-dependency analysis. Three factors that affect the accuracy of the inferred network were analyzed, i.e., the dynamics of the TF, the expression level of the target gene (pre-mRNA or mRNA), and the mRNA half-life of the target gene. (B) The dependency of the network inference accuracy on mRNA (or pre-mRNA) expression levels of target genes in four data sets. The dependency was quantified by using the P-value as in Supplemental Figure S7A, calculated by comparing the inference accuracies between targets of low or high expression levels for each network. Horizontal dashed line indicates P = 0.05. (C) Analogous to (B) for the mRNA half-life of the target gene. (D) The dependency of the network inference accuracy on the TF dynamics. The dynamics of TFs were approximated by using cell-to-cell variabilities of TF activities measured by public single-cell ATAC-seq data (Methods). TFs from all four data sets were sorted according to TF dynamics, and the fraction of TFs more accurately inferred by the pre-mRNA-based method than by the mRNA-based method was calculated for TFs above the indicated value of TF dynamics on the x-axis (see also Supplemental Fig. S7B,C).
Compared to the analysis of expression level, analyzing the effect of the target gene's mRNA half-life on inference accuracy is more challenging, as we do not have direct measurements of mRNA half-lives for the cell type of interest. We thus adopted mRNA half-life data from TimeLapse-seq measurement in K562 cells (Schofield et al. 2018), sorted target genes into two bins by their mRNA half-lives, and averaged the top-10-precision of each target within each bin (evaluated using Motif GRN). We found that in all four data sets, the mean top precision of the GRN inferred with the mRNA-based method exhibits a stronger dependency on the mRNA half-life compared to that of the pre-mRNA-based method (Fig. 6C), consistent with findings from simulated data sets (Fig. 3C).
We next focused on TFs, as the regulation time or dynamics of TFs have been shown to affect inference accuracy in our in silico results. To analyze the potential effect of TF dynamics in real data sets, we resorted to an indirect characterization of TF dynamics measured from single-cell ATAC-seq data (Buenrostro et al. 2015; Methods). Across four different data sets, we found that the advantage of the pre-mRNA-based method over the mRNA-based method becomes more obvious when TF activities are more dynamic (Fig. 6D; Supplemental Fig. S7B), consistent with findings from simulated data sets (Supplemental Fig. S4C). Moreover, such a comparison would be further facilitated by using cell type–specific ATAC-seq data (Supplemental Fig. S7C).
In summary, we applied factor-dependency analysis of GRN inference using scRNA-seq data sets. Our analyses indicated that the precision of GRN inferred with the pre-mRNA-based or the mRNA-based method could be explained by factors related to the target gene and the TF. This result supported the hypothesis that the advantage of the pre-mRNA-based method over the mRNA-based method in the experimental data sets arises because pre-mRNA levels could better report upstream TF activities in these data. Thus, GRN inferred using the pre-mRNA-based method generally has a higher precision compared to GRN inferred using the mRNA-based method for both simulated and experimental single-cell data.
Discussion
Researchers are leveraging the ever-growing single-cell transcriptome data for reconstructing GRNs, and many efforts have been devoted to the improvement of inference methods and algorithms (Aibar et al. 2017; Chan et al. 2017; Matsumoto et al. 2017; Pratapa et al. 2020; Qiu et al. 2020; Nguyen et al. 2021). A parallel line of work has been centered on integrating additional omics data with single-cell transcriptome to improve GRN inference (Welch et al. 2017; Stuart et al. 2019; Efremova and Teichmann 2020; Argelaguet et al. 2021; Hao et al. 2021). Although GRN inference using single-cell transcriptome data can indeed be improved by using new methods and algorithms, or by incorporating additional measurement modalities, it is critical to deciphering the limits of GRN inference using single-cell gene expression data alone, as it would provide key guiding principles for improving GRN inference. In this work, we provided a quantitative framework for delineating the effects of gene-level and network-level factors on the accuracy of GRN inference using single-cell transcriptome data, identified fundamental limitations of the inference accuracy, and described a new method that leverages typically disregarded pre-mRNA information for improving GRN inference using single-cell transcriptome measurement alone.
Both the kinetic model and the simulated single-cell data sets helped depict the fundamental limits of GRN inference using the mRNA-based method, which can be alleviated by leveraging pre-mRNA information. In particular, because mRNA is generally more long-lived compared to pre-mRNA, the level of mRNA exhibits a generally lower accuracy compared to pre-mRNA level for reporting the upstream regulator activity. Yet, the accuracy of the pre-mRNA-based method diminishes when the gene is transcribed at a low level, limiting the overall advantage of the pre-mRNA-based method over the mRNA-based method. More generally, whereas the pre-mRNA-based method overall outperforms the mRNA-based method for GRN inference, in silico results provided a systematic picture of the limitations of GRN inference using either method imposed by network backbones and motifs, gene expression level, and TF activity dynamics, demonstrating that inference accuracy is impacted by characteristics intrinsic to genes and that dynamics in the network can be leveraged by the pre-mRNA-based method for improved network inference compared to the mRNA-based method.
The overall advantage of the pre-mRNA-based method was also evident when inferring networks using experimental single-cell RNA-seq data sets. Such an advantage was demonstrated by using two independently sourced ground-truth GRNs (Aibar et al. 2017; Garcia-Alonso et al. 2019) to evaluate inferred networks, and was further supported by ground-truth-free evaluation of two related data sets. Notably, the factor-dependency analysis provided quantitative insights into the effects of gene-level factors on the accuracy of inferred networks, and the observed effects were consistent with findings from corresponding in silico studies. These results provided critical support for the conclusion that the pre-mRNA-based method can offer improved GRN inference with real data sets compared to the mRNA-based method, as we could explain how such improvements are achieved. Yet, several aspects of the proposed GRN inference method could be further improved. For example, to mitigate the generally low intronic read counts, it is possible to aggregate cells with similar transcriptomes; to improve the versatility of the method, it would be intriguing to explore the possibility of a hybrid method whereby both mRNA and pre-mRNA reads are leveraged; to improve the ground-truth networks, it would be helpful to leverage experimental data sets whereby many TFs are directly perturbed to alter cell fates (Hersbach et al. 2022; Joung et al. 2023).
More generally, this work highlights the importance of characterizing and understanding the temporal dynamics in gene regulatory networks. Fundamentally, GRN inference using single-cell gene expression is constrained by temporal mismatches between the target expression level and upstream TF expression level. Leveraging the pre-mRNA level of the target gene partially addresses such temporal mismatches, and the pervasively dynamic TFs in the GRN (Levine et al. 2013) further help alleviate the challenge. Nevertheless, existing approaches cannot address the challenge that temporal and dynamic TF activity may not be accurately captured by TF mRNA expression level; i.e., TF mRNA level may not be a good proxy of its activity. This challenge is reflected in the modest improvements of our new inference method on experimental data sets compared to the mRNA-based method (Fig. 4A–D), which also demonstrated the inherent limitations when using mRNA levels of TFs as a proxy of their activities as it ignores other factors affecting TF activities. It is thus conceivable that a better estimation of TF activity could be introduced to alleviate this challenge, such as by summing over pre-mRNAs from genes in the same regulons (Wu et al. 2022). Furthermore, to overcome the sparsity of intronic reads in typical scRNA-seq data sets, the single-cell transcriptome method using metabolic labeling can be leveraged to measure newly produced mRNAs, which has been shown to improve gene regulatory inference (Cao et al. 2020).
Methods
Kinetic model of gene expression
Details on model and model simulations
We used the kinetic model of gene expression in the literature (La Manno et al. 2018), in which both unspliced (pre-mRNA) and spliced mRNA species are included in the model (designated as u and s, respectively). In the first equation (Equation 1), the unspliced mRNA level (u) is increased by transcription at the rate of α and is reduced by splicing at the rate of βu. In the second equation (Equation 2), spliced mRNA level (s) is increased by splicing at the rate of βu and is reduced by degradation at the rate of γs. More specifically, in these equations, α denotes the transcription rate, β denotes the splicing rate constant of unspliced mRNA, and γ denotes the degradation rate constant of mRNA.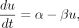 (1)
(1)
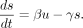 (2)
Because transcriptional regulation is often dynamic, i.e., the transcription factor is temporally activated and deactivated
(Wu et al. 2022), we needed to explore how such dynamics affect gene regulatory inference. To do so, we included TF dynamics in the above
equations by modeling a temporally changing transcription rate that is governed by TF activation and deactivation. More specifically,
we assumed that the transcription rate follows that:
(2)
Because transcriptional regulation is often dynamic, i.e., the transcription factor is temporally activated and deactivated
(Wu et al. 2022), we needed to explore how such dynamics affect gene regulatory inference. To do so, we included TF dynamics in the above
equations by modeling a temporally changing transcription rate that is governed by TF activation and deactivation. More specifically,
we assumed that the transcription rate follows that: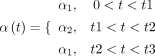 (3)
(3)
In Equation 3, α1 denotes the basal transcription rate without TF activation, α2 denotes the transcription rate upon TF activation, and t1, t2, and t3 denote the time points of step-like changes. More generally, we used g(t) in other places of the manuscript (e.g., Fig. 2A; Supplemental Fig. S1) to denote the temporal pattern of the transcription rate. Typical splicing rate constant and degradation rate constant are 0.1/min and 0.01/min, respectively, which were adopted from experimental values (Rabani et al. 2014). To evaluate the accuracy of gene regulatory inference, we solved these equations analytically and simulated the corresponding master equations. For stochastic simulations, we used the master equation to model the dynamics, which were solved by the “GillespieSSA” package in R (R Core Team 2022).
The calculation of inference accuracy
The inference accuracy is intended to describe the ability of pre-mRNA and mRNA levels to accurately infer upstream regulatory
activity. By quantifying inference accuracies under various model parameters, we can study how diverse factors affect gene
regulatory inference (as illustrated in Fig. 2A). In our model, the regulatory activity has two distinct states: ON and OFF. To calculate accuracy, we first binarize the
downstream expression levels (pre-mRNA or mRNA) by assigning the state ON if the expression level is larger than half of the
maximal value; otherwise, the state is considered OFF. After binarizing the downstream expression levels, we can compare them
with the upstream regulatory activity dynamics (which is a binary trace) to calculate the accuracy. The accuracy is defined
as the proportion of time when the downstream expression level (pre-mRNA or mRNA) can correctly match the upstream regulatory
activity: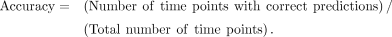 For example, in Supplemental Figure S1B, the mRNA level can correctly reflect the regulatory activity 55% of the time, whereas the pre-mRNA level can do so 95% of
the time. In Figure 2, we used the above metric to determine how well output signals (i.e., pre-mRNA dynamics or mRNA dynamics) track input signal
(i.e., upstream regulatory signal which is binary). As we illustrated, we thus needed to binarize target gene expression levels
based on half maximal value, which allows quantitative determination of whether input and output are matching in time.
For example, in Supplemental Figure S1B, the mRNA level can correctly reflect the regulatory activity 55% of the time, whereas the pre-mRNA level can do so 95% of
the time. In Figure 2, we used the above metric to determine how well output signals (i.e., pre-mRNA dynamics or mRNA dynamics) track input signal
(i.e., upstream regulatory signal which is binary). As we illustrated, we thus needed to binarize target gene expression levels
based on half maximal value, which allows quantitative determination of whether input and output are matching in time.
Evaluating network inference accuracy using simulated single-cell data sets
Generation of simulated single-cell data sets
To evaluate and compare mRNA-based and pre-mRNA-based methods in network inference, we generated synthetic single-cell gene expression data using stochastic simulations of predefined networks. These simulated data sets were generated by the dyngen package (Cannoodt et al. 2021). More specially, we selected four of the default backbones (linear, cycle, bifurcating, and converging), set the number of genes and cells, and generated a series of dynamical trajectories, which were then converted into expression matrices for network inference (note that the outputs of the simulation are mRNA and pre-mRNA counts, instead of intron and exon reads as in experimental data sets). It should be noted that the default splicing rate in the dyngen package (v0.4.0) was inappropriately set for mammalian cells, and we used the splicing rate from the experimentally measured value (Rabani et al. 2014) to replace the default values [splicing_rate = log(2)/splice_time, splice_time = 10 min]. For other parameters, we used the default values in the package.
Network inference for simulated single-cell data
To generate the single-cell expression matrix from the dynamical trajectories output by dyngen, we sampled the trajectories in equal time intervals (1 h) to obtain the input matrix for the GRN inference algorithm. Because experimental single-cell RNA-seq data is typically log normalized, we thus log-normalized the simulated expression counts [i.e., log(count + 1)] in order to better compare with experimental data. For network inference, we used a random forest regression method, GENIE3 (Huynh-Thu et al. 2010), which was developed for use with bulk transcriptomes and has become a standard method for GRN inference with single-cell transcriptomes. Note that in the input gene expression matrix, the information of transcription factor has been used and that only regulatory relationships from one TF to another gene (including other TFs) were considered for inference. For the mRNA-based method, because it is a typically used standard method, we followed the guideline provided in the package instruction and provided GENIE3 with mRNA expression matrices for both TFs and target genes. For the pre-mRNA-based method, we provided GENIE3 with the pre-mRNA expression matrix of the target genes and the mRNA expression matrix for the TFs, whereby the mRNA expression levels of TFs were used to regress the pre-mRNA expression levels of target genes by GENIE3.
Motif error analysis
For the analysis of motif-related errors, we followed the previous work (Marbach et al. 2010). The prediction confidence of a potential regulatory relationship is the rank of this relationship in the inferred network. The link with the highest weight in the inferred network has a prediction confidence of 100%, whereas the link with the lowest weight in the inferred network has a prediction confidence of 0. In Supplemental Figure S5, the links for all motifs were extracted, and the prediction confidence for those links was calculated. The distributions of prediction confidence were shown in the figure. As a comparison, the prediction confidence for background links is also calculated (for all links in the synthetic network, i.e., TRUE, and for links that are not in the synthetic network, i.e., FALSE). Fan-out error and cascade error were analyzed and the prediction confidence was calculated for motif errors and background (all true links and all false links).
Evaluating network inference accuracy using experimental single-cell data sets
Brief description of experimental data sets
We investigated a total of 30 scRNA-seq data sets, all acquired using the 10x Genomics platform (Zheng et al. 2017). These data include single cells of diverse types and states, ranging from human to mouse cells, from cells extracted from tissues to in vitro cultured cells, and from cells in differentiated states to cells with pluripotency. Detailed information regarding data sets can be found in the Supplemental Table S1.
Preprocessing of experimental data sets
Following the established convention (La Manno et al. 2018), we used exon read counts to quantify mRNA levels, and intron read counts to quantify pre-mRNA levels. For a few data sets, the expression matrices (including both intron and exon counts) have been provided by indicated publications, and we directly incorporated these matrices for downstream analysis. However, for most data sets, we needed to download the raw FASTQ files and calculated the expression matrices ourselves. Specifically, we used CellRanger (Zheng et al. 2017) (v3.02) to map the reads, and then used velocyto (La Manno et al. 2018) (v0.17.17) or UMI-tools (Smith et al. 2017) (v1.01) to quantify the unspliced mRNA and spliced mRNA levels. After obtaining the expression matrices, Seurat (Stuart et al. 2019) was used for performing quality control (to remove low-quality cells) and for normalizing expression matrices (LogNormalize).
Network inference procedure
Expression matrices were fed into GENIE3 for GRN inference. To enhance accuracy, we used the TF information (from RcisTarget [Aibar et al. 2017]) to label regulators and only regulatory relationships from one TF to another gene (including other TFs) were considered for inference. Because of the sparsity of scRNA-seq data, only genes expressed in more than 10% of cells were included for analysis. For the mRNA-based method, only spliced mRNA matrices were used to infer the network, and for the pre-mRNA-based method, both unspliced pre-mRNA and spliced mRNA matrices were used.
Ground-truth for network evaluation
We mainly used two databases as ground-truth GRNs for evaluating GRNs inferred from experimental data sets: (1) the GRN from DoRothEA (Garcia-Alonso et al. 2019), with only edges with confidence A and B included; (2) the GRN from RcisTarget (Aibar et al. 2017) (i.e., RcisTarget was used to identify enriched TF-binding motifs for all genes and to obtain candidate TFs). The rationale for choosing these two ground-truth GRNs was that they are complementary, i.e., the regulatory links in DoRothEA have high confidence but low coverage, whereas the regulatory links in RcisTarget have high coverage but low confidence. Besides these two ground-truth GRNs, we also used cell type–specific ChIP-seq for NEUROD2 (Bayam et al. 2015) to evaluate the network inferred from the human forebrain data set (Fig. 5C).
Metrics for evaluating network inference results
We used AUPR and AEP (average early precision) to evaluate the quality of the inferred GRNs, whereby AUPR has been widely used. We proposed to use AEP for networks inferred from typical single-cell RNA data, as explained below. More specifically, when comparing inferred GRN with ground-truth GRN, each link can be assigned into one of the four categories: TP (true positive), FP (false positive), TN (true negative), and FN (false negative). The precision and recall were defined as following: precision = TP/(TP + FP), recall = TP/(TP + FN). Because the output of the inferred GRN is a ranked list of potential regulatory links, a threshold should be chosen to determine the inference results. For each threshold, there is a pair of precision and recall values, and thus a curve can be plotted in the precision-recall plane. The area under the precision-recall curve is defined as AUPR. In this work, AUPR was adopted to evaluate simulated single-cell data sets. As for networks inferred from experimental single-cell data sets, although we also used AUPR, we believed that AEP is a more appropriate and informative metric, which focuses on the most confident links in the inferred network, analogous to the early precision ratio (EPR) used in a recent network inference evaluation study (Pratapa et al. 2020). Compared to EPR, which computes the ratio of the fraction of true positives in the top-k edges over the early precision for a random predictor, AEP measures the average precision in the top network. The top network includes links that have confidence levels ranked at a top threshold percentage, in which the threshold is set to be 10% or 1% when evaluated by DoRothEA or Motif GRN, respectively. The rationale for using AEP is that, by using the average, instead of a single data point as in EPR, AEP could, in principle, provide a more robust measure of early precision.
Factor-dependency analysis
In the networks inferred from four representative experimental data sets, we analyzed the effect of mRNA half-life, the expression level of mRNA or pre-mRNA, and TF dynamics on network inference accuracy by using Motif GRN as the ground-truth (Fig. 6). We calculated the top precision for each target gene, i.e., mean inference precision of the top 10 inferred TFs of each target, and divided all target genes into two groups according to mRNA half-life or expression level. We then compared the top precision of the two groups and calculated the P-values using Wilcoxon rank sum tests. The mRNA half-life data was from TimeLapse-seq in K562 cells (Schofield et al. 2018). For the effect of TF dynamics, we used the accessibility variability data (Buenrostro et al. 2015) to approximate the degree of TF dynamics, and the average variabilities of eight wild-type cells were used. More specifically, regarding the estimation of TF dynamics, Buenrostro and colleagues (Buenrostro et al. 2015) first estimated the activity of TFs in individual cells based on the accessibility of the motifs bound by these TFs (derived from single-cell ATAC-seq data), and then estimated the dynamics of TFs through evaluating the cell-to-cell variability in TF activity. We sorted all TFs by variability score and calculated the fraction of TFs (that are above each variability score) whose performance is better with the pre-mRNA-based method than with the mRNA-based method. The rationale is that by focusing on TFs that are more dynamic, we could ask whether the pre-mRNA-based method would be more advantageous in the presence of dynamic regulatory activities, as demonstrated in the kinetic model and simulated data sets (Supplemental Figs. S1E, S4C). We also further investigated the human forebrain data set using cell type–specific TF variability data (Trevino et al. 2020).
Software availability
The source codes of this study are freely available through GitHub (https://github.com/LingfengXue1999/NetworkInference) and as Supplemental Code. The associated data sets used for GRN inference are available through figshare (https://figshare.com/articles/dataset/Datasets/21342117) and as Supplemental Datasets S1 and S2.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
The authors thank the High-Performance Computing Platform of the Peking-Tsinghua Center for Life Sciences for computational support. Y.L. acknowledges support from the National Key R&D Program of China (grants no. 2020YFA0906900 and no. 2018YFA0900703) and the National Natural Science Foundation of China (grants no. 32088101 and no. 31771425).
Author contributions: L.X.: Conceptualization, investigation, methodology, software, writing – original draft preparation, writing – review and editing; W.Y.: methodology, writing – review and editing; Y.L.: conceptualization, funding acquisition, supervision, writing – original draft preparation, writing – review and editing.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277488.122.
- Received November 9, 2022.
- Accepted August 7, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








