
Cytosolic and mitochondrial translation elongation are coordinated through the molecular chaperone TRAP1 for the synthesis and import of mitochondrial proteins
- Rosario Avolio1,7,
- Ilenia Agliarulo2,7,
- Daniela Criscuolo1,
- Daniela Sarnataro1,
- Margherita Auriemma1,
- Sabrina De Lella1,
- Sara Pennacchio1,
- Giovanni Calice3,
- Martin Y. Ng4,
- Carlotta Giorgi5,
- Paolo Pinton5,
- Barry S. Cooperman4,
- Matteo Landriscina2,6,
- Franca Esposito1 and
- Danilo Swann Matassa1
- 1Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II, Naples 80131, Italy;
- 2Institute of Experimental Endocrinology and Oncology “G. Salvatore”–IEOS, National Research Council of Italy (CNR), Naples 80131, Italy;
- 3Laboratory of Preclinical and Translational Research, IRCCS, Referral Cancer Center of Basilicata, Rionero in Vulture 85028, Italy;
- 4Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, USA;
- 5Department of Medical Sciences, University of Ferrara, Ferrara 44121, Italy;
- 6Department Medical and Surgical Science, University of Foggia, Foggia 71122, Italy
-
↵7 These authors contributed equally to this work.
Abstract
A complex interplay between mRNA translation and cellular respiration has been recently unveiled, but its regulation in humans is poorly characterized in either health or disease. Cancer cells radically reshape both biosynthetic and bioenergetic pathways to sustain their aberrant growth rates. In this regard, we have shown that the molecular chaperone TRAP1 not only regulates the activity of respiratory complexes, behaving alternatively as an oncogene or a tumor suppressor, but also plays a concomitant moonlighting function in mRNA translation regulation. Herein, we identify the molecular mechanisms involved, showing that TRAP1 (1) binds both mitochondrial and cytosolic ribosomes, as well as translation elongation factors; (2) slows down translation elongation rate; and (3) favors localized translation in the proximity of mitochondria. We also provide evidence that TRAP1 is coexpressed in human tissues with the mitochondrial translational machinery, which is responsible for the synthesis of respiratory complex proteins. Altogether, our results show an unprecedented level of complexity in the regulation of cancer cell metabolism, strongly suggesting the existence of a tight feedback loop between protein synthesis and energy metabolism, based on the demonstration that a single molecular chaperone plays a role in both mitochondrial and cytosolic translation, as well as in mitochondrial respiration.
The flow of genetic information from DNA to protein entails multiple highly regulated steps from mRNA transcription, processing, and export to translation into proteins with subsequent folding, post-translational modification, and, eventually, degradation. Moreover, proteins imported or integrated into organelles are targeted to their destination on the basis of signals in the peptide sequence or even upstream through their mRNA localization (Weis et al. 2013). In the case of mitochondria, the coupling between protein import machinery, assembly and activity of respiratory-chain complexes, and regulation of mitochondrial protein synthesis adds a further level of complexity. In yeast, recent reports have shown that mitochondrial and cytosolic translations are rapidly, dynamically, and synchronously regulated (Couvillion et al. 2016). Thus, the nuclear genome coordinates mitochondrial and cytosolic translations to orchestrate the timely synthesis of oxidative phosphorylation complexes, representing a previously underestimated regulatory layer shaping the mitochondrial proteome (Couvillion et al. 2016). Accordingly, the mitochondrial protein import machinery has multiple connections to the respiratory chain (Kulawiak et al. 2013), and in turn, the yeast mitochondrial import receptor Tom20p mediates localization to the mitochondrial outer membrane of mRNAs encoding mitochondrial proteins in a translation-dependent manner (Eliyahu et al. 2010). At present, such a level of complexity is poorly explored in higher organisms, either in health or in disease. Contrary to conventional wisdom, nowadays outdated, functional mitochondria are essential for cancer cells (Zong et al. 2016). Although mutations in mitochondrial genes are common in cancer cells, they do not cause the inactivation of mitochondrial energy metabolism but rather alter the mitochondrial bioenergetic and biosynthetic status (Wallace 2012). Then communication with the nucleus through a mitochondrial “retrograde signaling” leads to modulation of signal transduction pathways, transcriptional circuits, and chromatin structure to meet the mitochondrial and nuclear requirements of the cancer cell. Among the numerous players involved in this process, the molecular chaperone TRAP1 has emerged in the past decade as a critical regulator of metabolic remodeling in cancer cells (Rasola et al. 2014). TRAP1 was initially described as a chaperone for the retinoblastoma protein during mitosis and after heat shock (Chen et al. 1996), as a TNF-receptor-associated protein (Song et al. 1995) and as a factor stabilizing PPID, which prevents permeability transition pore opening and thus apoptosis (Kang et al. 2007). However, in the past few years, TRAP1 has emerged as a modulator of mitochondrial respiration, both through direct binding to respiratory complex II (Chae et al. 2013; Sciacovelli et al. 2013) and III (Matassa et al. 2022) and through indirect modulation of complex IV activity (Yoshida et al. 2013). The regulation of cancer cell metabolism by TRAP1 appears to have contextual effects on cancer onset and progression, thus favoring the oncogenic phenotype in glycolytic tumors (Maddalena et al. 2020) while being negatively selected in tumors mostly relying on oxidative metabolism (Matassa et al. 2018). Conversely, TRAP1 also participates in biosynthetic pathways through the coupled regulation of protein synthesis and degradation (Matassa et al. 2013) and the remodeling of cholesterol homeostasis in ovarian cancer cells (Criscuolo et al. 2020). Indeed, TRAP1 is also partly localized on the endoplasmic reticulum membrane (Amoroso et al. 2012), facing the cytosol, where it binds translation factors (Matassa et al. 2013) and ribosomes (Matassa et al. 2014).
The present study aims to further characterize the molecular mechanisms involved in TRAP1 translational regulation inside and outside mitochondria and to evaluate possible mechanisms of coordinated regulations and feedback loops between protein synthesis and energy metabolism.
Results
TRAP1 is associated with both the cytosolic and mitochondrial protein synthesis machineries
We have previously shown that the extramitochondrial pool of TRAP1 is loosely associated to the endoplasmic reticulum membrane, facing the cytosol (Amoroso et al. 2012), and is bound to ribosomes and translation factors (Matassa et al. 2013). TRAP1 attenuates the rate of protein synthesis, thus allowing protein quality control and reducing cotranslational ubiquitination and degradation of nascent proteins (Matassa et al. 2013). We further characterized the association of TRAP1 to the translational machinery in HeLa cells by polysome profiling, followed by immunodetection of TRAP1 protein in the resulting fractions. Polysome profiling absorbance, measured at 254 nm (for details, see figure legend), shows that at least 0.7 ± 0.33% of total TRAP1 is found in the polysomal fractions (Fig. 1A). Treatment with the translation initiation inhibitor harringtonine, which leads to ribosome run-off, severely reduces the number of ribosomes found in the polysomal fraction and eliminates the presence of TRAP1 (Fig. 1B). This result confirms that the presence of TRAP1 in the collected fractions is linked to the presence of actively translating polysomes. Similar results were obtained by disassembling polysomes via EDTA treatment (Supplemental Fig. S1A) or by treating cells with the protein synthesis inhibitor puromycin (Supplemental Fig. S1B). The absence from polysomal fractions of cytosolic proteins that are not expected to bind ribosomes (GAPDH) confirms the quality of the fractionation. We observed the presence of other cellular components in the last fraction, similar to those documented by others (Sinha et al. 2020), which was therefore excluded from subsequent analyses. Polysome profiling of HT29 colorectal carcinoma cells yielded analogous results (Supplemental Fig. S1C,D), consistent with our previous observations in HCT116 (Matassa et al. 2013) and HEK293 (Matassa et al. 2014) cells. In both HeLa and HT29, the amount of cytosolic TRAP1 quantified by subfractionation and western blot was close to 7% of total protein (Supplemental Fig. S1E).
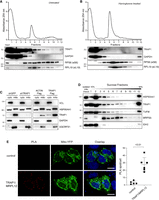
TRAP1 is associated with both cytosolic and mitochondrial ribosomes. (A,B) Polysome profiling absorbance, measured at 254 nm, of HeLa cell extracts, from untreated cells (A) or following a 5-min treatment with 2 µg/mL harringtonine (B). Fractions 1 and 2 are free cytosolic proteins or light complexes; fractions from 3 to 6, ribosomal subunits (60S, 40S) and monomer (80S); and fractions from 7 to 12, polysomes. Proteins from each fraction were analyzed by WB with the indicated antibodies. (C) Subcellular fractionation of HeLa cells showing the presence of indicated proteins into cytosolic (cyto) and mitochondrial (mito) fractions. VCL and GAPDH have been used as markers of cytosol and UQCRFS1 as a marker of mitochondria. (D) HeLa cell mitochondria were isolated, lysed, and loaded onto a 10%–30% linear sucrose gradient, followed by fractionation. Proteins were precipitated from the resulting fractions and subjected to western blot with indicated antibodies. (E) Representative image of PLA showing the interaction between TRAP1 and MRPL12 in HeLa mitochondria. Positive signals of interaction are shown as red dots; nuclei are stained with DAPI (blue); and mitochondria are marked by the mitochondria-directed YFP (green). The control PLA has been obtained by hybridizing with anti-TRAP1 only as primary antibody. Scale bar, 10 µm. The graph shows the average number of PLA spot/cell, with a P-value representing the statistical significance based on the Student's t-test (n = 6).
The predominant mitochondrial localization of TRAP1 protein (Fig. 1C) prompted us to verify whether TRAP1 interacts with the mitochondrial translation apparatus, analogously to what has been shown for the cytoplasmic translational machinery (Matassa et al. 2013, 2014; this work). To this end, we isolated and fractionated mitochondrial ribosomes (hereafter referred to as mitoribosomes) from HeLa cells and used immunoblot to verify the presence of TRAP1 in the mitoribosomal fractions, along with the mitochondrial ribosomal protein MRPS5 (also known as uS5m) and the mitochondrial translation elongation factor TUFM. The molecular chaperone HSP90AA1, highly homologous to TRAP1, also present in cancer cell mitochondria (Fig. 1C), is also visible in some of the mitoribosomal fraction but not at the same extent as TRAP1 (Fig. 1D). In contrast, the mitochondrial enzyme isocitrate dehydrogenase (NADP(+)) 2 (IDH2) is totally absent (Fig. 1D). As for the polysome profiling, the last collected fraction at the bottom of the gradient showed contamination of other cellular components; therefore, it was excluded from subsequent analyses (see next section). Additionally, we performed a proximity ligation assay (PLA) in HeLa cells using antibodies against TRAP1 and the mitochondrial ribosomal protein MRPL12 (also known as bL12m). Results showed that TRAP1 binds to mitochondrial ribosomes in HeLa cells, with an average of 28 spots/cell (Fig. 1E).
TRAP1 regulates protein synthesis both in cytosol and mitochondria
To elucidate the molecular mechanisms underpinning TRAP1 contribution to protein synthesis regulation, we characterized its function in a stable, inducible HeLa cell system. First of all, we confirmed in this model that modulation of TRAP1 expression levels, by either induction of shRNA-mediated silencing or overexpression of a TRAP1–GFP protein, affected the total cellular protein synthesis rate, as measured by incorporation of 35S Met/35S Cys. Specifically, TRAP1 silencing yielded increased incorporation, whereas TRAP1–GFP overexpression resulted in reduced incorporation compared with unfused GFP–expressing cells (Fig. 2A). Similarly, incorporation of puromycin into proteins is reduced upon TRAP1–GFP overexpression and increased upon shRNA-mediated silencing (Supplemental Fig. S2A). TRAP1 silencing with two different siRNA sequences in both HeLa and HT29 colorectal carcinoma cells yielded analogous results in fluorescent noncanonical amino acid tagging (FUNCAT) assays (Supplemental Fig. S2B).
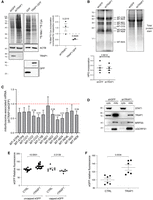
TRAP1 expression has opposite effects on total cell versus mitochondrial mRNA translation. (A) Representative autoradiography of total lysates from cells labeled with 35S Met/35S Cys, following tetracycline-induced induction of TRAP1-directed shRNA and control shRNA (72-h) cells or of TRAP1–GFP and unfused control GFP (24-h) cells, with relative densitometric band intensities and analysis (right). The P-values in the graph indicate the statistical significance based on the Student's t-test (n = 3). (B) Mito-FUNCAT-gel. Expression of GFP (control)-directed and TRAP1-directed shRNAs was induced in HeLa cells with tetracycline 72 h before labeling with 100 µM HPG-alkyne for 2 h. The resulting lysates were subjected to a click reaction with a TAMRA-azide, loaded for SDS-PAGE, and detected at 550 nm. (C) RT-qPCR performed on RNAs extracted from mitoribosomal fractions (4–11) isolated from HeLa cells 72 h after induction of shGFP/shTRAP1. The amount of mitoribosome-associated mRNA in the two samples has been normalized on 12S rRNA and corrected for its total expression level. Data are expressed as mean ±SEM (n = 5). Numbers above bars represent the statistical significance (P-value) based on the one-sample t-test. (D) Subcellular fractionation of HeLa cells showing the presence of indicated proteins into cytosolic (cyto) and mitochondrial (mito) fractions. (E) eGFP in vitro translation using wheat germ extracts. eGFP mRNA was added to reactions at a final concentration of 21.95 ng/μL. Where indicated, 0.3 μg/μL of TRAP1 recombinant protein (rTRAP1) was added to the reaction. Data are expressed as mean ± SEM (n = 14 for the translation of uncapped eGFP mRNA; n = 7 for the translation of capped eGFP mRNA). The two-tailed P-value represents the statistical significance based on the Student's t-test. (F) eGFP in vitro translation using E. coli extracts. eGFP mRNA was added to reactions at a final concentration of 21.95 ng/μL. Where indicated, 0.2 μg/uL of TRAP1 recombinant protein was added to the reaction. Data are expressed as mean ± SEM (n = 6). The P-value represents the statistical significance based on the Student's t-test.
Because TRAP1 is mainly localized into mitochondria (Fig. 1C; Felts et al. 2000; Amoroso et al. 2014), we further characterized its regulatory role in mitochondrial translation in whole cells by using the mitochondrial-specific FUNCAT (mito-FUNCAT) assay (Yousefi et al. 2021). The results showed that, in these conditions, the incorporation of the amino acid analog L-homopropargylglycine (HPG) in mitochondrial-encoded proteins is unaffected by TRAP1 silencing or overexpression (Fig. 2B; Supplemental Fig. S2C,D) on a background of overall equal mRNA abundance (Supplemental Fig. S2E). However, RT-qPCR performed on mRNAs extracted from isolated mitochondrial ribosomes in the absence of translation inhibitors (such as cycloheximide, emetine) showed that TRAP1 silencing reduces the association of the mitochondria-encoded transcripts to mitoribosomes (Fig. 2C), suggesting reduced translation. Quantification of mitoribosome-associated mRNAs was corrected to their relative expression levels and normalized to 12S rRNA, a structural constituent of the ribosome, which rules out a contribution from ribosome biogenesis. The validity of this procedure is further supported by the equal expression levels of the mitochondrial ribosomal protein MRPS5 in the mitochondria isolated from shGFP and shTRAP1 HeLa cells (Fig. 2D). For better resolution, we measured the association of four of the most regulated transcripts (MT-CO2, MT-CO3, MT-ND3, MT-ND4) and the transcript that does not show any change upon TRAP1 silencing (MT-ND1), by measuring the proportion of the total transcript that is associated with the different mitoribosomal fractions (Supplemental Fig. S3). The results confirmed the reduced association of MT-CO2, MT-CO3, MT-ND3, and MT-ND4 with the mitoribosomes, whereas MT-ND1 was unchanged, as expected. More specifically, the results showed that the difference between TRAP1 KD (shTRAP1) and control (shGFP) cells is restricted to the last two fractions of the sucrose gradient, suggesting that the “heavier” ribosomes are the ones mostly affected by the presence of TRAP1. One key difference between this approach and the mito-FUNCAT assay stems from the crucial requirement in the mito-FUNCAT assay that cytosolic translation be inhibited by cycloheximide/emetine. This impedes the detection of differences in mitochondrial translation upon TRAP1 knockdown/overexpression, given its dual role in the two compartments and in the cross talk between the two, as further explored in the next section. This observation is consistent with previous studies performed in yeast (Couvillion et al. 2016), showing a unidirectional cross talk between cytosolic and mitochondrial translation.
To further characterize TRAP1 effects on translation and to take advantage of our previous data on TRAP1 regulation of capped/uncapped mRNA translation (Amoroso et al. 2014), we evaluated the mechanistic contribution of TRAP1 to mRNA translation in an in vitro system upon the addition of a 5′ methylguanosine cap. As shown in Figure 2E, the addition of a recombinant mature TRAP1 protein in wheat germ extracts reduces the translation of an in vitro transcribed GFP mRNA when a 5′ methylguanosine cap is added to the transcript, but increases translation of the uncapped mRNA. This result not only supports the direct role of TRAP1 in translation but also supports that high expression of TRAP1 correlates with a higher IRES/cap-dependent translation ratio, in agreement with our previous observations (Matassa et al. 2014), although an effect on the folding of the nascent GFP can also contribute. In addition, we found, not surprisingly given the similarities between mitochondrial and prokaryotic translation, that TRAP1 addition increases GFP reporter protein synthesis in Escherichia coli extracts (Fig. 2F), showing that TRAP1 actually increases the translation of leaderless mRNAs. These findings also support the results indicating a reduced association of mitochondrial transcripts with mitoribosomes upon TRAP1 silencing (Fig. 2C).
TRAP1 couples cytosolic with mitochondrial translation
The mitochondrial respiratory complexes are composed by both nuclear-encoded and mitochondrial-encoded proteins. The former are synthesized into the cytosol and then have to be imported into the mitochondrion, whereas the latter are synthesized within the organelle through a mechanism largely dependent on the assembly of the complex and, therefore, on the availability of the nuclear-encoded components (Priesnitz and Becker 2018). Of note, in addition to TRAP1 involvement in both translation processes, previous results from a quantitative proteomic analysis (Avolio et al. 2018) showed the mitochondrial protein import channel component TOMM40 as the second most significant interactor among the TRAP1 protein partners in HeLa cells. Here, to provide further evidence for this interaction, two additional approaches were used: (1) a PLA, showing that endogenous TRAP1 and TOMM40 produce an average of 13 spots/cell (Fig. 3A), and (2) coimmunoprecipitation experiments, showing binding between TRAP1 and TOMM40 when the TRAP1–GFP protein is isolated from inducible HeLa cells by a GFP-trap using highly specific nanobodies. In contrast, no signal is observed with other components of the TIM–TOM complex (TOMM20, TIMM23, TIMM44), or mitochondrial proteins (PHB2), thus supporting the specificity of this binding (Fig. 3B). Similar results were obtained by PLA (Supplemental Fig. S4A). However, TRAP1 also interacts with mitochondrial proteins localized into the matrix, which, according to mass spectrometry data (Joshi et al. 2020; Cannino et al. 2022), includes the mitochondrial translation elongation factor TUFM. This latter interaction was confirmed by PLA in HCT116 cells (Fig. 3C) and HeLa cells (Supplemental Fig. S4B). The highly homologous HSP90AA1 does not show proximity ligation with TUFM (Supplemental Fig. S4B), further supporting the specificity of TRAP1–TUFM binding. The same approach also confirmed the association between TRAP1 and the cytosolic translation elongation factors EEF1A1 and EEF1G (Supplemental Fig. S4C). Association of TRAP1 with TOMM40, TUFM, and the cytosolic translation elongation factor EEF1A1 was further corroborated in HT29 colorectal cancer cell and MCF-7 breast cancer cells (Supplemental Fig. S4D). The binding of TRAP1 to both cytosolic and mitochondrial translation factors and ribosomes (Supplemental Fig. S4C,D; Matassa et al. 2013), as well as to protein import channel components, prompted us to evaluate whether these interactions are dependent on active protein synthesis. For this purpose, we treated cells with inhibitors specific for either cytosolic translation initiation (harringtonine) or elongation (emetine) or for mitochondrial translation initiation (linezolid) or elongation (chloramphenicol). Coimmunoprecipitation performed in these conditions showed that inhibition of cytosolic protein synthesis dramatically reduces the binding between TRAP1 and TOMM40, whereas the TRAP1–EEF1A1 and TRAP1–TUFM interactions increased (Fig. 3D). Conversely, inhibition of mitochondrial translation yielded no effect on the binding of either cytosolic or mitochondrial partners (Fig. 3E). The same emetine treatment is able to dissociate both TRAP1 and TUFM from mitoribosomes (Supplemental Fig. S5A) and to mimic TRAP1 silencing in reducing the association of mitochondrial-encoded transcripts to mitoribosomes (Supplemental Fig. S5B). Again, and in agreement with the observations in yeast (Couvillion et al. 2016), this result confirms the unidirectional nature of the process: Namely, cytosolic translation transduces a signal to the mitochondrial translation apparatus, whereas mitochondrial translation is unable to reverse a signal to the cytosolic ribosomes. TRAP1 takes part in this unidirectional mechanism.
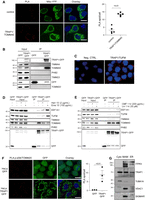
TRAP1 associates with the mitochondrial protein import machinery and could favor localized translation. (A) Representative images of PLA positivity between TRAP1 and TOMM40. Positive signals of interaction are shown as red dots; nuclei are stained with DAPI (blue); and mitochondria are marked by the mitochondria-directed YFP (green). Scale bar, 10 µm. The graph shows the average number of PLA spots/cell, with a two-tailed P-value representing the statistical significance based on the Student's t-test (n = 5). (B) Immunoprecipitation of unfused GFP and TRAP1–GFP performed in HeLa cells following 24-h induction of GFP and TRAP1–GFP. Total lysates were incubated with GFP-trap beads to isolate the proteins, and the resulting samples were immunoblotted with indicated antibodies. (C) Representative image of PLA showing the interaction of TRAP1 with TUFM in HCT116 cells. Positive signals of interaction are shown as red dots; nuclei are stained with DAPI (blue). A negative control has been obtained by hybridizing cells with TRAP1 antibody only. (D,E) Immunoprecipitation of unfused GFP and TRAP1–GFP performed in HeLa cells following 24-h induction of GFP and TRAP1–GFP. Where indicated, cells were treated for 15 min with emetine (100 µg/mL) or harringtonine (2 µg/mL) or for 1 hr with chloramphenicol (200 µg/mL) or linezolid (30 µM). Total lysates were incubated with GFP-trap beads to isolate the proteins, and the resulting samples were immunoblotted with indicated antibodies. (F) Representative image of PLA showing the interaction of TOMM20 with phosphorylated (active) ribosomal protein RPS6 in HeLa cells following 24 h induction of TRAP1–GFP or unfused GFP. Positive signals of interaction are shown as red dots; nuclei are stained with DAPI (blue). Scale bar, 10 µm. The graph shows the average number of PLA spots/cell, with a P-value representing the statistical significance based on the Student's t-test (n = 3). (G) Cytosolic, MAM, and ER fractions isolated from HCT116 cells probed with the indicated antibodies.
In yeast, Tom20p, another mitochondrial import channel component, is involved in the translation-dependent enrichment of mRNAs encoding mitochondrial proteins near mitochondria, facilitating protein transport into the organelle (Eliyahu et al. 2010). Considering the results shown above, we used a PLA to evaluate whether TRAP1 might be involved in both the import of mitochondrial proteins and localized protein synthesis. The results show that TRAP1–GFP induction in HeLa cells increases the number of proximity ligation spots between TOMM20 and actively translating ribosomes, as detected by phosphorylation of the ribosomal protein RPS6 (Fig. 3F). In contrast, TRAP1 interference by shRNAs decreased such proximity (Supplemental Fig. S5C). Moreover, we found that TRAP1 is indeed localized in the so-called mitochondria-associated membranes (MAMs), regions common to all cells in which the endoplasmic reticulum and mitochondria are physically connected (Fig. 3G). These results not only support our previous data on the TRAP1 role in protein synthesis control at the cytosol/mitochondria interface but also, for the first time, identify TRAP1 in the MAM compartment. Altogether, as MAMs are “hot spots” for the intracellular signaling of important pathways (Giorgi et al. 2015), the finding of TRAP1 physically associated to these structures provides further support for our previously described functions of TRAP1 in lipid biosynthesis (Criscuolo et al. 2020), calcium and endoplasmic reticulum homeostasis (Landriscina et al. 2010; Sisinni et al. 2014), reactive oxygen species generation (Gesualdi et al. 2007), and protein sorting (Pepe et al. 2017).
TRAP1 regulates translation elongation
To shed light on the mechanisms leading to TRAP1 translational control, we asked whether the binding to translation elongation factors could explain the TRAP1 role in the modulation of protein synthesis. This hypothesis was also supported by the evidence that the modulation of the translation elongation rate reduces cotranslational protein degradation (Conn and Qian 2013), and this would be consistent with the TRAP1 function in preventing ubiquitination of newly synthesized proteins (Amoroso et al. 2012, Matassa et al. 2013). Additionally, recent studies performed in yeast show that translation elongation stalling increases mRNA localization to mitochondria (Tsuboi et al. 2020). For this purpose, we first investigated TRAP1 binding to the translation elongation factor EEF1G by fluorescence lifetime imaging (FLIM) followed by confocal microscopy analysis, which allows verification of direct protein–protein interactions with high accuracy (Margineanu et al. 2016). The Förster resonance energy transfer (FRET)–based approach is a widely accepted method for this purpose, because it involves a donor fluorophore molecule that, when excited, transfers energy to an acceptor fluorophore molecule, provided that the two fluorophores are essentially fixed within a distance of 1–10 nm (Grecco and Bastiaens 2013), necessitating a direct interaction. Our results showed that TRAP1 binds EEF1G in HeLa cells directly, thus also confirming the presence of TRAP1 in the cytosolic compartment and its association with the cytosolic translational machinery (Fig. 4A). In agreement with its dual translational control, besides the binding to cytosolic translation elongation factors, TRAP1 also binds the mitochondrial translation elongation factor TUFM, as suggested by mass spectrometry data (Joshi et al. 2020; Cannino et al. 2022) and as shown above by PLA (Fig. 3C; Supplemental Fig. S4B–D) and immunoprecipitation (Fig. 3D,E). Therefore, we explored this issue deeply and found that the two proteins are actually bound directly with high efficiency, as confirmed by FLIM in HeLa TRAP1–GFP cells (Fig. 4B). In a related experiment using a stopped-flow fluorescence assay (Liu et al. 2014), we showed a direct interaction of TRAP1 with EF-Tu, the bacterial homolog of the mitochondrial EF-Tu (TUFM). This experiment monitors the interaction of a ternary complex (TC), aa-tRNA.EF-Tu-GTP, containing EF-Tu labeled with a fluorescence quencher, with a fluorescent-labeled ribosomal 70S initiation complex containing fMet-tRNAfMet in the ribosomal P-site, as part of the first elongation step, which results in formation of dipeptidyl-tRNA. In the positive control, entry of the quencher-labeled TC into the ribosomal A-site results in a rapid decrease in fluorescence, which was rapidly recovered upon the EF-Tu.GDP release from the ribosome (Fig. 4C). Addition of TRAP1 inhibits such release, showing the TRAP1:EF-Tu interaction. Inhibition of EF-Tu release or, more in general, EF1 release may be a more relevant factor for regulating the rate of cytoplasmic rather than mitochondrial protein synthesis at the elongation step (see Discussion).
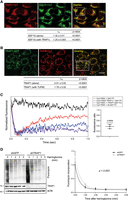
TRAP1 binds both cytosolic and mitochondrial translation elongation factors and slows down elongation rate. (A) Fluorescent confocal microscopy analysis of TRAP1-Cy3 (acceptor) and EEF1G-Cy2 (donor) in HeLa cells. Dipole–dipole energy transfer from the fluorescent donor to the fluorescent acceptor allowed calculating FRET efficiency (EFRET %) as described in the Methods section. The overlay images show the inset area in which FRET has been analyzed. Scale bar, 10 µm. τns values are expressed as mean ± SEM. The two-tailed P-value represents the statistical significance based on the Student's t-test. (B) Fluorescent confocal microscopy analysis of TRAP1–GFP (donor) and TUFM-Cy3 (acceptor) in TRAP1–GFP-inducible HeLa cells. Dipole–dipole energy transfer from the fluorescent donor to the fluorescent acceptor allowed calculating FRET efficiency (EFRET %) as described in the Methods section. The overlay images show the inset area in which FRET has been analyzed. Scale bar, 10 µm. τns values are expressed. (C) TRAP1 inhibits EF-Tu release from the 70S initiation complex (70SIC) in stopped-flow assays. TRAP1 recombinant protein was preincubated with a ternary complex (TC) in which EF-Tu is labeled with the QSY9 fluorescence quencher (QSY-TC; purple trace), and the resulting solution was rapidly mixed with a Cy3-labeled 70S initiation complex (Cy3-70SIC; blue trace). The change in Cy3 fluorescence was monitored using a stopped-flow fluorometer. Upon entering in the A site, the quencher-labeled EF-Tu decreases the Cy3-labeled ribosome fluorescence, whereas its dissociation from the ribosome allows Cy3 fluorescence recovery. Black trace indicates negative control; red trace, positive control. The graph on the right shows the ratio of fluorescence increase in the presence of added TRAP1 to the fluorescence increase in the absence of TRAP1 in three independent experiments. The P-value represent the statistical significance based on the one-sample t-test. (D) Seventy-two hours after tet-induction shGFP-directed (control) or TRAP1-directed shRNAs, HeLa cells were treated with harringtonine (2 µg/mL) for the indicated times (0, 1, 2, 3, and 5 min) and subsequently treated with puromycin (10 μg/mL) for 10 min. Cells were lysed and subjected to immunoblotting with antipuromycin antibody. The graph shows densitometric intensity of the puromycin labeling, normalized on the total protein content (quantified by no-stain labeling; see Methods). Data are represented as mean ± SEM from eight independent experiments, with trend lines showing exponential one-phase decay analysis. P-value on the graph represents the statistical significance based on the extra sum-of-squares F-test.
To evaluate the relevance of these findings in human cells, we set up a SunRiSE assay (Argüello et al. 2018) to assess whether such a mechanism reflects changes in the elongation rate. Briefly, following inhibition of translation initiation with harringtonine, we measured puromycin incorporation into nascent peptide chains at different time intervals. The decay of puromycin labeling following harringtonine treatment was found to be steeper in TRAP1 knockdown cells compared with the shGFP controls (Fig. 4D), with a significant lower decay rate constant (KshGFP = 1.401; KshTRAP1 = 1.268; P = 0.003). This indicates a higher speed of run-off of ribosomes from mRNAs following harringtonine, consistent with a higher processing speed of the ribosomes along mRNAs in the absence of TRAP1 and, therefore, an increased elongation rate. Accordingly, a 2-min treatment with harringtonine caused a lower decrease in polysomes and an increase in monosomes in TRAP1-FLAG overexpressing cells compared with the control ACTIN-FLAG cells (Fig. 5A), whereas shTRAP1 cells displayed faster run-off compared with shGFP controls (Fig. 5B). Decrease in the heavy-to-light polysome ratio following harringtonine indeed follows different decay constants, which inversely correlate with TRAP1 expression (Supplemental Fig. S5D). Again, these findings are consistent with the hypothesis that TRAP1 regulates translation at the elongation step. The reduction of translation elongation rate is also consistent with the reduced pulse labeling shown in Figure 2A and Supplemental Figure S2A. Considering the dual role in the regulation of cytosolic and mitochondrial protein synthesis, we measured the translation speed of transcripts encoding several mitochondrial ATP synthase proteins, synthesized by either mitochondrial (MT-ATP6 and MT-ATP8) or cytoplasmic (ATP5 subunits) ribosomes. For this purpose, we set up a run-off experiment and determined the ratio between polysome-associated and monosome-associated mRNAs following harringtonine treatment. Control (shGFP) and shTRAP1 HeLa cells were treated for 2 min with harringtonine and fractionated, and the number of transcripts associated with the polysomal and monosomal fractions of both treated and untreated cells was estimated by qPCR. Figure 5C shows the polysome/monosome ratio for each transcript upon harringtonine treatment compared with the untreated samples. For the ATP5-encoding transcripts, this ratio is much more reduced in the shTRAP1 cells than in the shGFP controls. In contrast, no significant reduction occurs when mitochondrial-encoded transcripts are analyzed, as expected, because harringtonine does not inhibit mitochondrial ribosomes. In the absence of harringtonine, the impact of TRAP1 silencing per se on the association of the same transcripts with polysomes is not significant (Supplemental Fig. S5E), consistent with overall comparable ribosome loading of shGFP and shTRAP1 cells (Fig. 5B). Transcripts encoding cytosolic proteins like the ribosomal protein RPL36A (also known as eL42) respond to harringtonine by reducing their polysome/monosome ratio but do not display differences in this reduction between shGFP and shTRAP1 cells (Fig. 5C).
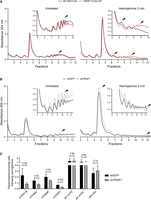
TRAP1 slows down translation elongation by cytoplasmic ribosomes. (A,B) Polysome profiling absorbance, measured at 254 nm, of extracts from ACTIN-FLAG (control) and TRAP1-FLAG overexpressing HeLa cells (A) and from shGFP (control) and shTRAP1 HeLa cells (B), 24 h (A) or 72 h (B) after induction, in the absence (left) or presence (right) of 2 µg/mL of harringtonine (cells were treated for 2 min and then blocked with cycloheximide). Inset areas show magnification of regions of interest. (C) Ratio between polysome-associated and monosome-associated mRNAs following harringtonine treatment (2 µg/mL, 2 min), normalized on the respective untreated samples. The amount of the associated transcripts was measured by RT-qPCR performed on RNAs extracted from pooled monosomal and polysomal fractions, both corrected for an external reference spike-in RNA (luciferase). Data are represented as mean ± SEM from three independent experiments. Numbers above bars represent the statistical significance (P-value) calculated by a multiple t-test.
TRAP1 is a putative chaperone linked to protein synthesis
Previous results identified and functionally characterized a selective group of chaperones linked to protein synthesis (CLIPSs), coexpressed with components of the translational machinery in yeast (Albanèse et al. 2006). Our findings suggest that TRAP1, owing to its role in protein synthesis regulation, might behave as the first CLIPS (or CLIPS-like) identified in higher organisms. As a preliminary test of this hypothesis, we performed a coexpression analysis using the software COXPRESdb (Obayashi et al. 2019) and found that TRAP1 is significantly coexpressed with several components of the mitochondrial translation apparatus, with 10% of the most 100 coexpressed genes encoding mitochondrial ribosomal proteins (highlighted in Supplemental Table S1). When a network is constructed with this subset of genes, it builds up discrete clusters of proteins related to mitochondrial translation and metabolism connected by TUFM, mostly constituted by mitochondrial ribosomal proteins, electron transport chain components and assembly factors, and organelle biogenesis and metabolism (Fig. 6A). Accordingly, a Gene Ontology analysis of biological processes, performed through Enrichr (Xie et al. 2021) on the list of coexpressed genes, showed that all top five enriched pathways are correlated with ribosomal RNA and tRNA processing and mitochondrial translation (Fig. 6B). For instance, TUFM is the second most correlated gene (Fig. 6C), and the correlation between TRAP1 and TUFM is conserved when protein levels are compared (Fig. 6D). Consistently, CLUH, which encodes an RNA-binding protein involved in the proper cytoplasmic distribution of mitochondria, is one of the most TRAP1-coexpressed genes (Fig. 6E). CLUH specifically binds mRNAs of nuclear-encoded mitochondrial proteins in the cytoplasm and regulates the transport or translation of these transcripts close to mitochondria, playing a role in mitochondrial biogenesis (Gao et al. 2014). Although preliminary and speculative, this analysis suggests the possibility that TRAP1 could actually behave as a mammalian CLIPS, which deserves to be experimentally investigated in the future.
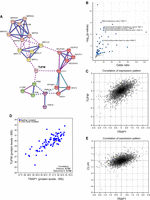
TRAP1 is coexpressed with the mitochondrial translational machinery. (A) Network analysis of the top 100 TRAP1-coexpressed genes, generated by STRING using a Markov cluster (MCL) algorithm. Network nodes represent proteins; edges represent protein–protein associations, by coexpression (black line), experimentally determined association (pink line), or database-curated association (blue line). Edges between clusters are represented by dotted lines. The red cluster is enriched in mitochondrial electron transport components; pink and yellow clusters are constituted by structural component of the mitochondrial ribosome (small and large subunit, respectively); the green cluster contains the mitochondrial prohibitin complex; and the purple cluster is constituted by the association between the mitochondria protein import inner membrane translocase subunit PAM16 and the mitochondrial-processing peptidase subunit alpha PMPCA. (B) Gene set enrichment analysis on the list of genes significantly coexpressed with TRAP1 in human tissues. (C) Coexpression analysis performed with COXPRESdb between TRAP1 and TUFM. (D) Coexpression analysis between TRAP1 and TUFM at protein level according to the Provisional database. (E) Coexpression analysis performed with COXPRESdb between TRAP1 and CLUH.
Discussion
TRAP1, the main mitochondrial member of HSP90 protein family, interacts in mitochondria with respiratory complexes and contributes to the regulation of cellular respiration in cancer cells (Rasola et al. 2014). In addition, we and others have shown that this protein is also partially localized on the outer side of the endoplasmic reticulum, where it is involved in the regulation of protein synthesis through the binding to components of the translational machinery (Matassa et al. 2013; this study). Interest in TRAP1 has considerably grown in the past decades owing to its contextual effects in different tumor types: It is highly expressed in several cancers and correlated with drug resistance, but is down-regulated in specific tumors with predominant oxidative metabolism (Lettini et al. 2017). The interplay between protein synthesis and mitochondrial respiration has been recently investigated in yeast (Couvillion et al. 2016), but the involvement of this phenomenon in humans is almost unexplored. Although the coordination between the mitochondrial and the nuclear genomes can be mediated at the transcription level (Barshad et al. 2018), mRNA localization and localized translation are also emerging as important regulatory layers for the expression of mitochondrial metabolism components, which are coordinated by retrograde signaling from the organelle (Wallace 2012). To allow coordination of oxidative phosphorylation complexes assembly and activity, mitochondrial and cytosolic translations must be rapidly, dynamically, and synchronously regulated (Couvillion et al. 2016). This represents a unique challenge for the cells, because respiratory complex subunits are encoded by both the nuclear and the mitochondrial genomes. In this study, we explored the hypothesis that TRAP1 may function as a “moonlighting” protein, regulating mRNA translation in both the cytosol and mitochondria, upstream to the previously shown modulation of activity respiratory complexes II (Sciacovelli et al. 2013), III (Matassa et al. 2022), and IV (Yoshida et al. 2013). Such regulation would require coordination between the synthesis of mitochondria-destined proteins in the cytosol, their import into the organelle, and the synthesis of mitochondrial-encoded proteins in the matrix. This latter synthesis is restricted to the translation of only 13 transcripts, exclusively producing components of the electron transport chain. Here we provide the first clear evidence that TRAP1 specifically binds the mitochondrial translation apparatus, facilitating translation of mtDNA-encoded proteins. However, TRAP1 not only participates in both mitochondrial and cytosolic protein synthesis but also is involved in protein import into mitochondria: TRAP1 specifically binds TOMM40 in a translation-dependent manner, and the resulting signal is transduced to the organelle, where TRAP1–TUFM binding is also affected by the activity of cytosolic translation. Of note, we show for the first time that TRAP1 is localized in the contact sites between the endoplasmic reticulum and mitochondria (the MAM compartment) (Giorgi et al. 2015), not only supporting the TRAP1 role in protein synthesis control at the cytosol/mitochondria interface but also providing a further support of our previously described functions of TRAP1 in lipid biosynthesis (Criscuolo et al. 2020), calcium homeostasis (Landriscina et al. 2010), reactive oxygen species generation (Gesualdi et al. 2007), and protein sorting (Pepe et al. 2017). We also find that TRAP1 expression is directly correlated to the amount of active ribosomes in the proximity of channel component TOMM20. TOMM20 is known to mediate the localization of mRNAs encoding mitochondrial proteins to mitochondria in a translation-dependent manner (Eliyahu et al. 2010). Relevant to this topic, we previously showed that TRAP1 expression is crucial as well for the correct localization of the prion protein SPRN at the interface between the endoplasmic reticulum and mitochondria in neuronal cells (Pepe et al. 2017).
Moreover, we present evidence that TRAP1 behaves as a translation regulator via direct binding to translation elongation factors. Further studies are needed to compare the specific contribution of these interactions to additional mechanisms involving cotranslational chaperoning on nascent protein chains. Although translation control has been traditionally attributed mostly to the regulation of the initiation step, evidence is accumulating on the importance of elongation rate control in the homeostasis of the translation process and its coordination with related pathways (Sinha et al. 2020). Relevant for our findings is the recent demonstration that translation rate affects the localization of mRNAs coding for mitochondrial proteins to this organelle and that translation elongation stall impacts such localization (Tsuboi et al. 2020). We therefore propose that TRAP1 reduces the translation elongation rate of transcripts encoding mitochondrial proteins on the cytosolic side, thus favoring cotranslational import. In turn, this stimulates the activation of mitochondrial translation for the coupled synthesis of mtDNA-encoded components of the respiratory complexes. Regulation of translation elongation inside the organelle could also assist the cotranslational assembly of the complexes. The correlation between translation elongation rate and protein yield is not trivial. Because eukaryotic mRNAs are circularized, potentially allowing terminating ribosomes to preferentially reinitiate on the same transcript, increased elongation rates can result in increased protein yield without changes in ribosome density on a specific mRNA (Rogers et al. 2017). Although in a linear model of translation, protein abundance is mainly determined by initiation rate, in a closed-loop model, the continuous reinitiation on the same transcript results in protein yield being significantly determined by elongation rate. This model well fits with our data on protein synthesis regulation by TRAP1, showing that it binds a translation elongation factor, inhibits its release from the ribosome, and reduces the incorporation rate of amino acids into proteins, without changing the global ribosome loading and, therefore, the total amount of active polysomes in the cell. Indeed, the presence of TRAP1 only seems to reduce the incorporation of amino acids during the translation of capped transcripts, which can be easily circularized. Of note, although mRNA circularization can occur in mammalian mitochondria, it does not appear to play a role in making translatable mRNAs (Mance et al. 2020). Because uncapped mRNAs may or may not be subject to circularization (Komar and Hatzoglou 2011), the model also fits with the evidence indicating that uncapped transcripts are more efficiently translated in the presence of TRAP1, because in this case, protein yield would be mainly determined by the initiation rate. Additionally, in other cell models, we have already shown that TRAP1-expressing cells show increased levels of EIF2A phosphorylation (which represses only cap-dependent initiation of translation) (Thakor and Holcik 2012) and a higher ratio of IRES-dependent versus cap-dependent translation (Matassa et al. 2014). The latter mechanism is relevant in cancer development because, among 70 experimentally verified cellular IRES elements (Mokrejš et al. 2010), a large number are found in cancer-related genes (Walters and Thompson 2016). Slowdown of elongation phase also contributes to cotranslation protein quality control, contributing to reducing cotranslational ubiquitination and degradation of nascent peptides and ensuring stability; sometimes, “less is more” in protein synthesis (Sherman and Qian 2013). In this regard, it is noteworthy that we have previously shown how the binding between TRAP1 and the proteasome regulatory subunit component PSMC4 in the cytosol reduces global cotranslational ubiquitination (Amoroso et al. 2012), which critically tunes the expression level of the mitochondria-destined proteins ATP5F1B and SRI isoform B (Matassa et al. 2013). The proteasome is absent from mitochondria, and to the best of our knowledge, similar ribosome-bound quality-control mechanisms triggering degradation of nascent peptides synthesized into the mitochondria have never been described.
Finally, we show that a correlation exists between TRAP1 levels and the concomitant expression of the mitochondrial protein synthesis apparatus in human tissues, and, in general, of proteins involved in mitochondrial biogenesis and metabolism. TRAP1 is also coexpressed with a subset of protein partners, including the translation factors EIF2A and EEF1A1. Up-regulation of this subset is characteristic of a cohort of metastatic colorectal carcinomas with a significantly shorter overall survival (Maddalena et al. 2017).
Taken together, the present results show an unprecedented level of complexity in the regulation of cancer cell metabolism, in which both mitochondrial and cytosolic protein synthesis are coregulated and coordinated with energy metabolism through the contribution of a common molecular chaperone, TRAP1. As cell metabolism is a complex network of interdependent pathways and as knowledge of the correlations between protein synthesis and mitochondrial respiration in higher organisms is still in its infancy, the highlighting of bioenergetic pathways, with an eye toward protein synthesis, may provide a novel approach for therapeutic development in multiple human disease, as evidenced by growing interest in the field (Boczonadi and Horvath 2014; Webb et al. 2020). Although limited by lack of structural details of the interactions between TRAP1, the protein synthesis machinery, and the protein import channel, these novel insights add a relevant piece to the complex puzzle of TRAP1 chaperone functions in cancer cells. Further studies are needed to identify the specific transcripts whose translation is affected by TRAP1-dependent regulations and to dissect the molecular mechanisms involved in localized translation and coupling between cytosolic and mitochondrial translation.
Methods
Cell cultures
Human HT29 and HCT116 colon carcinoma cells, human cervical carcinoma HeLa cells, and human breast cancer MCF-7 cells were purchased from American Type Culture Collection (ATCC) and cultured in McCoy's 5A medium (HCT116, HT29) and DMEM (HeLa, MCF7). Both culturing media contain 10% fetal bovine serum, 1.5 mmol/L glutamine. The authenticity of the cell lines was verified by STR profiling, in accordance with the ATCC product description. The HeLa Flp-In T-REx (FITR) cell line was kindly provided by Dr. Matthias Gromeier (Duke University Medical Center). Generation of the HeLa FITR stable cell lines expressing the eGFP- or FLAG-fusion proteins or the short hairpin RNA was performed as described in the manufacturer's protocol (FITR; Invitrogen). HeLa FITR cells were cultured in DMEM supplemented with 10% fetal bovine serum, 1.5 mmol/L glutamine, and appropriate selective antibiotics. The addition of tetracycline induces proteins as described previously (Castello et al. 2012).
Plasmid generation and transfection procedures
For TRAP1-eGFP and TRAP1-FLAG plasmids generation, HeLa cDNA library, eGFP, and FLAG plasmids were used as templates for fusion PCR. The resulting chimeric cDNAs were cloned into pCDNA5/FRT/TO. pFRT-U6tetO is a kind gift from Prof. John J Rossi. Inducible short hairpin (sh) RNAs were generated as previously described (using BglII/KpnI as restriction sites) (Aagaard et al. 2007). Short hairpin sequences used were as follows: GFP = agatctGCACAAGCTGGAGTACAACTACCTGACCCATAGTTGTACTCCAGCTTGTGCTTTTTggtacc; TRAP1 = agatctGCCCGGTCCCTGTACTCAGAAACCTGACCCATTTCTGAGTACAGGGACCGGGCTTTTTggtacc.
Transient silencing was performed with two different siRNAs targeting TRAP1 (Qiagen SI00115150, target sequence CCCGGTCCCTGTACTCAGAAA; Qiagen SI00115164, target sequence CCGCTACACCCTGCACTATAA), and nontargeting control siRNA (Qiagen SI03650318). All the transfections were performed using Lipofectamine 2000 (Thermo Fisher Scientific), according to the manufacturer's protocol.
Polysome profiling
Plates (3 × 10 cm) of cells were incubated 15 min at 37°C with fresh medium supplemented with 100 μg/mL of cycloheximide (Sigma-Aldrich). Where indicated, cells were treated with 2 μg/mL of harringtonine or 100 μg/mL of puromycin. Cells were then washed with ice-cold PBS supplemented with 100 μg/mL cycloheximide and resuspended in 1 mL lysis buffer (10 mM Tris-HCl at pH 7.4, 100 mM KCl, 10 mM MgCl2, 1% Triton X-100, 1 mM DTT, 10 U/mL RNaseOUT [Invitrogen], 100 μg/mL of cycloheximide). After 5 min of incubation on ice, cell lysate was centrifuged for 10 min at 14,000 rpm at 4°C. Where indicated, the cell extract was treated with 30 mM EDTA to disassemble polysomes as a negative control. The supernatant was collected, and the absorbance was measured at 260 nm with a NanoDrop. Eight A260 units were loaded onto a 10%–60% sucrose gradient obtained by adding 6 mL of 10% sucrose over a layer of 6 mL of 60% sucrose prepared in lysis buffer without Triton X-100 and containing 0.5 mM DTT, in a 12-mL tube (Polyallomer; Beckman Coulter). Gradients were prepared using a gradient maker (gradient master; Biocomp). Polysomes were separated by centrifugation at 35,000 rpm for 2 h using a Beckmann SW41 rotor. Twelve fractions of 920 µL were collected, and polysomes were monitored by following the absorbance at 254 nm. Total protein was retrieved by 20% trichloroacetic acid (TCA) precipitation performed overnight, washed with acetone, and analyzed by SDS-PAGE followed by western blot. RNA extraction was performed by adding TRI reagent (Merck T9424) to the fractions at a 1:1 v/v ratio and then followed the manufacturer's protocol.
Cell fractionation and isolation of mitoribosomes
Mitochondria and cytosolic fractions were purified as previously described (Choi and Barrientos 2021) with a few modifications. Mitochondrial extracts were loaded onto a 10%–30% sucrose gradient prepared as described in the previous section. Mitoribosomes were separated by centrifugation at 38,000 rpm for 210 min using a Beckmann SW41 rotor. MAM isolation was performed as previously described (Wieckowski et al. 2009).
Western blot and immunoprecipitation
Equal amounts of protein from cell lysates were subjected to SDS-PAGE and transferred to a PVDF membrane (Millipore). HeLa GFP and HeLa TRAP1–GFP cells were treated with either 2 μg/mL of harringtonine and 100 μg/mL of emetine for 15 min at 37°C to inhibit cytosolic translation or 30 µM linezolide or 200 ng/mL chloramphenicol for 1 hr at 37°C to inhibit mitochondrial translation. eGFP-fusion proteins were immunoprecipitated with GFP-trap magnetic agarose beads (GFP-trap_MA Chromotek) according to the manufacturer's instructions. The following antibodies were used for WB, immunofluorescence, and immunoprecipitation: anti-TRAP1 (Santa Cruz Biotechnology sc-13557; Genetex GTX102017), anti-β-ACTIN (ACTB; Santa Cruz Biotechnology sc-69879), antipuromycin (Merck MABE343), anti-HSP90 (HSP90AA1; Santa Cruz Biotechnology sc-1057), anti-tubulin (TUBA1A; Sigma-Aldrich T9026), anti-TOM40 (Genetex GTX133780), anti-TIM44 (Santa Cruz Biotechnology sc-390755), anti-TIM23 (Santa Cruz Biotechnology sc-514463), anti-TOM20 (Santa Cruz Biotechnology sc-17764), anti-GAPDH (Santa Cruz Biotechnology sc-69778), anti-PHB2 (Santa Cruz Biotechnology sc-133094), anti-TUFM (Genetex GTX101763), anti-eEF1a (Millipore 05-235), anti-IDH2 (Genetex GTX133078), anti-Rieske FeS (UQCRFS1; Santa Cruz Biotechnology sc-271609), anti-STAT1 (Santa Cruz Biotechnology sc-346), anti-RPS6 (eS6; Santa Cruz Biotechnology sc-74459), anti-RPL19 (eL19; Santa Cruz Biotechnology sc-100830), anti-MRPS5 (uS5m; Genetex GTX103930), anti-GFP (Santa Cruz Biotechnology sc-81045), antivinculin (VCL; Santa Cruz Biotechnology sc-73614), anti-ATP5B (ATP5F1B; Santa Cruz Biotechnology sc-16690), anti-MRP-L10 (uL10m; Santa Cruz Biotechnology sc-377196), and anti-UQCRC2 (Genetex GTX114873). Total protein normalization has been performed by using No-Stain protein labeling reagent (Thermo Fisher Scientific A44717). Images were acquired with a Chemidoc MP imaging system (Bio-Rad), and where indicated, protein levels were quantified by densitometric analysis using the software ImageJ (Schneider et al. 2012).
35S Met/35S Cys labeling–puromycilation assay
HeLa FITR was seeded in a six-well plate. HeLa GFP and HeLa TRAP1–GFP cells were induced with 1 μg/mL tetracycline for either 24 h (HeLa GFP and HeLa TRAP1–GFP) or 48 h (HeLa shGFP and shTRAP1). 35S Met/35S Cys labeling was performed as follows. Following induction of overexpression or silencing of TRAP1, cells were incubated in cysteine/methionine-free medium (Sigma-Aldrich) for 15 min at 37°C, followed by incubation in cysteine/methionine-free medium containing 50 µCi/mL 35S-labeled cysteine/methionine (PerkinElmer) for 30 min. Cells were then washed with PBS and lysed. Ten micrograms of total protein extract was analyzed by SDS-PAGE and autoradiography. For the puromycilation assay, following induction, cells were treated with puromycin (1 µg/mL) for 10 min to allow for the puromycin to be incorporated into newly synthesized proteins. Cells were then harvested, and total lysates were used for western blot.
FUNCAT and mito-FUNCAT
HeLa shGFP and shTRAP1 cells were seeded in four 15-cm plates. HPG (Thermo Fisher Scientific) labeling was performed as follows. Following silencing of TRAP1 for 72 h upon induction with 1 μg/mL tetracycline, cells were incubated in cysteine/methionine-free medium (Sigma-Aldrich) for 45 min at 37°C, and then, in the case of mito-FUNCAT, treated with 100 μg/mL of cycloheximide and 100 μg/mL of emetine for 15 min at 37°C to inhibit cytosolic translation. Subsequently, 100 μM of HPG was added to the media for 1 h (FUNCAT) or 2 h (mito-FUNCAT) at 37°C. For mito-FUNCAT, after harvesting the cells, mitochondria were isolated by differential centrifugation, as previously described (Choi and Barrientos 2021). Freshly isolated mitochondria were resuspended in 50 µL of Click-iT lysis buffer (50 mM TRIS-HCl at pH 8, 1% SDS, 250 U/mL Universal Nuclease [Thermo Fisher Scientific]) and incubated on ice for 15 min. Mitochondrial extracts were centrifuged at 18,000 rcf for 5 min, and protein concentrations were determined by BCA assay (Thermo Fisher Scientific). One hundred eighty micrograms of mitochondrial proteins were subjected to a click reaction using a commercial kit (Click-iT cell reaction buffer kit; Thermo Fisher Scientific), with 40 µM tetramethylrhodamine (TAMRA)-azide (Sigma-Aldrich). According to the manufacturer's protocol, proteins were purified from the mixture using a MeOH/chloroform approach, after the end of the click reaction. The extracted pellet was dissolved in 20 µL of Click-iT lysis buffer containing 3% SDS, and protein concentrations were determined by BCA assay. Equal amounts of proteins were loaded on a 15% Tris-glycine gel. Fluorescent signals in the gel were analyzed using a ChemiDoc MP imaging system (Bio-Rad). Total protein levels were detected using the no-stain protein labeling reagent (Thermo Fisher Scientific). Click chemistry and immunostaining of fixed cells have been performed as follows. After HPG labeling, coverslips were fixed in 4% paraformaldehyde (PFA) and prepermeabilized with a 0.0005% digitonin solution. Then cells were fully permeabilized with 0.1% Triton X-100 for 5 min at room temperature and subsequently incubated with the click reaction buffer for 30 min according to the Click-iT cell reaction buffer kit (Thermo Fisher Scientific). After washing with 2% BSA in PBS solution, mitochondria were immunostained with anti-TOM20 primary antibody (Santa Cruz Biotechnology sc-17764), and then, cells were subsequently labeled with Alexa Fluor 488–conjugated secondary antibody (Invitrogen A21202). The coverslips were mounted using mowiol, and images were acquired using confocal microscope Zeiss LSM700.
In vitro translation assay (including transcription and capping of the GFP mRNA)
The wheat germ extract kit used to synthesize eGFP and the RTS 100 E. coli HY kit used to synthesize EmGFP were purchased from Promega (L4380) and Biotechrabbit (BR1400102), respectively. eGFP mRNA has been transcribed in vitro using a pLEXY-eGFP vector, and EmGFP has been transcribed from pV94F-emGFP. For the wheat germ translation assay, we assembled each reaction as follows: 5 µL of wheat germ extract, 1 mM of amino acids mixture minus methionine, 1 mM of amino acids mixture minus leucine, 8 U of RNasin ribonuclease inhibitor, in a final volume of 10 µL. For the E. coli transcription/translation assay, we assembled each reaction as follows: 2.4 µL of E. coli lysate, 2 µL of reaction mix, 2.4 µL of amino acids, 0.2 µL of methionine, 1 µL of reconstitution buffer, in a final volume of 10 µL. mRNAs were added to reactions at a final concentration of 21.95 ng/µL. Where indicated, TRAP1 recombinant protein was added to the reaction at a final concentration of 0.3 µg/µL in the wheat germ translation assay and 0.2 µg/µL in the E. coli transcription/translation assay. eGFP and EmGFP fluorescence increases were recorded at 535 nm, at 1-min intervals for 5 h at 32°C and at 30-sec intervals for 3 h at 30°C, respectively. mRNAs were obtained using the MEGAscript kit (Thermo Fisher Scientific AM1334) and prepared according to the manufacturer's manual. Capped eGFP transcript was synthesized by adding a cap analog directly to the MEGAscript reaction.
RNA extraction and RT-qPCR analysis
Total RNA extraction was performed using the TRI reagent (Merck T9424) following the manufacturer's instruction. For first-strand synthesis of cDNA, 1 µg of RNA was used in a 20-µL reaction mixture by using a SensiFAST cDNA synthesis kit (Bioline). For real-time PCR analysis, 0.4 µL of cDNA sample was amplified by using the SensiFAST Syber (Bioline) in an CFX Opus 96 real-time PCR instrument (Bio-Rad Laboratories). The reaction conditions were 5 min at 95°C followed by 45 cycles of 15 sec at 95°C and 1 min at 60°C. The sequence of the oligos used are listed in Table 1 in 5′-3′ direction.
The sequence of the oligos used
FRET assay by FLIM
In FRET experiments, the TRAP1–GFP fusion protein or a Cy2 conjugated to a secondary antibody was used as donor, and Cy3 conjugated to a secondary antibody was used as acceptor. The cells were fixed in 2% PFA, mounted on a slide, and analyzed using a TCS SMD FLIM Leica SP5 microscope (Leica) equipped with a 63×/1.4 NA objective to measure FRET efficiency (EFRET). EFRET varies as the sixth power of the distance (r) between the two molecules according to the following formula: EFRET = 1/[(1 + r/R0)6], where R0 is the distance corresponding to EFRET = 50%, which can be calculated for any pair of fluorescent molecules. For distances less than R0, FRET efficiency is close to maximal, because of the 1/r6 dependence, whereas for distances greater than R0, the efficiency is close to zero. EFRET by FLIM was calculated with the following formula: EFRET: 1 − (tDA/tD), where tDA is the donor lifetime in the presence of the acceptor, and tD is the lifetime of the donor alone.
Duolink in situ PLA
Duolink in situ PLA (Navinci NF.MR.100) was performed according to the manufacturer's instructions. Briefly, cells were seeded on coverslips, fixed, permeabilized, and hybridized with primary antibodies. For negative controls, only one of the two primary antibodies has been used. After 1 d, cells were hybridized with secondary antibodies conjugated with the PLA probes (PLUS and MINUS) and then subjected to ligation and rolling circle amplification using fluorescently labeled oligonucleotides. Cells were washed and mounted on slides using a mounting media with DAPI to detect nuclei, and the signal was detected by confocal microscopy analysis. For PLAs, the following antibodies were used: anti-TRAP1 (sc-13557 and GTX102017), anti-TUFM (GTX101764), anti-MRPL12 (Genetex GTX114731), anti-TOM40 (GTX133780), anti-TOM20 (sc-17764), anti-PHB2 (sc-133094), anti-HSP90 (sc-1057), anti-TIM44 (sc-390755), and anti-phospho-S6 ribosomal protein (Ser240/244; Cell Signaling Technology 2215).
Image acquisition was performed by confocal laser-scanning microscopy using a Zeiss 510 LSM from Carl Zeiss Microimaging or by using the Leica thunder imaging system (Leica Microsystems) equipped with a Leica DFC9000GTC camera and a PlanApo 63× oil-immersion (NA 1.4) objective lens. A fluorescence LED light source and appropriate excitation and emission filters were used. Images were acquired taking Z-slices from the top to the bottom of the cell by using the same setting (LED source power, exposure time) and the small volume computational clearing (SVCC) mode for the different cell lines and in all experimental conditions.
Stopped-flow FRET assay
The 70S initiation complex labeled with Cy3 on protein L11 (Cy3-70SIC) and E-348C-EF-Tu labeled with the QSY9 fluorescence quencher (QSY-EF-Tu) was prepared as previously described (Liu et al. 2014). The TC was obtained by incubating in buffer QSY-EF-Tu, yeast Phe, GTP, EP, PK for 5 min at 37°C. Measurements of Cy3-L11 fluorescence were performed at stopped-flow fluorometer in the absence of TRAP1 (red trace, positive control) and with TRAP1 preincubated with either Cy3-70SIC (blue trace) or QSY-TC (purple trace). A negative control (black trace) was performed using a wild-type EF-Tu that is unable to quench Cy3 fluorescence.
Gene coexpression, gene set enrichment, and network analyses
Gene coexpression analyses, defined as similarity of changes in gene expression patterns between genes of interest, was performed by using COXPRESdb (Obayashi et al. 2019; https://coxpresdb.jp). A Pearson correlation coefficient was used as a measure of gene coexpression. Gene set enrichment analyses of the coexpression gene list (Supplemental Table S1) were performed using Enrichr (Xie et al. 2021; https://amp.pharm.mssm.edu/Enrichr/). Network analysis on the 100 genes with the highest degree of coexpression with TRAP1 has been performed by using STRING (Szklarczyk et al. 2021).
Statistical analyses
Statistical analyses were performed by using GraphPad Prism software. The two-tailed unpaired Student's t-test was used to establish the statistical significance of changes in gene expression levels compared with controls in qPCR experiments and of changes of densitometric band intensity in western blots and FUNCAT assays. As for the SunRiSe assay, densitometric quantification of the puromycin incorporation at different time points following harringtonine treatment has been fitted by the least square regression method. The statistical significance in the decay rate of the two curves has been analyzed by the extra sum-of-squares F-test by assuming plateau = 0.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We acknowledge Prof. John J. Rossi for the pFRT-U6tetO plasmid. We acknowledge Elena Dobrikova and Matthias Gromeier (at Duke University Medical Center) for the establishment of the HeLa Flp-In T-REx cell line. The eGFP alone cloned into pcDNA5/FRT/TO (Invitrogen) was kindly provided by Prof. Matthias Hentze, EMBL/Heidelberg University, molecular medicine partnership unit. The inducible Actin-FLAG HeLa cell line was kindly provided by Prof. Alfredo Castello, University of Glasgow. We acknowledge the microscopy facility at the Department of Molecular Medicine and Medical Biotechnology, University of Napoli “Federico II,” for the support in microscopy analyses. This work was supported by National Center for Gene Therapy and Drugs based on RNA Technology, PNRR MUR-CN3 UNINA: E63C22000940007, POR CAMPANIA FESR 2014/2020 (project Sviluppo di Approcci Terapeutici INnovativi per patologie neoplastiche resistenti ai trattamenti [SATIN]), and National Institutes of Health NIH-GM080376 to B.S.C., Lega Italiana per la Lotta contro I Tumori: 2020 5×1000 LILT grant to M.L., and a Finanziamento della Ricerca in Ateneo (FRA 2020) grant to D.S.M.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277755.123.
-
Freely available online through the Genome Research Open Access option.
- Received January 31, 2023.
- Accepted July 19, 2023.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








