
Leveraging family data to design Mendelian randomization that is provably robust to population stratification
- 1Department of Computer Science, University of California Los Angeles, Los Angeles, California 90095, USA;
- 2Department of Statistics, University of California Los Angeles, Los Angeles, California 90095, USA;
- 3Department of Computational Medicine, University of California Los Angeles, Los Angeles, California 90095, USA;
- 4Department of Human Genetics, University of California Los Angeles, Los Angeles, California 90095, USA
-
↵5 These authors contributed equally to this work.
Abstract
Mendelian randomization (MR) has emerged as a powerful approach to leverage genetic instruments to infer causality between pairs of traits in observational studies. However, the results of such studies are susceptible to biases owing to weak instruments, as well as the confounding effects of population stratification and horizontal pleiotropy. Here, we show that family data can be leveraged to design MR tests that are provably robust to confounding from population stratification, assortative mating, and dynastic effects. We show in simulations that our approach, MR-Twin, is robust to confounding from population stratification and is not affected by weak instrument bias, whereas standard MR methods yield inflated false positive rates. We then conduct an exploratory analysis of MR-Twin and other MR methods applied to 121 trait pairs in the UK Biobank data set. Our results suggest that confounding from population stratification can lead to false positives for existing MR methods, whereas MR-Twin is immune to this type of confounding, and that MR-Twin can help assess whether traditional approaches may be inflated owing to confounding from population stratification.
Mendelian randomization (MR) is a widely used analytical tool that uses genetic variants (“genetic instruments”) to determine whether one trait (the “exposure”) has a causal effect on another (the “outcome”). With the availability of massive biobank data sets such as the UK Biobank (Bycroft et al. 2018), MR analyses have become increasingly powerful and have been used to identify causal relationships between numerous pairs of traits (Haase et al. 2012; Haycock et al. 2017; Lyall et al. 2017; Hemani et al. 2018; Wade et al. 2018). The validity of MR rests on three key assumptions (Lawlor et al. 2008): (1) that the genetic instrument is significantly associated with the exposure, (2) that the genetic instrument is independent of confounders of the exposure–outcome relationship, and (3) that the genetic instrument affects the outcome only through its effect on the exposure.
Unfortunately, the latter two assumptions are often violated in practice, owing to several factors, including horizontal pleiotropy, population stratification (and related phenomena such as assortative mating and dynastic effects), and batch effects. Even when these assumptions are met, the weak effects of typical genetic instruments on the exposure coupled with spurious correlation between genetic instruments and confounders (Burgess et al. 2011) can bias the results of MR analyses (“weak instrument bias”). The problem of population stratification has been extensively studied in the Genome-Wide Association Study (GWAS) literature, and approaches for mitigating its effects have been developed, including the usage of principal component analysis (PCA) and linear mixed models (LMMs) (Price et al. 2010). These approaches have generally been found to be effective at reducing the confounding introduced by population stratification (Price et al. 2010).
However, recent studies have shown that, with sample sizes as large as those found in modern biobanks, even a small amount of residual population stratification can cause a considerable amount of bias (Berg et al. 2019; Haworth et al. 2019; Brumpton et al. 2020; Cook et al. 2020) and may even cause false positives in MR analysis (Haworth et al. 2019; Cinelli et al. 2022). In addition, although the confounding effects of population stratification are well known, less attention has been directed toward confounding from other phenomena such as (cross-trait) assortative mating and dynastic effects, which can also cause MR false positives (Hartwig et al. 2018; Brumpton et al. 2020). Recent work has shown that cross-trait assortative mating is widespread and substantially inflates genetic correlation estimates between many trait pairs (Border et al. 2022).
It has recently been proposed that family-based genetic data sets could be used in MR studies to avoid confounding from population stratification (Pingault et al. 2018; Brumpton et al. 2020). A recent suite of methods has been developed for this purpose and was shown to reduce the bias from this type of confounding (Brumpton et al. 2020). However, like other MR methods, these methods are susceptible to weak instrument bias, which can be substantial for small family-based data sets (Brumpton et al. 2020). In this paper, we introduce MR-Twin, a test for causal effects between pairs of traits that is able to leverage family-based genetic data to provably control for population stratification and to use publicly available summary statistics estimated in large biobank data sets to achieve power competitive with the top existing methods for the same sample size. We develop versions of MR-Twin for trio, parent–child duo, and sibling data; evaluate MR-Twin's ability to control false positives owing to population stratification and weak instrument bias; and compare it with existing methods.
Results
Methods overview
In an MR analysis, we wish to determine whether one phenotype (the “exposure”) has a causal effect on another phenotype (the “outcome”) using genetic instrumental variables, which can be either single-nucleotide polymorphisms (SNPs), a polygenic score, or other genetic features. Under the assumption that the genetic instruments are associated with the exposure and are independent of the outcome given the exposure, the MR effect estimate of the exposure on the outcome will be valid even if there are unobserved confounders of the exposure–outcome relationship. The independence assumption, however, is often violated by population stratification (Fig. 1A) or horizontal pleiotropy, as these phenomena cause the genetic instruments to be correlated with the outcome through pathways other than those through the exposure.
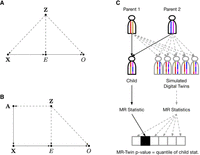
Illustrations of Mendelian randomization (MR) assumptions and the MR-Twin framework. (A) Directed acyclic graph (DAG) depicting variables and their relationships in a typical MR study, where X is the genotypic instrument, E is the exposure trait, and O is the outcome trait. An external confounder Z, such as population stratification, can cause violations of the MR assumptions. (B) If we have the parental haplotypes A, then X is independent of Z given A. (C) Illustration of the MR-Twin workflow. Digital twin genotypes are sampled from the parental genotypes. MR-Twin is a conditional randomization test, conditioned on A and therefore immune to confounding from Z, in which the P-value is computed based on the quantile of the true offspring's MR-Twin statistic compared with the digital twins’ statistics.
MR-Twin is a method that uses family-based genetic data to construct a test for whether the exposure has an effect on the outcome that is immune to confounding from population structure. It is based on the key idea that the genotypes of observed individuals are independent of population structure given the genotypes of the individuals’ parents, because the mechanisms by which genetic information is passed from parents to offspring are known (Fig. 1B). In other words, conditioned on the parental genotypes, population structure provides no additional information about the distribution of the offspring's genotypes. Thus, by conditioning on the parental genotypes, confounding from population stratification can be avoided (Fig. 1C), along with confounding from other phenomena such as cross-trait assortative mating and dynastic effects that operate through the parental genotypes (see Fig. 1 of Brumpton et al. 2020).
We now outline the algorithm in the context of a trio design in which we have genetic data on the parents and the offspring.
Let X and O denote the genotypes and outcome phenotype values, respectively, for some individual, and let 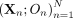 denote these across N trios. Also let P1 and P2 denote the genotypes of the parents of the individual with genotypes X, and let A: = (P1, P2) refer to the set of parental genotypes. Let Z denote the set of external confounders measured on the same individual, which we define as the set of confounders that satisfy
X⫫Z|A. Thus, population stratification is an external confounder (as are assortative mating and dynastic effects), whereas horizontal
pleiotropy is not. The key idea is that we can formulate a hypothesis test of a causal effect conditional on the parental
haplotypes A. Bates et al. (2020) show that such a test is also a test of the stronger null hypothesis of a causal effect conditional on (A, Z).
denote these across N trios. Also let P1 and P2 denote the genotypes of the parents of the individual with genotypes X, and let A: = (P1, P2) refer to the set of parental genotypes. Let Z denote the set of external confounders measured on the same individual, which we define as the set of confounders that satisfy
X⫫Z|A. Thus, population stratification is an external confounder (as are assortative mating and dynastic effects), whereas horizontal
pleiotropy is not. The key idea is that we can formulate a hypothesis test of a causal effect conditional on the parental
haplotypes A. Bates et al. (2020) show that such a test is also a test of the stronger null hypothesis of a causal effect conditional on (A, Z).
The way that this is accomplished is through a conditional randomization test, similar to the digital twin test (DTT) proposed
by Bates et al. (2020) in the context of GWAS (Candès et al. 2018). The idea is to sample so-called “digital twins” 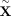 from each set of parents A such that
from each set of parents A such that 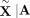 has the same distribution as
has the same distribution as 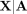 , which can easily be accomplished using the laws of Mendelian inheritance (Methods). We construct B such random samples across all trios,
, which can easily be accomplished using the laws of Mendelian inheritance (Methods). We construct B such random samples across all trios, 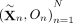 , and for each set b of twins, we compute a test statistic
, and for each set b of twins, we compute a test statistic 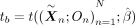 , representing the strength of association between the genetically predicted exposure and the outcome. We also compute a test
statistic for the true offspring of the trios,
, representing the strength of association between the genetically predicted exposure and the outcome. We also compute a test
statistic for the true offspring of the trios, 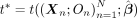 .
.
We can then obtain a P-value for a nonzero causal effect of the exposure on the outcome, 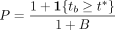 . The set of B statistics derived from the digital twins represents a null distribution conditioned on the parental genotypes. If there
is a true nonzero effect of the exposure on the outcome, we expect the statistic derived from the true offspring to be stronger
than statistics derived from digital twins whose genotypes are randomly sampled from the parental genotypes. The test statistic
and algorithm are explained in more detail in the Methods section.
. The set of B statistics derived from the digital twins represents a null distribution conditioned on the parental genotypes. If there
is a true nonzero effect of the exposure on the outcome, we expect the statistic derived from the true offspring to be stronger
than statistics derived from digital twins whose genotypes are randomly sampled from the parental genotypes. The test statistic
and algorithm are explained in more detail in the Methods section.
MR-Twin controls for arbitrarily strong population stratification confounding in simulations
Simulate genotypes under population structure
1: procedure SimGeno(FST, groups, N,M) ▷ FST is the fixation index, groups is the number of populations, N is number of samples, M is number of SNPs.
2: Initialize the average 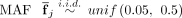 for each SNP j.
for each SNP j.
3: for k ≤ groups do
4: 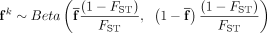
5: Generate genotype matrix X(k) of population k such that 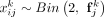 for each individual i and SNP j.
for each individual i and SNP j.
6: end for
7: X = [X(1); . . . ;X(groups)] . ▷ Stack the rows of each genotype matrix
8: return Genotype matrix X
9: end procedure
Simulate population-stratified phenotypes
1: procedure getPheno(X, U, h2, αE, γue, γuo) ▷ X is the normalized genotype matrix, U is a vector with the fixed population label for each sample, h2 is heritability of exposure E, αE is effect of E on outcome O, γue and γuo are fixed confounding effects of U on E and O.
2: Generate genetic coefficient 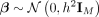
3: Compute 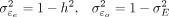
4: Simulate E = Xβ + γueU + ɛe where 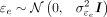
5: Simulate O = αEE + γuoU + ɛo where 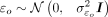
6: return (E,O)
7: end procedure
We compared the performance of MR-Twin to other MR methods via simulations consisting of two populations with allele frequency differences modeled according to the standard Balding–Nichols model (Balding and Nichols 1995), following the method of previous works (Pritchard et al. 2000; Price et al. 2006; Hubisz et al. 2009; Chen et al. 2015; Conomos et al. 2016; Ochoa and Storey 2021). The procedure for simulating the genotypes is outlined in Algorithm 1. We use this algorithm to simulate “external” samples (nontrio data, e.g., from a biobank), as well as the parents for the trios. The offspring genotypes for the trios can then be easily sampled given the parental genotypes (Methods). For each sample, we retain the population label, a binary variable indicating to which population each sample belongs. Unless otherwise specified, each simulation had 50,000 (false-positive rate [FPR] simulations) or 100,000 (power simulations) external samples and 1000 trio samples evenly split between two populations with a fixation index FST = 0.01 and 100 SNPs, 50 of which were causal for the exposure trait. Unless otherwise specified, the heritability of the exposure trait was set to h2 = 0.2.
Next, we simulate both the exposure and outcome phenotypes following a linear model, as outlined in Algorithm 2. This model allows the population labels from the first step to have an effect on the exposure and outcome phenotypes, which models population stratification that violates the MR assumptions. We use this setting to assess the FPR of methods under population stratification, allowing the effects of the population labels on the exposure and outcome phenotypes to range from zero (no confounding) to 0.8 (substantial confounding). In a separate set of simulations to assess power, we set the confounding effect to zero and varied the causal effect.
We performed 1000 simulation replicates under these settings, each time simulating a set of external and trio genotypes and phenotypes according to the chosen parameters, performing linear regression between each SNP and the exposure and outcome phenotypes, and using the resulting association statistics as input to each of the MR methods. We excluded SNPs with association P-values of above 0.05/M (M=100) with the exposure phenotypes in the external data in order to limit weak instrument bias (Burgess et al. 2011). The methods we assessed include the trio mode of MR-Twin, standard inverse-variance weighted (IVW) MR (Burgess et al. 2013), MR-Egger (Bowden et al. 2015), the weighted median estimator (Median) (Bowden et al. 2016), the mode-based estimator (Mode) (Hartwig et al. 2017), and a method introduced by Brumpton et al. (2020) to use family data to control for confounding owing to population stratification and other population-related effects. Brumpton et al. (2020) provide a suite of methods for different family data sets, following previous work such as that by Fulker et al. (1999); here we focus on the trio-based method they describe, and simply refer to that method as “Brumpton” below.
The trio mode of MR-Twin maintained a calibrated FPR irrespective of the strength of confounding (Fig. 2A). Non-family-based methods such as IVW, Egger, Median, and Mode all displayed substantially inflated FPR in the face of confounding, consistent with their sensitivity to potential residual population stratification. The Brumpton method also displayed slightly inflated FPR, which increased with the strength of the confounding effect, likely owing to weak instrument bias (Brumpton et al. 2020). To mitigate the impact of weak instrument bias, we applied a common approach used in MR studies (Burgess et al. 2011) that involves filtering out variants for which the F-statistic of the association signal is low (F < 10 following previous recommendations). This rendered the FPR inflation negligible but also rendered Brumpton substantially less powerful than MR-Twin, whereas the “unfiltered” mode had similar power to MR-Twin (Fig. 2B). Results with confidence intervals are shown in Supplemental Figure S10. We further investigated the weak instrument bias by running simulations with no SNP filtering based on external data and with increasing numbers of SNPs—settings expected to generate large numbers of weak instruments—and confirmed that Brumpton had greater FPR inflation in these settings, whereas MR-Twin remained calibrated (Supplemental Fig. S11) and did not lose power (Supplemental Fig. S12).
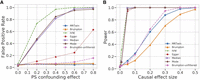
False-positive rate (FPR) and power comparison between various methods run on simulated data. (A) FPR (y-axis) under varying levels of confounding owing to population stratification (PS), with the x-axis describing the magnitude of the confounding effect of population labels on the exposure and outcome trait. (B) Power (y-axis) as a function of the magnitude of the causal effect of the exposure on the outcome trait (x-axis) in a setting with no confounding. Results are averaged over 1000 simulation replicates.
The standard MR methods (IVW, Egger, Median, and Mode), when run on the external data, had substantially higher power than the family-based methods, MR-Twin and Brumpton (Fig. 2B). We performed additional simulations to understand if the lower power of MR-Twin was owing to the smaller number of trios as opposed to methodological limitations. When applied to the offspring in each trio (Fig. 3), the standard MR methods still had substantially inflated FPR (Fig. 3A) but similar power to MR-Twin and Brumpton (Fig. 3B). We also evaluated the FPR and power of these methods under varying number of trios (Supplemental Fig. S1). We observed that increasing the number of trios increased power for all methods, as expected, suggesting that the family-based methods can be expected to obtain increased power as more genetic family data are ascertained in the future. The relative power of the methods remained roughly consistent across these experiments.
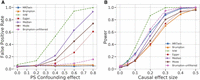
FPR and power comparison between various methods run on simulated trio data. This is similar to Figure 2 except that IVW, Egger, Median, and Mode are run on the offspring of the trio data set instead of the large “external” group of unrelated individuals, such that all methods have the same sample size. (A) FPR (y-axis) under varying levels of confounding owing to population stratification (PS), with the x-axis describing the magnitude of the effect of the population labels on the exposure and outcome trait. (B) Power (y-axis) as a function of the causal effect size (x-axis). Results are averaged over 1000 simulation replicates.
We also evaluated the area under the receiver operating characteristic curve (AUC-ROC) (Supplemental Figs. S5, S6). Comparing the two main family-based approaches, MR-Twin generally had higher AUC than Brumpton (filtered). Predictably, the AUC of standard MR methods drops sharply when there is confounding, and MR-Twin outperforms these methods in most such cases, although Egger was a notable exception in our findings and remained competitive with family-based approaches even under confounded settings. Similar to our findings in Figures 2 and 3, family-based methods are more competitive with standard MR methods when run on similar sample sizes. As an additional sensitivity analysis, we also assessed the FPR (Supplemental Fig. S7) and power (Supplemental Fig. S8) of methods in settings in which there are very few instruments or very low heritability. The trends were broadly similar to those seen in Figures 2 and 3. MR-Twin maintained a calibrated FPR in all settings, although it did suffer a loss of power when heritability was very low (Supplemental Fig. S8B,D).
We performed simulations increasing the magnitude of population structure as measured by the FST (without necessarily increasing the confounding strength), and observed that increasing the population structure leads to further FPR inflation for standard MR methods (Supplemental Fig. S2). We observed inflated FPR for standard MR methods even when there is no confounding (stratification) for large values of FST (Supplemental Fig. S2) likely owing to correlation or linkage disequilibrium (LD) among the genetic variants induced by population structure. The standard implementation of IVW and Egger (Yavorska and Burgess 2017; Broadbent et al. 2020) allows the user to pass in a variant correlation matrix, which removed the FPR inflation with no stratification (Supplemental Fig. S2); other methods such as Median and Mode do not currently have this option.
Next, we assessed the runtime of methods run on the trio data (Supplemental Fig. S3). Brumpton, along with non-trio-based methods (e.g., IVW), had similar run times (<1–5 sec per simulation replicate); for succinctness, only Brumpton is shown. MR-Twin (with 100 simulated digital twins) took ∼1 min per simulation replicate under the simulation settings described above, with time increasing to up to 4 min if the number of families or SNPs was increased. The number of digital twins to simulate for MR-Twin involves a trade-off between speed and stability of results. We assessed the stability of MR-Twin with different numbers of digital twins, with the results shown in Supplemental Figure S9. We interpreted these findings as indicating that 100 digital twins are likely stable enough for simulations for which many replicates are run and speed is a priority, but 1000 or more digital twins are recommended for one-off real data analysis. Therefore, we simulated 100 digital twins in our simulations and 1000 in our real data analysis. We note, however, that although the MR-Twin runtime increases linearly with the number of digital twins simulated, the generation and statistic computation for the digital twins can be performed in parallel, so many twins can be simulated efficiently given multiple compute cores or nodes. For clarity of results, we did not take advantage of this in our runtime assessment.
Finally, MR-Twin also enables users to use parent–child duo or sibling data sets (Methods). We assessed the performance of these modes versus the trio mode of MR-Twin (Supplemental Fig. S4). We found that the duo and sibling modes, although having lower FPR than most standard MR methods, did not maintain a calibrated FPR at high levels of confounding, which is expected because the precise sampling of offspring genotypes from parents is not possible when either or both of the parental genotypes are not available.
Application to trio data in the UK Biobank
To assess the results given by MR-Twin relative to other approaches in a real data context, we next applied MR-Twin and four other MR methods (IVW, Egger, Median, and the Brumpton et al. method) (Brumpton et al. 2020) to 144 real trait pairs in the UK Biobank (Bycroft et al. 2018). These consisted of all pairwise combinations of 12 metabolic, anthropometric, and socioeconomic traits that were widely measured among the UK Biobank participants (listed in Supplemental Table S1). We isolated 955 White British genetic trios from the full UK Biobank data set (Supplemental Materials) and used PLINK (Purcell et al. 2007) to run linear regression on the remaining unrelated White British individuals for these 12 traits, including the top 20 principal components (PCs), age, and sex as covariates. The genetic instruments selected for each analysis were the SNPs with genome-wide significant P-values ( < 5.0 × 10−8) for the exposure trait, after LD pruning was performed so that none of these instruments were in substantial LD with one another (Supplemental Materials). Ignoring the degenerate cases in which the exposure and outcome were the same trait or in which there were no significant SNPs for the exposure trait (as was the case for the Townsend deprivation index [TDI]), there were 121 usable trait pairs.
Table 1 shows the results for six selected trait pairs (excluding Median for brevity because it gave similar results to IVW), and Supplemental Table S2 shows the full set of analyses. Brumpton was run with several different variant filtering settings to assess the impact of potential weak instrument bias (Supplemental Materials); results for all runs are given in Supplemental Table S2. For Table 1, we selected six analyses: two positive controls representing causal effects that are true by definition (LDL cholesterol → total cholesterol and weight → body mass index [BMI]), two negative controls that represent seemingly implausible effects (glucose → TDI and height → body fat), and two trait pairs with unclear or conflicting evidence (BMI → diastolic blood pressure [DBP] and BMI → TDI). In particular, previous studies have identified a significant effect for BMI → DBP (Lyall et al. 2017) and for BMI → TDI in women (Tyrrell et al. 2016) with IVW analysis, although Egger analysis did not replicate the significant findings in either case (Tyrrell et al. 2016; Lyall et al. 2017).
Traditional MR results and MR-Twin results on selected trait pairs from the UK Biobank
All methods performed as expected on the controls, with highly significant P-values for positive controls and insignificant P-values for negative controls. For BMI → DBP, IVW and Brumpton yielded significant results, whereas Egger and MR-Twin did not. For BMI → TDI, IVW and Egger yielded significant results, whereas Brumpton and MR-Twin did not. In general, IVW tended to yield much stronger P-values than other methods, and the family-based methods (Brumpton and MR-Twin) tended to be conservative (Supplemental Table S2), in line with our simulation results. In particular, of the 121 usable trait pairs, IVW identified 78 as significant, Egger identified 56 as significant, Brumpton identified 20 as significant, and MR-Twin identified 19 as significant.
Discussion
We introduced MR-Twin, a method for testing causal effects between pairs of traits within an MR framework, which is provably robust to confounding of any strength resulting from population stratification. Our primary contributions are the following: (1) developing a DTT, originally proposed by Bates et al. (2020) in the context of genetic association studies, for MR, coupled with a novel statistic for this test; (2) showing that, by leveraging trio data, our proposed framework is robust to confounding owing to population stratification and to biases from the inclusion of genetic instruments with weak effects; (3) extending our framework to the setting of sibling data, a setting not considered by Bates et al; and (4) conducting the first (to our knowledge) large-scale evaluation of the DTT framework in comparison with existing methods for MR. We showed that existing MR methods, including those designed to correct for confounding resulting from horizontal pleiotropy, are prone to false positives when there is confounding from population stratification.
Although population stratification was the focus of this paper, the MR-Twin framework also provides immunity to several other types of confounding effects. Theory dictates that MR-Twin is immune to confounding from familial effects such as assortative mating and dynastic effects because these effects operate through the parental genotypes (see Fig. 1 of Brumpton et al. 2020), although we do not explicitly test this in this paper. As recently shown, cross-trait assortative mating is pervasive and impacts many common genetic analyses (Border et al. 2022), including MR (Hartwig et al. 2018), so this represents another valuable aspect of MR-Twin even if population stratification is believed to be well controlled in a particular study. In general, MR-Twin is immune to any confounder that is independent of the genotypes of offspring given the genotypes of their parents. We note that when we refer to “immunity” we mean this in a theoretical sense, for instance, under the assumption that the model for Mendelian inheritance is correct. In our particular implementation, we assume that the genetic instruments have been selected to be roughly independent, and thus, we can sample digital twin genotypes from the parental genotypes using a binomial model. In practice, of course, genetic variants on the same chromosome are never perfectly independent, although with appropriate caution, the dependence is weak enough that the effect on calibration should be negligible. More complex models of meiosis will also rely on other factors such as haplotype phasing accuracy.
In addition to population and familial effects, we highlight two underappreciated sources of bias in MR studies, both of which MR-Twin avoids without requiring the user to modify any parameters or arguments. The first is weak instrument bias (Burgess et al. 2011), which can bias the effect estimate of standard MR methods, including the Brumpton approach (Brumpton et al. 2020). This accounts for the Brumpton method yielding inflated FPRs when the confounding effects were strong (Fig. 2A). One of the most common ways to control for weak instrument bias is by filtering out variants with a weak association signal, often with a threshold of F < 10 for the association between a variant and the exposure trait. However, this procedure has been criticized (Burgess et al. 2011) and may not fully correct for weak instrument bias. Other MR methods may also be affected by this bias. In two-sample study designs, the direction of the bias is toward the null rather than the confounded exposure–outcome association estimate (Lawlor 2016), but the bias remains.
Additionally, we found that standard MR methods (IVW, Egger, Median, Mode, etc.) may have inflated FPR when there is population structure that induces correlation between variants, even in the absence of stratification (Supplemental Fig. S2). The reason for the induced correlation is that, even though the variants were simulated independently, they were correlated with one another through the population labels. For example, suppose we have two variants, X1 and X2, and a population label U. The causal diagram for these three variables is X1 ← U → X2, so X1 and X2 are correlated. Our findings corroborate earlier findings that correlation between SNPs can cause calibration issues in MR methods (Burgess et al. 2013). This phenomenon should be taken into account when performing MR simulations or when applying MR to real data sets where variants may be correlated. In the latter case, users should obtain SNP correlations from an appropriately population-matched (Peterson et al. 2019) and sufficiently large (Benner et al. 2017) reference panel.
MR-Twin avoids both of these issues, without requiring the user to specify an SNP correlation matrix or apply various approaches to mitigate weak instrument bias. First, both MR-Twin and Brumpton avoid the correlated-variant issue because they condition on parental genotypes, severing the link between the offspring genotypes and the population structure. Second, MR-Twin would not lose FPR calibration owing to weak instrument bias, because this phenomenon has nothing to do with the aspects of the MR-Twin test that guarantee immunity from confounding owing to population and familial effects (sampling digital twin genotypes conditioned on parental genotypes). Theoretically, it is possible that the bias in the MR effect estimate used in the MR-Twin statistic (Methods) could lower power, but because the MR effect estimate equally affects both the digital twin statistics and the true offspring statistics, a reduction in power seems unlikely and was not observed empirically (Supplemental Fig. S12).
There is extensive literature on family-based methods for avoiding confounding owing to population structure in genome-wide association studies or linkage analysis (Spielman et al. 1993; Thomson 1995; Fulker et al. 1999; Abecasis et al. 2000; Laird and Lange 2006; Weiner et al. 2017). One prominent example is the transmission disequilibrium test (TDT) (Spielman et al. 1993) and the more recent polygenic TDT (pTDT) (Weiner et al. 2017). Bates et al. (2020) compare the DTT to the TDT and show that the DTT is a generalization of the TDT and highlight some of its benefits. Because it is not immediately obvious how to adapt the TDT and pTDT to MR, we do not evaluate their potential use in this context.
There are several considerations that come into play when applying the MR-Twin method, which we note here. First, the number of digital twins simulated involves a trade-off between speed and precision (Supplemental Fig. S9). Although MR-Twin was slower than competing MR methods (Supplemental Fig. S3), it still ran in a few minutes or less per run on both simulated and real data analyses, justifying the use of a fairly large number of digital twins if possible. Consequently, we recommend 1000 or more digital twins for real data analysis, which should be computationally feasible and precise (and, again, parallelization can make this quite efficient). One hundred digital twins are likely sufficient in simulations in which there are many replicates and speed is the paramount concern. Second, the populations of the external and family data sets should be similar. This is natural for biobanks like the UK Biobank but can be more challenging when attempting to combine separate data sets. Third, care should be taken to ensure that the normalization method used and covariates controlled for are similar in the external and trio data sets in order to avoid potential loss of power.
Although the genetic trio offspring used in our UK Biobank analysis were all adults (as all participants in this data set were aged 40–69 at collection time) (Bycroft et al. 2018), other trio data sets may contain young children. This is a potential issue because some commonly analyzed traits such as height and weight may not have the same relationship in youths or adolescents as they do in adults, and variants that affect these traits may not yet have realized their full effect in the children yet. Dealing with such time-varying exposures in the context of MR is an area of ongoing research (Labrecque and Swanson 2019), and it is not clear how this would impact MR-Twin results. Even when the offspring of the trios are all adults, it may be difficult to adequately sample certain traits. For example, we were not able to perform MR analysis for complex traits such as heart disease, because none of the offspring in our sample had developed heart disease, largely because all offspring in our sample were aged 40–49.
We note a few trends seen across many trait pairs in the real data results, reflecting some practical considerations. First, all standard MR methods identified substantially more trait pairs than did either family-based approach. Given our simulation results showing a large power difference in the methods when run with different sample sizes (Fig. 2) but similar power when run with the same sample size (Fig. 3), along with the fact that the UK Biobank has many more unrelated individuals than trios, we believe that this difference is largely because of the difference in the available sample sizes between unrelated and trio data. The number of trios available as part of public data sets is currently relatively small, limiting the power of family- or trio-based methods, but future increases in the number of available trios will lead to increases in the power of MR-Twin and other family-based methods. Second, some trait pairs had quite different results when the exposure and outcome traits were switched. For example, none of the standard MR methods had significant P-values for DBP → weight, but all were significant for weight → DBP (Supplemental Table S2). This may be owing to one causal direction being correct while the other is incorrect but may also be affected by factors such as differences in the heritability and/or polygenicity between the two traits.
Several extensions to the methods presented here are also possible. Although we explored continuous traits in this paper, further work needs to be performed to apply MR-Twin to binary phenotypes such as disease labels. First, a different statistic such as binary cross entropy (rather than our negative squared loss statistic) may be more appropriate. Second, the use of the external effect size estimates in the statistic may have to be modified, depending on the regression method used and the interpretation of the estimates. For example, it would be inappropriate to replace the effect size estimates in our statistic with odds ratios produced by logistic regression. Even for linear data, it is possible that a different statistic than the one we proposed would be more powerful in some situations. Finding the most powerful statistics for a given significance threshold is a direction for future work. Future work could also improve upon the sibling mode of MR-Twin by using population-based priors to infer parental genotypes with a greater level of accuracy, thereby obtaining superior control of false positives. This approach could, in principle, be developed for and applied to more extended pedigrees.
In the DTT paper, Bates et al. (2020) propose using a hidden Markov model (HMM) to simulate digital twins from the parental haplotypes, the latter being generated by phasing the parental genotypes. For the simplicity of avoiding this phasing step and because genetic instruments in MR studies are usually selected to be roughly independent (Burgess et al. 2013), we used a simpler method for simulating digital twins using binomial draws from the parental genotypes (Methods). However, the variants used may not be independent even if they appear to be (Burgess et al. 2013), or one may wish to include correlated variants to increase power. Extending MR-Twin to perform the HMM-based digital twin simulation could therefore increase power.
Finally, a preprint from Tudball et al. (2022) proposes a randomization-based approach to MR that, although being conceptually similar, differs from MR-Twin in a few practical aspects. First, Tudball et al. (2022) do not discuss the use of external summary statistics to increase power, whereas this is a core part of the MR-Twin approach (as well as in the DTT of Bates et al. 2020). Second, Tudball et al. develop family-based propensity scores for individual SNPs and suggest aggregating them with Fisher's method or another P-value aggregation method, which is substantially different from our proposed sum-of-squares statistic over all SNPs (Methods). Finally, Tudball et al. used the HMM-based digital twin simulation model, whereas (as discussed above) we use the simpler binomial model. Nevertheless, the broad conceptual similarities between the two methods highlight the promise of randomization-based approaches to make MR findings more robust and the value of continued development to extend these approaches to more complex pedigrees.
Methods
The MR-Twin framework
We first introduce the standard MR model, without any confounding. Suppose that for a collection of N individuals we obtain their genotypes at M SNPs, as well as a phenotypic measure for an exposure trait and an outcome trait. For a given individual n, we denote the genotype vector as Xn, the genotype at some SNP j as Xnj, the exposure trait as En, and the outcome trait as On. Let 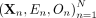 denote the collection of these genotypes and traits over all N individuals, where (Xn) is an (N × M) matrix, and (En) and (On) are (N × 1) vectors. Finally, let X, E, and O refer to the genotype vector, exposure trait, and outcome trait for a generic individual.
denote the collection of these genotypes and traits over all N individuals, where (Xn) is an (N × M) matrix, and (En) and (On) are (N × 1) vectors. Finally, let X, E, and O refer to the genotype vector, exposure trait, and outcome trait for a generic individual.
MR uses the genetic “instrument” X to estimate the effect of an “exposure” trait E on an “outcome” trait O. This estimate is valid regardless of any confounder U of the association between E and O, assuming that the following conditions hold (Lawlor et al. 2008):
-
The genetic instrument X is significantly associated with the exposure trait E;
-
The genetic instrument X is independent of any variables (such as those in U) that confound the relationship between E and the outcome trait O; and
-
The genetic instrument X is not associated with O except owing to its association with E.
The latter two criteria can be captured by the independence statement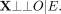 (1)
Assuming these conditions hold and assuming a linear model for the relationships between the genotypes and phenotypes (a typical
assumption in MR analyses), we can test the null hypothesis that there is no direct causal effect of E on O:
(1)
Assuming these conditions hold and assuming a linear model for the relationships between the genotypes and phenotypes (a typical
assumption in MR analyses), we can test the null hypothesis that there is no direct causal effect of E on O: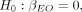 (2)
where βEO is not obtained by direct regression but rather via instrumental variables estimators such as the ratio estimator
(2)
where βEO is not obtained by direct regression but rather via instrumental variables estimators such as the ratio estimator 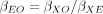 (when a single instrument is used) or by two-stage least squares or inverse-variance weighting (when multiple instruments
are used) (Burgess et al. 2013).
(when a single instrument is used) or by two-stage least squares or inverse-variance weighting (when multiple instruments
are used) (Burgess et al. 2013).
However, in the case in which we have residual population stratification, denoted Z (Fig. 1A), this independence assumption is violated. This is because, using terminology from Pearl's graphical formalism (Pearl 1995), X ← Z → O is a backdoor path between X and O, so the two are not marginally independent. Conditioning on E fails to block this backdoor path (i.e., see Fig. 1A). Residual population stratification generally cannot be controlled for directly, although approaches such as PCAs and LMMs have been used to reduce its effect (Price et al. 2010).
MR-Twin (Fig. 1C) is a method that uses family-based genetic data to avoid this confounding. Suppose that we also observe, corresponding
to each individual's genotypes X, the genotypes P1 and P2 of their parents (we later relax the trio assumption to allow for parent–child duo or sibling data). Let A: = (P1, P2). According to the graphical criteria for d-separation developed by Pearl (1995), A d-separates X from Z (Fig. 1B):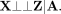 (3)
This means that, assuming X does not affect some unmeasured variable which in turn affects O (i.e., no horizontal pleiotropy),
(3)
This means that, assuming X does not affect some unmeasured variable which in turn affects O (i.e., no horizontal pleiotropy),
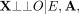 (4)
thereby satisfying the MR conditions regardless of any residual population stratification.
(4)
thereby satisfying the MR conditions regardless of any residual population stratification.
As shown by Bates et al. (2020), the DTT framework outlined in Algorithm 3 can be used to perform a hypothesis test conditioned on A. The resulting test involves computing the test statistic 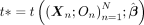 (we give the statistic used in this paper in the subsection “MR-Twin test statistic incorporating external weights”). To
perform a test, we construct B random samples
(we give the statistic used in this paper in the subsection “MR-Twin test statistic incorporating external weights”). To
perform a test, we construct B random samples 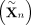 where each
where each 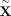 is a random sample given A with the same distribution as X given A (such a sample can be easily constructed using Mendelian inheritance; see subsection “Generating digital twins”). We refer
to these samples as “digital twins.” For each such sample b, we then compute
is a random sample given A with the same distribution as X given A (such a sample can be easily constructed using Mendelian inheritance; see subsection “Generating digital twins”). We refer
to these samples as “digital twins.” For each such sample b, we then compute 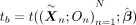 , representing a null distribution of genotypes conditioned on the parental genotypes. This, in turn, gives us a P-value for
, representing a null distribution of genotypes conditioned on the parental genotypes. This, in turn, gives us a P-value for 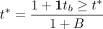 , where B is the total number of permutations we perform. The MR-Twin test is therefore a kind of conditional randomization test (Candès et al. 2018; Bates et al. 2020).
, where B is the total number of permutations we perform. The MR-Twin test is therefore a kind of conditional randomization test (Candès et al. 2018; Bates et al. 2020).
Importantly, the proposed algorithm can leverage effect size estimates 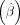 from any external GWAS data sets (even GWAS data sets in which such estimates might be biased owing to population stratification)
while providing valid tests. The proposed algorithm is robust to any external confounder satisfying Equation 3, such as population stratification, assortative mating, and dynastic effects.
from any external GWAS data sets (even GWAS data sets in which such estimates might be biased owing to population stratification)
while providing valid tests. The proposed algorithm is robust to any external confounder satisfying Equation 3, such as population stratification, assortative mating, and dynastic effects.
Outline of MR-Twin
1. Input: Effect sizes for SNPs: 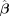 , trio data
, trio data 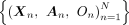
2. Compute the MR-Twin test statistic 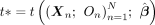
3. For b = 1 to B:
(a) Sample digital twins 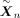 given their ancestors An.
given their ancestors An.
(b) Compute the MR-Twin test statistic 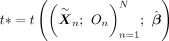
4. 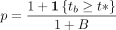
Output: P-value: P
Next, we detail the MR-Twin test statistic, digital twin generation algorithms, and formal proofs of the exchangeability of digital twins with each other and their real counterparts.
Conditional randomization test for MR
The MR-Twin test is related to the DTT (Bates et al. 2020) and, likewise, is a kind of conditional randomization test (Candès et al. 2018). Like the DTT, MR-Twin leverages the fact that offspring genotypes are conditionally independent of “external” confounders such as population structure given the parental genotypes, and uses a conditional randomization test to test the weaker, but equivalent, null hypothesis of no effect conditioned upon the parental genotypes.
Let X be a vector of offspring genotypes, and let A be the genotype vectors of the two parents of the offspring. A may be directly observed, as in trio data, or inferred using parent–child duo or sibling data (see subsection “Generating
digital twins”). Let Z be one or more “external” confounders, defined (Bates et al. 2020) as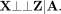 (5)
Thus, population structure is an external confounder, whereas horizontal pleiotropic traits are not. We therefore have
(5)
Thus, population structure is an external confounder, whereas horizontal pleiotropic traits are not. We therefore have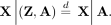 (6)
Assuming that all confounders are external and that X is significantly associated with E, O is independent of X given A under the MR null hypothesis that E has no effect on O. This is because X would not have any effects on O mediated by E (because E does not affect O under the MR null hypothesis), and all paths not through E are blocked by conditioning on A as shown in Equation 6. We therefore want to test
(6)
Assuming that all confounders are external and that X is significantly associated with E, O is independent of X given A under the MR null hypothesis that E has no effect on O. This is because X would not have any effects on O mediated by E (because E does not affect O under the MR null hypothesis), and all paths not through E are blocked by conditioning on A as shown in Equation 6. We therefore want to test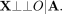 (7)
If this holds, then we cannot rule out that either X has no effect on E or E has no effect on O. We test this null hypothesis via a conditional randomization test (Candès et al. 2018).
(7)
If this holds, then we cannot rule out that either X has no effect on E or E has no effect on O. We test this null hypothesis via a conditional randomization test (Candès et al. 2018).
In testing this null hypothesis, it is helpful to be able to leverage SNP effect sizes estimated from large, external data
sets (such as publicly released summary statistics for resources like the UK Biobank) (Bycroft et al. 2018), as this will often yield more statistically significant variants and better effect size estimates than those generated
using small genetic family data sets. We therefore note that the following property also holds: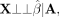 (8)
where we use the shorthand
(8)
where we use the shorthand 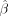 to refer to the estimated effect sizes of each SNP on the exposure and outcome traits.
to refer to the estimated effect sizes of each SNP on the exposure and outcome traits.
We construct “digital twins” 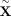 sampled from the parental genotypes via Mendelian inheritance (see subsection “Generating digital twins”) such that
sampled from the parental genotypes via Mendelian inheritance (see subsection “Generating digital twins”) such that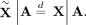 (9)
Given Equations 7, 8, and 9, we have the following under the null hypothesis:
(9)
Given Equations 7, 8, and 9, we have the following under the null hypothesis: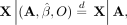 (10)
(10)
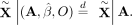 (11)
It follows from Equations 9, 10, and 11 that the digital twins are exchangeable under the null hypothesis:
(11)
It follows from Equations 9, 10, and 11 that the digital twins are exchangeable under the null hypothesis: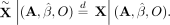 (12)
Therefore, given some statistic
(12)
Therefore, given some statistic 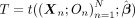 , where N is the number of families,
, where N is the number of families,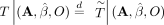 (13)
under the null, where
(13)
under the null, where 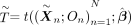 . We can then use the procedure outline in Algorithm 3 to obtain a P-value for this test statistic (Candès et al. 2018).
. We can then use the procedure outline in Algorithm 3 to obtain a P-value for this test statistic (Candès et al. 2018).
MR-Twin test statistic incorporating external weights
We construct a test statistic based on a negative sum of squares loss when using X to predict O via an MR estimate for the effect of E on O. First, we leverage the effect sizes from the external data set of the genotype on the exposure trait 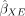 to obtain the genetically predicted exposure trait values:
to obtain the genetically predicted exposure trait values: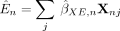 (14)
for each individual n and SNP j. We then compute the MR estimate for the effect of the exposure trait on the outcome trait,
(14)
for each individual n and SNP j. We then compute the MR estimate for the effect of the exposure trait on the outcome trait, 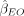 . This estimate may be a conventional IVW estimate (Burgess et al. 2013) or various statistics designed to be robust to pleiotropy such as the Egger-based statistic (Bowden et al. 2015), the weighted median statistic (Bowden et al. 2016), or others. We then predict the outcome trait for each individual n as
. This estimate may be a conventional IVW estimate (Burgess et al. 2013) or various statistics designed to be robust to pleiotropy such as the Egger-based statistic (Bowden et al. 2015), the weighted median statistic (Bowden et al. 2016), or others. We then predict the outcome trait for each individual n as 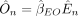 . Finally, we compute the negative squared error of these predictions
. Finally, we compute the negative squared error of these predictions 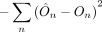 , summed across all individuals. The full statistic is then
, summed across all individuals. The full statistic is then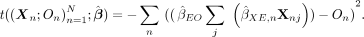 (15)
(15)
Generating digital twins
We have assumed that trio data are available thus far for simplicity. However, the MR-Twin framework can also be used when parent–child duo data or sibling data are available. Here we discuss the algorithms used to generate digital twins given trio, parent–child duo, or sibling data.
Trio and duo modes
We assume that the SNPs used in the MR instrument are independent, a common assumption when multi-SNP instruments are used
in MR (Burgess et al. 2013). Therefore, we separately sample the genotype of each SNP of the digital twin given the parent and/or offspring genotypes
at that SNP. Let (Dn) be the (N × M) matrix of digital twin genotypes we will sample, corresponding to the true “offspring” genotypes in (Xn). Further, let n index some family and j index some SNP, such that P1nj (e.g.) is the genotype for one parent in family n at SNP j. If we have both parents available, sampling Dnj is straightforward. Because the SNPs are considered independent, we do not need to know the parental haplotypes. If a parental
genotype P1nj is zero or two, respectively, then a zero or one, respectively, is inherited by Dnj. If the parent genotype is one, then either zero or one is inherited with 50% probability each. Dnj inherits alleles from the two parents independently. This can be summarized as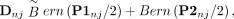 (16)
where Bern stands for the Bernoulli distribution, for each family n and SNP j.
(16)
where Bern stands for the Bernoulli distribution, for each family n and SNP j.
If we only have one parent genotype available, then following the method of Bates et al. (2020), we fix the offspring's haplotype from the unobserved parent and only simulate a random draw from the observed parent's haplotype.
If the observed parent is homozygous, then the allele inherited from that parent is fixed as well, so Dnj = Xnj. Otherwise, the allele inherited from this parent will be Bern(0.5). In principle, 0.5 could be replaced with some value based on population allele frequencies. Similar to that above,
the model for the allele from the other parent can be written as Bern(Xnj/2). Thus, if the parent is a heterozygote, we have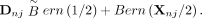 (17)
(17)
Sibling mode
In the case in which we observe sibling genotypes but not the genotypes of their parents, we assessed two potential approaches. In either case, the observed sibling information is used to infer the probabilities of digital twin genotypes based on the fact that the sibling genotypes give information about the probabilities of various parental genotypes. For instance, a child with a two genotype at an SNP guarantees that neither parent has a zero genotype at that SNP and makes it more likely that the parents have two genotypes than one genotypes. Most simply, if one sibling has a two genotype at an SNP and the other sibling has a zero, then the parents must both be heterozygotes. In all other cases, approximation is needed.
The first approach is straightforward and involves randomly drawing two haplotypes from the observed sibling haplotypes to generate a digital twin. This shuffling approach gives a rough approximation of the likelihood of digital twin genotypes given the information the observed siblings provide. The second approach, described in the Supplemental Materials, involves using the sibling data to infer a distribution over the possible parents and then performing a weighted random draw of digital siblings based on those parents. In practice, we found that the shuffling approach was faster and yielded lower FPR than the probabilistic approach while achieving similar power, so we used the shuffling approach for the results in this paper.
Software availability
The code implementing the MR-Twin package can be found at GitHub (https://github.com/nlapier2/MR-Twin) and as Supplemental Code. Scripts and instructions for repeating the experiments in this paper can be found at GitHub (https://github.com/nlapier2/MRTwin-replication) and as Supplemental Code. Please note that UK Biobank genotypes are not publicly released, so those wishing to replicate the experiments will first have to get access to that data via the UK Biobank (https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This research has been conducted using the UK Biobank Resource under application number 33127. B.F. and S.S. were supported in part by National Institutes of Health (NIH) R35GM125055 and National Science Foundation (NSF) CAREER-1943497, III-2106908. E.E. and N.L. are funded by NSF award 2106908 and NIH awards U01HG011715 and R56HG010812. We thank Matthew J. Tudball for productive discussions on potential future work and our respective efforts to develop family-based MR methods.
Author contributions: S.S. and E.E. conceived of and supervised the project. N.L., B.F., and S.S. developed the methods and wrote the manuscript. N.L., B.F., and S.T. wrote the software code and performed the analyses. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277664.123.
- Received January 5, 2023.
- Accepted April 16, 2023.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








