
Abstract
A growing number of recent genomic studies report asexual parthenogenetic reproduction in a wide range of taxa, including vertebrate species from the reptile, bird, and fish lineages. Yet, self-fertilization (selfing) has been recorded only in a single vertebrate, the mangrove killifish Kryptolebias marmoratus. In cichlid fishes, sex determination is notably diverse and can be influenced by the environment, and sequential hermaphroditism has been reported for some species. Here, we present evidence for a case of facultative selfing in the cichlid fish Benitochromis nigrodorsalis, which is otherwise known as biparentally reproducing ovophilic mouthbrooder from Western Africa. Our laboratory observations revealed that a wild-caught individual produced repeatedly viable offspring in absence of a mating partner. By analyzing genome-wide single-nucleotide polymorphism (SNP) data, we compare that individual and two of its offspring to shed light on its reproductive mode. First, our results confirm uniparental reproduction. Second, overall heterozygosity is reduced in the offspring compared with outbred individuals. Retained maternal heterozygosity in the offspring is ∼51%, which is close to the theoretically expected value of a heterozygosity reduction of 50% by selfing. Heterozygosity patterns along individual chromosomes do not point to alternative parthenogenetic reproductive mechanisms like automixis by terminal or central fusion. Facultative selfing may represent an adaptive strategy ensuring reproduction when mating partners are absent and, hence, contribute to the cichlids’ enormous evolutionary success.
Although vertebrates reproduce predominantly sexually (Neaves and Baumann 2011), recent studies report evidence for asexual parthenogenetic reproduction in a number of reptiles, birds, and fishes (Chapman et al. 2007; Booth et al. 2012; Fields et al. 2015; Warren et al. 2018; Card et al. 2021; Ryder et al. 2021), suggesting that vertebrates’ modes of reproduction are more variable than commonly assumed. Still, simultaneous hermaphroditism (i.e., individuals produce male and female gametes at the same time) and self-fertilization (selfing) seem to be almost absent in vertebrates. This is interesting considering that sequential hermaphroditism (changing sex during lifetime) is common, for example, in fishes (Warner 1975). The only recorded self-fertilizing hermaphroditic vertebrate is the mangrove rivulus (Kryptolebias marmoratus) (Harrington 1961; Tatarenkov et al. 2009; Kelley et al. 2016), which can, however, also outcross.
In sexual reproduction, haploid gametes are produced via meiosis. The fusion of female and male gametes, that is, of egg and sperm, results in diploid offspring, which carry new allelic mixtures through segregation and recombination of the parental genetic information (Engelstädter 2017). Selfing is the fusion of meiotic products resulting from independent meiosis events of the same individual, occurring in oogenesis and spermatogenesis, respectively. As an extreme form of inbreeding, selfing reduces the degree of heterozygosity by 50% each generation.
Contrastingly, asexual parthenogenetic reproduction relies on the development of unfertilized gametes. Parthenogenesis includes clonal reproduction, generating offspring that are genetically identical to their mother. Offspring identical to the parent can result from functionally mitotic parthenogenesis and from meiotic parthenogenesis (Fig. 1). However, meiotic parthenogenesis can also lead to offspring that are genetically distinct from their parent (Fig. 1). Such automictic systems rely on a modified meiosis that leads to diploid eggs without the need of fertilization by a mating partner. The meiotic modifications guarantee the maintenance of the ploidy level under automictic reproduction. In automictic parthenogenesis with terminal fusion, the second polar body fuses with the egg nucleus by the end of meiosis II to form the zygote. Because of the assortment of sister chromatids, this leads to offspring being homozygous (Lampert 2008). Offspring production by an aborted meiosis II and gamete duplication has the same genetic consequences (see Fig. 1; Asher 1970). In automixis with central fusion, products of meiosis I fuse and give rise to offspring genomes, which retain all of the maternal heterozygosity, owing to the assortment of nonsister chromatids. The same pattern results from offspring generated by an aborted meiosis I (Asher 1970; see Fig. 1 in the work of Smith et al. 2019). Although terminal fusion seems to be the predominant automictic reproductive mode in parthenogenetic vertebrates, such as recently shown for the king cobra (Card et al. 2021), central fusion is more widespread among invertebrate species, such as in the crustacean Artemia parthenogenetica (Nougué et al. 2015).
Overview of sexual and asexual reproduction modes. Sexual reproduction includes the fusion of male and female gametes, which can originate from two different individuals (biparental reproduction) or from the same individual (selfing). Asexual reproduction includes mitotic and meiotic parthenogenesis. Consequences for offspring heterozygosity are presented for each mode (for further explanation, see text).
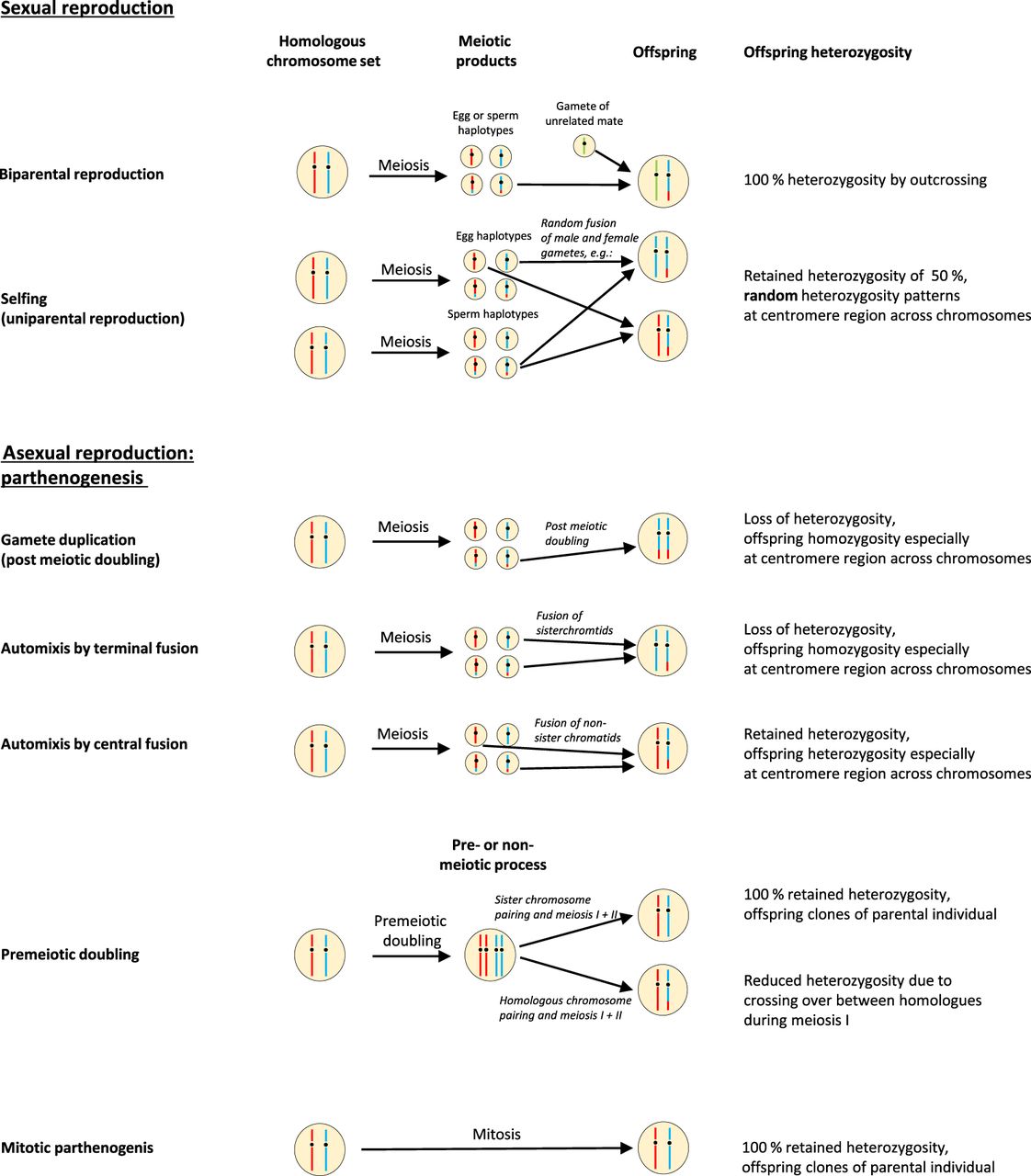
For both automixis based on central fusion and on terminal fusion, recombination may affect chromosomal heterozygosity patterns in the offspring. As a consequence, the actual heterozygosity found in the offspring may be higher (in case of terminal fusion) or lower (in case of central fusion) than expected in the absence of recombination. Recombination rates have been suggested to correlate with chromosome positions (Sardell and Kirkpatrick 2020) with higher rates at the telomere (Haenel et al. 2018) and crossovers being suppressed at the centromere (Talbert and Henikoff 2010; Sardell et al. 2018). If recombination events are more likely at telomeric regions (Haenel et al. 2018), alleles become randomly distributed among sister and nonsister chromatids at chromosomal peripheries. Resultantly, here maternal alleles are sampled randomly without replacement, and thus, after one of the meiotic products is chosen first, two out of the three remaining products (i.e., 67%) contain the alternate allele. Consequently, analyzing the heterozygosity distribution along chromosomes can disentangle the two forms of automixis (i.e., central fusion and terminal fusion) and potentially differentiate between automixis by terminal fusion and selfing. Self-fertilization, as well as automixis by terminal fusion, reduces offspring heterozygosity. Yet, under self-fertilization, patterns of retained maternal heterozygosity are independent from the chromosomal position (equivalent to sampling with replacement because taken from independent meiotic events). Accordingly, the heterozygotic state of the centromere can be used to distinguish terminal fusion, central fusion, and selfing: Under the two forms of automixis, all centromeric regions within an individual should have the same state (i.e., under terminal fusion they have all lost heterozygosity, under central fusion they have all retained heterozygosity), whereas under selfing and hence random chromosome pairing, 50% of all centromeres are expected to lose heterozygosity (Svendsen et al. 2015).
Another meiotic parthenogenesis reproduction mode is based on premeiotic doubling of the chromosomes (Fig. 1). Bivalent formation of sister chromosomes and subsequent meiosis I and II lead to full retention of heterozygosity in the offspring. Bivalent formation occurring between nonsister chromosomes leads to loss of heterozygosity in 50% of the progeny with no relation to centromere position (see Fig. 3 in the work of Archetti 2010); so far, this reproductive mode lacks evidence from nature. Recent genome-wide studies of facultative and obligate automictic parthenogenetic species disentangled the underlying meiotic processes and provided insight into the genome's trajectory under the particular type of reproduction (Booth et al. 2012; Card et al. 2021).
We here set out to elucidate possible facultative selfing in the West African cichlid fish Benitochromis nigrodorsalis, which has been described as bisexually (biparentally) reproducing ovophilic mouthbrooder (Lamboj 2004). B. nigrodorsalis is found in the coastal rivers and creeks of Cameroon and Equatorial Guinea. Here, we report the laboratory observation that an isolated wild-caught individual repeatedly produced viable offspring, which can be explained either by parthenogenesis or by self-fertilization. Because classic microsatellite analyses are insufficient to differentiate between the different forms of automictic parthenogenesis and selfing (Svendsen et al. 2015), we applied a whole-genome sequencing approach to compare the parent and two offspring at the full genomic level. We also integrated data from a presumed unrelated individual caught in the wild from the same source population at the same time as the parental individual and kept under the same conditions.
Results
One wild-caught individual of the species B. nigrodorsalis repeatedly produced offspring in absence of a mating partner. To examine the underlying mode of reproduction, we compared the genomes of the parent and two of its offspring and another wild-caught individual (referred to as wildI in the following).
A first inspection of the raw sequencing data via k-mer-spectra analysis revealed a similar genome size across all four sequenced individuals (mean size across k-mer sizes 20, 30, and 40: 800–817 Mb) and pointed toward lower estimates of genome-wide heterozygosity in both offspring (mean heterozygosity across k-mer sizes 20, 30, and 40; wildI: 0.097%, parent: 0.119%, offspring1: 0.084%, offspring2: 0.079%; note that sequencing coverage used here is lower than recommended by the applied k-mer method, and hence, the estimates might be only rough approximations) (Supplemental Table S1; Supplemental Figure S1). Accordingly, genome-wide heterozygosity estimates based on all accessible sites after mapping the reads to the reference genome of the Nile tilapia (Oreochromis niloticus) and variant identification were higher for the two wild-caught fish (parent: 0.0634%, wildI: 0.0631%) than those of the offspring (offspring1: 0.0425%, offspring2: 0.0423%) (Supplemental Table S2), Overall, the genome-wide estimates of heterozygosity resembled those of other wild-caught cichlid fish (Ronco et al. 2021), albeit being at the lower edge of so-far-observed values.
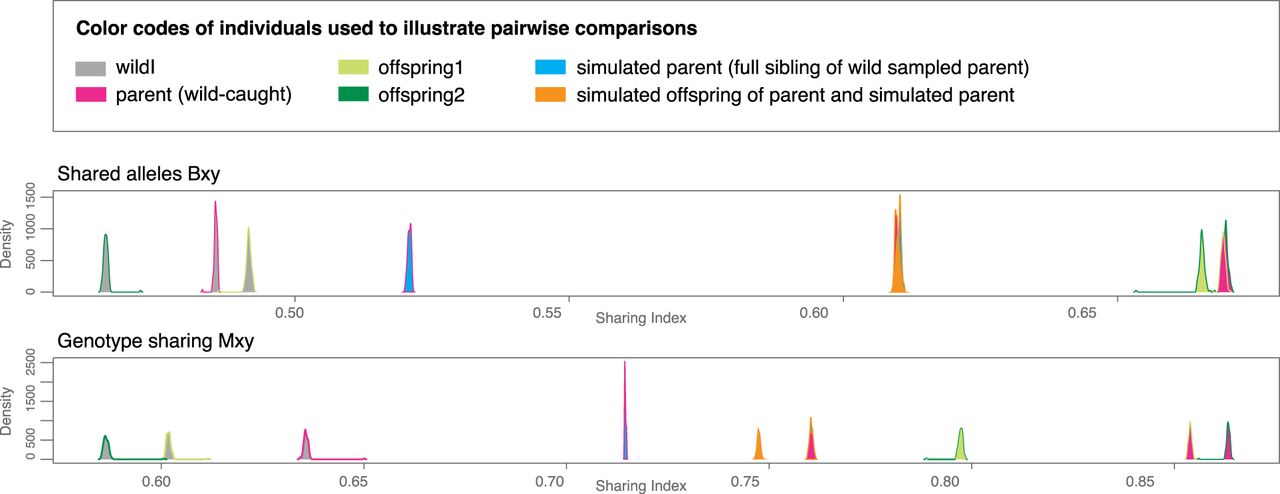
To shed light on the potential uniparental origin of the investigated offspring, we next analyzed allele and genotype sharing among our focal individuals similar to other studies that showed uniparental offspring in vertebrates (e.g., Card et al. 2021). Indeed, supporting their uniparental origin, both offspring showed a higher proportion of allele and genotype sharing with their parent even compared with simulated full-sib offspring (generated as offspring from simulated siblings to the parent individual and the parent individual), that is, closest relatives possible as resulting from brother–sister mating (Fig. 2).
Allele and genotype sharing. Bootstrap densities of observed pairwise indices of shared alleles (top) and genotypes (bottom) based on 100 bootstrap replicates are shown for the experimentally accessed individuals (wildI, parent, offspring1, and offspring2) together with the same indices calculated for 10 simulated full-siblings to the parent individual and 10 simulated offspring derived from crossing each simulated parent and the parent fish.
We next set out to investigate genotype distrubutions along the chromosomes and across the offspring (based on biallelic variants placed on chromosomes; see Methods), revealing that for offspring1 and offspring2 71.48% and 71.73%, respectively, of the sites were identical to the parent, whereas offspring1 had 3.8% sites and offspring2 had 3.4% sites with one nonmatching allele to the parental genotype and offspring1 had 0.82% sites (2779) and offspring2 had 0.83% sites (2816) with genotypes completely incompatible with the parental genotype. We suspected, in particular for the latter, that those sites were the result of genotyping error. An inspection of quality metrics indeed revealed significantly lower quality in the offspring (Supplemental Fig. S2A) as well as significantly lower depth and genotype quality in the parent for sites that would disagree with uniparental origin (Supplemental Fig. S2B). We excluded from further analyses any sites from the data set that had both alleles not matching the parental genotype.
We next inspected genotype distributions along chromosomes at the remaining sites in all four individuals (Supplemental Fig. S3). Clearly, although the overall heterozygosity estimates of the two offspring were very similar (see above), also supported by the genotype indices (Fig. 2), the offspring differed in genotypes on a per site basis and subsequently also in their distribution of heterozygous sites along the genome. However, this genotype visualization revealed a need for further in depth inspection of genotype quality. Largely, the patterns of retained and lost heterozygosity appeared block-like, as would be expected, with a small number of recombination events (for cichlids, we roughly anticipate one to four recombination events per chromosome) (Palaiokostas et al. 2013; Feulner et al. 2018). However, individual interspersed sites did not show the expected genotype given their neighboring single-nucleotide polymorphisms (SNPs) (i.e., isolated homozygous sites in a block of heterozygous sites and vice versa). This would require an unlikely mutation or recombination rate, and we thus suspected wrong genotypes among the individuals. Our individual genotype quality filter at GQ20 corresponds to a 1% error rate. Our individual depth filter was set to a minimum of three reads with a median depth of 16–17 reads per individual (Supplemental Fig. S4).
Based on this depth distribution and previous recommendations (the data set of Carson et al. [2014] favored a DP of eight), we assessed the impact of different depth filters on patterns and values of retained heterozygosity. We did so under the assumption that variants with unusually high coverage might represent wrong variants based on faulty read assignment from paralogs or repetitive genome regions owing to the usage of a reference genome from another species or simply repetitive regions. Variants with unusually low coverage, especially in the parent, on the other hand, might represent false homozygous calls with too low sequence support for the nonrepresented allele. Indeed, on average, heterozygous sites had significantly higher coverage than homozygous sites (Supplemental Figs. S5, S6).
More narrow depth filters (Supplemental Figs. S7–S17) made patterns of heterozygosity more block-like as would be expected with a limited number of recombination events per chromosome. Imposing upper limits on the depth reduced the total amount of heterozygous sites in the data set, but imposing only lower limits removed homozygous sites (Supplemental Table S3). The filters affected both offspring very similarly. Balancing the signal-to-noise level, we continued the subsequent analyses with an additional filter of DP > 8 < 20.
Theoretically, the modifications of meiosis underlying parthenogenetic reproduction can be detected when analyzing the resulting offspring's genotypes. Parthenogens resulting from central fusion tend to fully retain maternal heterozygosity (assortment of nonsister chromatids), whereas offspring based on terminal fusion obtain close to fully homozygous genotypes (assortment of sister chromatids). Given that recombination is more likely to happen in regions more distant to the centromere, these patterns are expected to be especially valid in vicinity to the centromeres.
Upon investigating sites that are genotyped heterozygous in the parent (Fig. 3; Supplemental Fig. S18), we did not see a close-to-complete loss of heterozygosity in the offspring, which clearly excludes automixis with terminal fusion as mode of reproduction; retained heterozygosity was 51.4% for offspring1 and 50.3% for offspring2, indicating a substantial loss of heterozygosity rather than close-to-complete retention.
Retained heterozygosity profiles. Panels show per chromosome individual genotypes at variant sites at which the parental individual was heterozygous. Each bar represents a variant site, and colors of the bars indicate genotypes as depicted in the inset.
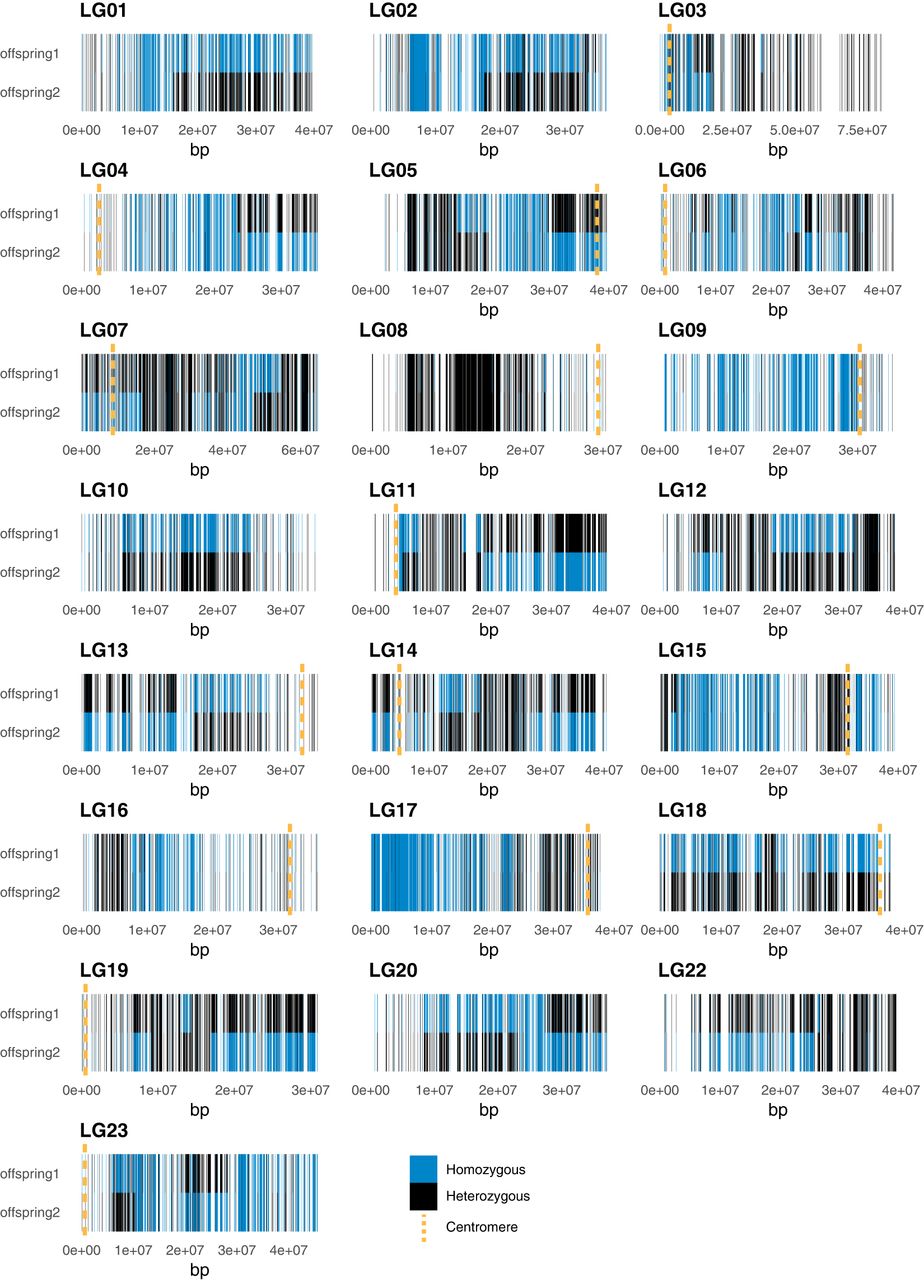
The inspection of sites that are heterozygous in the parent further revealed different patterns of recombination and (haplotype) inheritance between the offspring and among chromosomes (Fig. 3; Supplemental Fig. S18): There are chromosomes that either (1) are mostly homozygous (LG01, LG2, LG09, LG10, LG15, LG16, LG18 in offspring1; LG04, LG06, LG09, LG16 in offspring2), (2) have mostly retained heterozygosity (LG03, LG08, LG19, LG22 in offspring1; LG08, LG10, and LG18 in offspring2), or (3) display one or more recombinant patterns compared with the parental genotype (remaining LGs). Recombination events did not seem to be limited to telomeric regions only and differed among the offspring.
Most of the 16 centromeric regions we could infer were located toward the ends of the chromosome (Fig. 3), matching the expectations of many acrocentric chromosomes in the reference genome (Ferreira et al. 2010). Inferred centromeric regions differed in the heterozygous state among the offspring and were both heterozygous and homozygous, depending on the chromosome (Supplemental Table S4; Fig. 3). We observed more heterozygous than homozygous centromeres. Note that because of their location and probably because of their sequence content, centromeric regions were poorly covered with SNP information in several cases.
Discussion
Here, we report that a separately housed individual of the cichlid fish B. nigrodorsalis repeatedly produced viable offspring, indicating uniparental reproduction. Facultative parthenogenetic reproduction has been reported in several vertebrate species (Fields et al. 2015). We compared the genomic data from the parent individual and two of its offspring individuals to elucidate the underlying mode of reproduction. Because our whole-genome-based analyses clearly indicated that the offspring were not genetically identical to the parent, we could exclude any clonal forms of parthenogenesis. The genomic comparison of the parental individual and the two offspring individuals revealed an important reduction in genome-wide heterozygosity of variant sites from the parent (62% heterozygous sites out of all variant sites) to the two offspring (41%). Retained maternal heterozygosity in the two offspring was ∼51% in both offspring, which, however, should be taken with some caution because we show that this can be impacted by SNP read depth filtering. Heterozygosity reduction is expected to result from specific modes of automictic parthenogenesis as well as from selfing. In theory, heterozygosity patterns along the chromosomes can be used to differentiate between different modes of automixis (Svendsen et al. 2015) as follows: Automixis by terminal fusion would lead to a close to fully homozygous genotype with decreasing homozygosity with increasing distance to the centromere. In contrast, automixis with central fusion would lead to fully retained heterozygosity, especially at the centromeres, and to increasing homozygosity with increasing distance to the centromere. We did not find clear evidence for heterozygosity patterns supporting terminal fusion in the two offspring individuals or for supporting central fusion given the important reduction of heterozygosity we observed and the varying state of heterozygosity of the inferred centromeres. Likewise, the levels of retained heterozygosity contradict premeiotic doubling with exclusive sister chromosome pairing, which would result in 100% heterozygosity retention as shown, for instance, in parthenogenetic lizards (Lutes et al. 2010). Premeiotic doubling with (exclusive) homolog (i.e., nonsister) chromosome pairing has to the best of our knowledge not been reported. Under random sister and nonsister chromosome pairing, we would expect that half of the chromosomes retain full parental heterozygosity, a pattern we did not observe. We think that the amount and distribution of heterozygosity loss and the genotypic state of centromeres we observed are best explained by random pairing of sister and nonsister chromatids from independent meiotic events and with recombination, as would be the case under selfing.
Our results further show that the absolute levels of heterozygosity are impacted by data filtering such that certain filter conditions (especially on lower depth) would lead to an overrepresentation of heterozygous sites in the data set, also affecting rates of retained heterozygosity. Hence, the rates in our study, and probably other studies based on sequencing data only, should be taken with a grain of caution. Under most filter settings, the overall observed decline in heterozygosity was lower than expected by selfing. Similar results were reported in maize (Roessler et al. 2019) and in eucalyptus (Hedrick et al. 2016).
Until recently, investigations of reproductive modes were limited to marker sets (e.g., microsatellites) or reduced representation sequencing lacking the level of resolution whole-genome sequencing can provide. Although we clearly show that such data can be used to support the uniparental origin of offspring and identify recombined chromosomes, further sequencing at a larger depth and of more offspring would substantiate our drawn conclusions. Even under stringent filtering conditions, we observed impossible genotype combinations of the parent and the offspring (sites not matching the allelic state of neighboring sites). This might be explained by varying sequence coverage of heterozygous and homozygous sites; all individuals had on average higher sequence coverage at heterozygous sites. We further assume that this is in part driven by sequence information derived from repetitive regions. Because of our aim to analyze genotypes on the chromosomal level, we relied on a good-quality reference genome and hence had to refer to the genome of another species. Although on the large scale, different cichlid species have comparable karyotypes (Ozouf-Costaz et al. 2017); smaller-scale rearrangements are frequent (Conte et al. 2019). This could lead to further false read assignment by wrong paralog mapping owing to using a reference genome from another species and hence inducing noise in genotype assignment.
The used reference genome has a karyotype of 2n = 44, whereas the known karyotypes for Chromidotilapiini (that B. nigrodorsalis is part of) consist of 2n = 48 acrocentric chromosomes, with one typically very large chromosome pair, as has the reference (Post 1965; Ozouf-Costaz et al. 2017). Our centromere inferences would support acrocentric chromosomes for the majority of the Nile tilapia chromosomes, matching its karyotype data. We assume that, at least to some extent, centromere locations are conserved between our focal species and the reference species, but note that this needs further investigation. Also, centromere locations in the reference were inferred by a similarity search with a centromeric repeat region, which was not conclusive for six of the chromosomes. Hence, at this stage, we cannot provide a fully conclusive picture of the heterozygosity state of the centromeric regions. In general, in the focal species, this will also be hindered by the fact that the centromeres with their position toward the ends of chromosomes are often located in data-deficient regions. Keeping this in mind, we observed loss and retention of heterozygosity at centromeric regions, which is incompatible with central and terminal fusion automixis. Also, incompatible with these two fusion modes is our observation of retained heterozygosity (and, of course, complementary loss of heterozygosity), considering both offspring jointly can cover most of the chromosome; offspring differ in their locations of retained heterozygosity and do not have the same pattern of heterozygosity state at centromeric regions.
Furthermore, we observed that recombination events in the offspring were not limited to telomeric regions. Because, in general, recombination is more likely in regions distal to the centromeres (Sardell et al. 2018), this pattern is in agreement with a predominantly acrocentric chromosome type.
In a documented spawning event in our laboratory, only half of a dozen offspring reached the juvenile stage, indicating a high mortality rate or very low fertilization success (provided that this species under regular sexual reproduction usually spawns at least 70 eggs), which could point to inbreeding depression. Especially, a high mortality rate could result from even a few recessive lethals, that is, alleles that when present in homozygous status cause lethality. Persistent selfing/inbreeding can reduce genetic load and inbreeding depression at the population level (purging effect) and has been shown in both experimental laboratory studies (Pekkala et al. 2012) and wild populations (Xue et al. 2015; Robinson et al. 2018; Grossen et al. 2020). Although the effectiveness of purging seems to differ between and within species, the consensus is that obligate selfing is selected against in the long term (Glémin et al. 2019). Still, facultative selfing may contribute to purging deleterious alleles at the population level and allows reproduction in the absence of a mating partner. The natural habitat of the here-studied fish in the Moliwe River is highly structured and characterized by rapids and small and bigger waterfalls and pools (Langen et al. 2011). It is not unlikely that individuals get separated from conspecifics, for example, after heavy rainfalls. Population genetic examinations revealed that the population of the sympatrically occurring cichlid Pelvicachromis taeniatus was structured accordingly (Langen et al. 2011). Alternatively, explorative individuals may migrate to new habitats alone. Selfing following long-term separation from conspecifics may lead to extreme founder effects under natural conditions, and if mating among resulting offspring is maintained across generations, this could lead to stable founder subpopulations potentially driving population divergence and diversification. In contrast to clonal reproduction, selfing allows recombination with the associated selective benefits, which may facilitate the establishment of stable populations. The before-mentioned P. taeniatus Moliwe population is highly inbred (Langen et al. 2011), and laboratory experiments revealed active kin-mating preferences in this species, indicating that inbreeding is tolerated and even might be beneficial under certain conditions (Thünken et al. 2007). To compare the performance of offspring resulting from selfing and outcrossing would be mandatory to estimate the costs of selfing in the specific B. nigrodorsalis system.
Both wild-caught individuals showed levels of heterozygosity in the expected range for cichlid fish (Ronco et al. 2021), suggesting that biparental sexual reproduction is the regular mode of reproduction in this species. An alternative explanation would be cross-mating between different offspring groups with each originating from independent lines of selfing, that is, each with loss of heterozygosity but potentially homozygous for different alleles.
In vertebrates, selfing has so far only been reported in the mangrove killifish K. marmoratus, in which males are rare and in which most individuals are hermaphrodites reproducing by self-fertilization (Kelley et al. 2016); fertilization is internal and ovotestes simultaneously produce eggs and sperm. In cichlids, sex determination is highly flexible, and sex change has been reported in several species (Oldfield 2005). In this context, it is interesting that a laboratory-bred F1-hybrid cross of two closely related cichlid fish species within the Haplochromis genus developed into a functional hermaphrodite that produced offspring most likely by selfing (Svensson et al. 2016). Whether B. nigrodorsalis can change its sex is unknown. Because the time interval between the different reproduction events reported in the present study were quite long (>1 yr), it is rather unlikely that we observed reproduction at a transformational stage when both female and male gametes are present. Rather, social stress caused by changes in the social environment (i.e., social isolation) may cause changes or disturbance in the hormonal balance. Cortisol is a hormone whose level is highly sensitive to stress and has been suggested to alter steroidogenesis (Goikoetxea et al. 2017), which may play a role in the development of hermaphroditic individuals.
In conclusion, we here provide clear evidence that at least one individual cichlid fish from the species B. nigrodorsalis is capable of reproducing uniparental and most likely via selfing. Future research should aim to examine the environmental and social conditions that trigger this reproductive mode and the underlying proximate mechanisms mediating the hermaphroditic development. Ultimately, it would be interesting to screen the natural population for the presence of individuals that originate from selfing to realistically estimate the potential of selfing as strategy driving the evolution of animal mating systems.
Methods
Ethics statement
The present study follows Animal Behavior Society guidelines for the use of animals in research and was conducted in accordance with German laws for animal experiments. Experiments were approved by the regional office for nature, environment, and consumer protection in North-Rhine Westphalia (LANUV NRW, reference nos. 84e02.04.2015.A580 and 81-02.04.2021.A199).
Sample collection and sequence data generation
Juvenile B. nigrodorsalis individuals were derived from a field work trip at Moliwe River (Southwest Cameroon) in 2006 as described by Langen et al. (2011). Individuals were kept at the Institute of Evolutionary Biology and Ecology at the University of Bonn and used for behavioral experiments (Meuthen et al. 2016). Fish were first kept as a group. Because of high levels of inter-individual aggression, individuals were kept in separate tanks starting from about 2007. Visual contact to the conspecifics was ensured. Tanks (50 × 35 × 30 cm, length × width × height) were equipped with sand, Java moss, and a filter (gully filter, Hobby). Tanks were covered with glass plates. Fish were fed daily in excess with defrosted adult Artemia and mosquito larvae. Tanks were illuminated by fluorescent tubes (True-Light T8 natural daylight 5500 K). The water temperature was kept at 23 ± 2°C, and the light–dark cycle was set to 12-h:12-h light–dark. One-third of the water was exchanged every 2 wk. Because of the monomorphic appearance, sexes of individuals could not be distinguished. One individual repeatedly produced offspring after being kept in isolation for ∼4 yr to ∼10 yr. The first breeding attempt failed, and the small number of offspring died at an early juvenile stage. Because we could not rule out—although highly unlikely—that the sperm of fish from nearby tanks ended up in that individual's tank by accident during the regular water change or tank cleaning, from that time point particular care was exercised when cleaning that tank, and the individual was monitored on a regular basis. From the second clutch, six offspring individuals survived, which were subsequently moved to another tank and kept in a group under the same conditions as the parent. A further clutch was removed already at the egg stage (at least 73 eggs were spawned) because we wanted to rear the eggs artificially. This approach, however, failed although ex situ raising is possible in other mouth brooding cichlids (see, e.g., Heule et al. 2014).
For the experiment conducted here, tissue samples were obtained from the parent individual, two of its offspring (sex unknown, juvenile samples), and another adult individual derived from the same original field collection event. Tissue samples were taken from dead individuals and stored in 95% ethanol. DNA was extracted with a Qiagen DNeasy blood and tissue kit following the manufacturer's instructions. DNA concentration and integrity were assessed with a fluorometer (Quantus, Promega) and a fragment analyzer (Agilent).
Four individual TruSeq DNA PCR-free libraries for whole-genome Illumina sequencing were constructed by Macrogen and sequenced on an Illumina HiSeq 2000 in paired-end mode, generating a total read number of 103,465,584 for the parent, 105,717,920 for offspring individual one (offspring1), 102,902,068 for offspring individual two (offspring2), and 103,237,228 for the wild individual (wildI). Adapter sequences were removed from FASTQ files with Trimmomatic v0.39 (Bolger et al. 2014). Cleaned FASTQ files were assessed with Genomescope2 (v2) (Ranallo-Benavidez et al. 2020) based on individual k-mer catalogs generated with FastK (https://github.com/thegenemyers/FASTK, vApril 18 2021) and k-mer sizes 20, 30, and 40.
Trimmed files were mapped with BWA-MEM v2.1 (Vasimuddin et al. 2019) against the best-assembled cichlid reference genome available at the time of this study, the Nile tilapia O. niloticus (assembly version GCF_001858045.2), which was previously determined as a suitable reference genome for analyzing cichlid whole-genome sequencing Illumina data, including the genus Benitochromis (Matschiner et al. 2020).
Variant identification
The resulting alignments (average read mapping depth: 10.2819 parent, 10.6514 offspring1, 10.1311 offspring2, and 10.2599 wildI) were converted to BAM format with SAMtools v1.8 (Li et al. 2009) and duplicates marked with Picard tool's v2.22.0 MarkDuplicates (http://broadinstitute.github.io/picard/). After this step and before variant calling, we assessed the final mapping rate with the flagstat command of SAMtools v1.8, which was >90% for all samples and in the range of previously reported mapping rates for cichlid genomes used in population genomic analyses (individual mapping rates; wildI: 91.46%, parent: 91.43, offspring1: 91.96%, offspring2: 91.59%), delivering a reliable input data set for the subsequent analyses.
SNPs were identified with the Genome Analysis Toolkit (GATK) v4.1.4.1 suite (McKenna et al. 2010) by running HaplotypeCaller per individual followed by merging per chromosome with CombineGVCFs and subsequently genotyping with GenotypeGVCFs. Resulting files were merged with GatherVCFs into a single variant call format (VCF). GenotypeGVCF was run in two modes: first in standard mode to derive only variant sites (in the following referred to as OnlyVariants file) and second with using the “‐‐include-nonvariant-sites” flag to derive callable sites (in the following referred to as AllSites file).
Next, we removed indels with GATK's SelectVariants from both files. Resulting files were quality inspected based on the outputs of GATK's VariantsToTable and Vctools v0.1.16 stats in RStudio v1.3.1093 with R v4.0.3 (R core team 2020). Subsequently, both files were filtered for site-wise quality with GATK's VariantFiltration and SelectVariants, applying the following hard-quality filters and removing sites outside the following ranges: mapping quality (MQ) < 40.0, strand bias (FS) > 40.0, quality by depth (QD) < 2.0, depth (DP) < 20, DP > 200, strand odds ratio (SOR) > 9.0, mapping quality rank sum (MQRankSum) < −12.5, and read position rank sum (ReadPosRankSum) < −8.0. We further masked individual genotypes with a read depth below three or a genotype quality below 20 with BCFtools v1.10.2 (http://samtools.github.io/bcftools/bcftools.html) and only retained sites with no missing genotypes with VCFtools. We determined all sites within regions of the used reference genome in which read mapping could be ambiguous based on the output of SNPable (http://lh3lh3.users.sourceforge.net/snpable.shtml) by dividing the reference genome into overlapping k-mers of 100 nt and aligning them back to the reference genome with BWA-ALN, BWA v0.7.17 (Li and Durbin 2010). We subsequently excluded from both VariantsOnly and AllSites files all sites from regions in which fewer than 90 out of 100 fragments mapped back correctly with VCFtools.
This resulted in a total of 455,197,170 accessible sites (final AllSites file), of which 454,586,028 were located on chromosomes (final AllSites file after removal of unlocated contigs). We calculated the overall percentage of heterozygous sites out of all accessible sites and out of all variable sites (VariantsOnly) with BCFtools stats: [heterozygous SNP sites/(heterozygous SNP sites + homozygous reference sites + homozygous alternative sites)].
Heterozygosity and relatedness estimates
From the variants only file, we further removed multiallelic SNPs with VCFtools, as well as those that contained spanning deletions as alternative alleles and all invariant sites across samples, that is, removing all sites that were homozygous alternative with respect to the reference genome in all samples with BCFtools. This resulted in a total of 368,453 SNPs (final VariantsOnly file).
We focused on biallelic sites and excluded all SNPs on unplaced scaffolds, as well as on the mitochondrial scaffolds, in the following analyses: (1) genotype inspection of sites with genotype incompatible with parental genotype, (2) indices of shared alleles and genotypes including offspring simulations, and (3) patterns of genotype and heterozygous sites. For these analyses, we further derived from the AllSites file the actual number of accessed sites per nonoverlapping 10-kb window along each chromosome with the aim to exclude regions with low information. We only kept SNPs in windows, which contained at least more sites than the third quartile number of sites over all windows (i.e., we only kept windows with at least 1677 accessible sites).
We calculated indices of shared alleles (Bxy) (Li and Horvitz 1953) and genotypes (Mxy) (Blouin et al. 1996) with the R packages Demerelate v0.9.3 (Kraemer and Gerlach 2017) and Rhh v1.0.2 (Alho et al. 2010) as previously described (scripts modified from Card et al. [2021]; for R scripts, see Supplemental Data file GenericCode.zip) on the VariantsOnlyMulti file, including 100 bootstrap replicates.
Simulation study to compare observed patterns of allele and genotype sharing to crossing with full sibling
Full siblings (i.e., brothers and sisters) share, on average, the same alleles at ∼50% of all their loci. Under this assumption, we simulated a full sibling to the parent fish such that, based on the filtered VariantFile after removing sites placed in low-quality windows, that sibling was at a random selection of 50% of all sites identical to the parent fish and at the other 50% of sites either 0|0, 0|1, or 1|1 at the same proportions as the parent fish (i.e., homozygous reference 0|0 20.4%, homozygous alternative 1|1 19.6%, heterozygous 0|1 60%) but not identical to the parent. We next simulated 10 offspring from that sibling and the parent individual by randomly selecting one allele of each parent at every site. We repeated this operation 10 times to obtain a total of 100 simulated F1 offspring. We calculated for these offspring Mxy and Bxy as described above.
Retained heterozygosity
In preparation for the analysis of patterns of retained heterozygosity, we first accessed genotypes of all sites at which the offspring individuals had two alleles incompatible with the parental genotype (e.g., parent 1/1 and offspring 0/0), as well as all sites with one nonmatching allele (e.g., parent 1/1 and offspring 0/1 or 0/0; note that this includes the first category); inspected their quality metrics in R; and evaluated statistical differences with a Wilcoxon two-sample test (Hollander and Wolfe 1973). We then inspected individual depth distributions across these variants in R.
We next inspected genotype distributions in both offspring at individual heterozygous sites in the parent (Fig. 3). For this, we derived estimates of centromere positions in the reference genome using the O. niloticus core centromere repeat previously described (Melters et al. 2013) in a BLASTN search against the O. niloticus genome with default parameters (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed 09/2021).
BLAST results were imported into R and centromere core positions per chromosome inferred as the midpoint of all BLAST hits of the centromere sequence on each chromosome. We obtained putative centromere positions for 16 out of 22 chromosomes in total (positions detailed in Supplemental Data file EstimatedCentromerPositions.txt).
Based on the depth distribution and previous recommendations favoring a DP of eight (Carson et al. 2014), we next applied lower depth filters starting from our initial DP of three to a DP of more than eight to DP 15 and the same lower filters in combination with additional filtering below an upper limit of DP 20, 25, and 30. Applying each filter, we excluded all sites in which any of the analyzed individuals failed the filter. We then calculated retained offspring heterozygosity and visualized genotype and depth distributions. We continued our analysis with an additional filter of DP > 8 and DP < 20.
Data access
The whole-genome sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA832128. Source code and auxiliary and intermediate data to reproduce the analyses are available as Supplemental Code and as Supplemental Data, respectively.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
A.B., I.C., Z.O., S.M., and S.K. were supported by funding from the Leibniz Association and Deutsche Forschungsgemeinschaft (DFG) grant 492407022 to A.B. This study and S.V. and T.T. were supported by DFG grants TH 1615/3-1, 3-2, and 3-3 to T.T. and the Institute of Evolutionary Biology and Ecology of the University of Bonn. We thank Harald Kullmann for his ichthyologic enthusiasm during the field trip in Cameroon. We further thank Kamil S. Jaron and Jeramiah Smith for valuable comments on the manuscript, as well as Christoph Haag for insightful discussions on meiotic modifications.
Author contributions: A.B. and T.T. designed the study to analyze the mode of reproduction. S.V., D.M., and T.T. planned the housing conditions and monitored and documented the reproductive performance of the fish. S.K. extracted DNAs. A.B., Z.O., and I.C. analyzed sequencing data aided by S.M. and T.T. S.M. provided bioinformatic support. A.B., Z.O., and T.T. wrote the manuscript.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277368.122.
References
- ↵Alho JS, Välimäki K, Merilä J. 2010. Rhh: an R extension for estimating multilocus heterozygosity and heterozygosity–heterozygosity correlation. Mol Ecol Res 10: 720–722. 10.1111/j.1755-0998.2010.02830.x
- ↵Archetti M. 2010. Complementation, genetic conflict, and the evolution of sex and recombination. J Hered 101: S21–S33. 10.1093/jhered/esq009
- ↵Asher JH. 1970. Parthenogenesis and genetic variability. II. One-locus models for various diploid populations. Genetics 66: 369–391. 10.1093/genetics/66.2.369
- ↵Blouin M, Parsons M, Lacaille V, Lotz S. 1996. Use of microsatellite loci to classify individuals by relatedness. Mol Ecol 5: 393–401. 10.1111/j.1365-294X.1996.tb00329.x
- ↵Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170
- ↵Booth W, Smith CF, Eskridge PH, Hoss SK, Mendelson JRIII, Schuett GW. 2012. Facultative parthenogenesis discovered in wild vertebrates. Biol Lett 8: 983–985. 10.1098/rsbl.2012.0666
- ↵Card DC, Vonk FJ, Smalbrugge S, Casewell NR, Wüster W, Castoe TA, Schuett GW, Booth W. 2021. Genome-wide data implicate terminal fusion automixis in king cobra facultative parthenogenesis. Sci Rep 11: 7271. 10.1038/s41598-021-86373-1
- ↵Carson AR, Smith EN, Matsui H, Brækkan S, Jepsen K, Hansen JB, Frazer KA. 2014. Effective filtering strategies to improve data quality from population-based whole exome sequencing studies. BMC Bioinformatics 15: 125. 10.1186/1471-2105-15-125
- ↵Chapman DD, Shivji MS, Louis E, Sommer J, Fletcher H, Prodöhl PA. 2007. Virgin birth in a hammerhead shark. Biol Lett 3: 425–427. 10.1098/rsbl.2007.0189
- ↵Conte MA, Joshi R, Moore EC, Nandamuri SP, Gammerdinger WJ, Roberts RB, Carleton KL, Lien S, Kocher TD. 2019. Chromosome-scale assemblies reveal the structural evolution of African cichlid genomes. GigaScience 8: giz030. 10.1093/gigascience/giz030
- ↵Engelstädter J. 2017. Asexual but not clonal: evolutionary processes in automictic populations. Genet Breed 206: 993–1009. 10.1534/genetics.116.196873
- ↵Ferreira IA, Poletto AB, Kocher TD, Mota-Velasco JC, Penman DJ, Martins C. 2010. Chromosome evolution in African cichlid fish: contributions from the physical mapping of repeated DNAs. Cytogenet Genome Res 129: 314–322. 10.1159/000315895
- ↵Feulner PGD, Schwarzer J, Haesler MP, Meier JI, Seehausen O. 2018. A dense linkage map of lake Victoria cichlids improved the Pundamilia genome assembly and revealed a major QTL for sex-determination. G3 (Bathesda) 8: 2411–2420. 10.1534/g3.118.200207
- ↵Fields AT, Feldheim KA, Poulakis GR, Chapman DD. 2015. Facultative parthenogenesis in a critically endangered wild vertebrate. Curr Biol 25: R446–R447. 10.1016/j.cub.2015.04.018
- ↵Glémin S, François CM, Galtier N. 2019. Genome evolution in outcrossing vs. selfing vs. asexual species. Methods Mol Biol 1910: 331–369. 10.1007/978-1-4939-9074-0_11
- ↵Goikoetxea A, Todd EV, Gemmell NJ. 2017. Stress and sex: Does cortisol mediate sex change in fish? Reproduction 154: R149–R160. 10.1530/REP-17-0408
- ↵Grossen C, Guillaume F, Keller LF, Croll D. 2020. Purging of highly deleterious mutations through severe bottlenecks in Alpine ibex. Nat Commun 11: 1001. 10.1038/s41467-020-14803-1
- ↵Haenel Q, Laurentino TG, Roesti M, Berner D. 2018. Meta-analysis of chromosome-scale crossover rate variation in eukaryotes and its significance to evolutionary genomics. Mol Ecol 27: 2477–2497. 10.1111/mec.14699
- ↵Harrington RWJr. 1961. Oviparous hermaphroditic fish with internal self-fertilization. Science 134: 1749–1750. 10.1126/science.134.3492.1749
- ↵Hedrick PW, Hellsten U, Grattapaglia D. 2016. Examining the cause of high inbreeding depression: analysis of whole-genome sequence data in 28 selfed progeny of Eucalyptus grandis. New Phytol 209: 600–611. 10.1111/nph.13639
- ↵Heule C, Göppert C, Salzburger W, Böhne A. 2014. Genetics and timing of sex determination in the east African cichlid fish Astatotilapia burtoni. BMC Genet 15: 140. 10.1186/s12863-014-0140-5
- ↵Hollander M, Wolfe DA. 1973. Nonparametric statistical methods. John Wiley & Sons, New York.
- ↵Kelley JL, Yee M-C, Brown AP, Richardson RR, Tatarenkov A, Lee CC, Harkins TT, Bustamante CD, Earley RL. 2016. The genome of the self-fertilizing mangrove rivulus fish, Kryptolebias marmoratus: a model for studying phenotypic plasticity and adaptations to extreme environments. Genome Biol Evol 8: 2145–2154. 10.1093/gbe/evw145
- ↵Kraemer P, Gerlach G. 2017. Demerelate: calculating interindividual relatedness for kinship analysis based on codominant diploid genetic markers using R. Mol Ecol Res 17: 1371–1377. 10.1111/1755-0998.12666
- ↵Lamboj A. 2004. The cichlid fishes of Western Africa. Birgit Schmettkamp Verlag, Bornheim, Germany.
- ↵Lampert KJ. 2008. Facultative parthenogenesis in vertebrates: reproductive error or chance? Sex Dev 2: 290–301. 10.1159/000195678
- ↵Langen K, Schwarzer J, Kullmann H, Bakker TCM, Thünken T. 2011. Microsatellite support for active inbreeding in a cichlid fish. PLoS One 6: e24689. 10.1371/journal.pone.0024689
- ↵Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26: 589–595. 10.1093/bioinformatics/btp698
- ↵Li C, Horvitz DJ. 1953. Some methods of estimating the inbreeding coefficient. Am J Hum Genet 5: 107.
- ↵Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352
- ↵Lutes AA, Neaves WB, Baumann DP, Wiegraebe W, Baumann P. 2010. Sister chromosome pairing maintains heterozygosity in parthenogenetic lizards. Nature 464: 283–286. 10.1038/nature08818
- ↵Matschiner M, Böhne A, Ronco F, Salzburger W. 2020. The genomic timeline of cichlid fish diversification across continents. Nat Commun 11: 5895. 10.1038/s41467-020-17827-9
- ↵McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. 10.1101/gr.107524.110
- ↵Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby JG, Sebra R, Peluso P, Eid J, Rank D, 2013. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol 14: R10. 10.1186/gb-2013-14-1-r10
- ↵Meuthen D, Baldauf SA, Bakker TCM, Thünken T. 2016. Conspecific alarm cues affect interspecific aggression in cichlid fishes. Hydrobiologia 767: 37–49. 10.1007/s10750-015-2473-0
- ↵Neaves WB, Baumann P. 2011. Unisexual reproduction among vertebrates. Trends Genet 27: 81–88. 10.1016/j.tig.2010.12.002
- ↵Nougué O, Rode N, Jabbour-Zahab R, Ségard A, Chevin LM, Haag CR, Lenormand T. 2015. Automixis in Artemia: solving a century-old controversy. J Evol Biol 28: 2337–2348. 10.1111/jeb.12757
- ↵Oldfield RG. 2005. Genetic, abiotic and social influences on sex differentiation in cichlid fishes and the evolution of sequential hermaphroditism. Fish Fish 6: 93–110. 10.1111/j.1467-2979.2005.00184.x
- ↵Ozouf-Costaz C, Coutanceau JP, Bonillo C, MerCot H, Fermon Y, Guidi-Rontani C. 2017. New insights into the chromosomal differentiation patterns among cichlids from Africa and Madagascar. Cybium 41: 35–43. 10.26028/cybium/2017-411-004
- ↵Palaiokostas C, Bekaert M, Khan MG, Taggart JB, Gharbi K, McAndrew BJ, Penman DJ. 2013. Mapping and validation of the major sex-determining region in Nile tilapia (Oreochromis niloticus L.) using RAD sequencing. PLoS One 8: e68389. 10.1371/journal.pone.0068389
- ↵Pekkala N, Knott K, Kotiaho J, Nissinen K, Puurtinen M. 2012. The benefits of interpopulation hybridization diminish with increasing divergence of small populations. J Evol Biol 25: 2181–2193. 10.1111/j.1420-9101.2012.02594.x
- ↵Post VA. 1965. Vergleichende untersuchungen der chromosomenzahlen bei Süßwasser-teleosteern. Z Zool Syst Evol Forsch 3: 47–93. 10.1111/j.1439-0469.1965.tb00426.x
- ↵Ranallo-Benavidez TR, Jaron KS, Schatz MC. 2020. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11: 1432. 10.1038/s41467-020-14998-3
- ↵R Core Team. 2020. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/.
- ↵Robinson JA, Brown C, Kim BY, Lohmueller KE, Wayne RK. 2018. Purging of strongly deleterious mutations explains long-term persistence and absence of inbreeding depression in island foxes. Curr Biol 28: 3487–3494.e4. 10.1016/j.cub.2018.08.066
- ↵Roessler K, Muyle A, Diez CM, Gaut GR, Bousios A, Stitzer MC, Seymour DK, Doebley JF, Liu Q, Gaut BS. 2019. The genome-wide dynamics of purging during selfing in maize. Nat Plants 5: 980–990. 10.1038/s41477-019-0508-7
- ↵Ronco F, Matschiner M, Böhne A, Boila A, Büscher HH, El Taher A, Indermaur A, Malinsky M, Ricci V, Kahmen A, 2021. Drivers and dynamics of a massive adaptive radiation in cichlid fishes. Nature 589: 76–81. 10.1038/s41586-020-2930-4
- ↵Ryder OA, Thomas S, Judson JM, Romanov MN, Dandekar S, Papp JC, Sidak-Loftis LC, Walker K, Stalis IH, Mace M, 2021. Facultative parthenogenesis in California condors. J Hered 112: 569–574. 10.1093/jhered/esab052
- ↵Sardell JM, Kirkpatrick M. 2020. Sex differences in the recombination landscape. Am Nat 195: 361–379. 10.1086/704943
- ↵Sardell JM, Cheng C, Dagilis AJ, Ishikawa A, Kitano J, Peichel CL, Kirkpatrick M. 2018. Sex differences in recombination in sticklebacks. G3 (Bathesda) 8: 1971–1983. 10.1534/g3.118.200166
- ↵Smith HA, Moya A, Cantin NE, Van Oppen MJ, Torda G. 2019. Observations of simultaneous sperm release and larval planulation suggest reproductive assurance in the coral Pocillopora acuta. Front Mar Sci 6: 362. 10.3389/fmars.2019.00362
- ↵Svendsen N, Reisser CM, Dukić M, Thuillier V, Ségard A, Liautard-Haag C, Fasel D, Hürlimann E, Lenormand T, Galimov Y, 2015. Uncovering cryptic asexuality in Daphnia magna by RAD sequencing. Genetics 201: 1143–1155. 10.1534/genetics.115.179879
- ↵Svensson O, Smith A, García-Alonso J, Van Oosterhout C. 2016. Hybridization generates a hopeful monster: a hermaphroditic selfing cichlid. R Soc Open Sci 3: 150684. 10.1098/rsos.150684
- ↵Talbert PB, Henikoff S. 2010. Centromeres convert but don't cross. PLoS Biol 8: e1000326. 10.1371/journal.pbio.1000326
- ↵Tatarenkov A, Lima SM, Taylor DS, Avise JC. 2009. Long-term retention of self-fertilization in a fish clade. Proc Natl Acad Sci 106: 14456–14459. 10.1073/pnas.0907852106
- ↵Thünken T, Bakker TCM, Baldauf SA, Kullmann H. 2007. Active inbreeding in a cichlid fish and its adaptive significance. Curr Biol 17: 225–229. 10.1016/j.cub.2006.11.053
- ↵Vasimuddin M, Misra S, Li H, Aluru S. 2019. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. In 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), pp. 314–324. Institute of Electrical and Electronics Engineers, Piscataway, NJ. 10.1109/IPDPS.2019.00041
- ↵Warner RR. 1975. The adaptive significance of sequential hermaphroditism in animals. Am Nat 109: 61–82. 10.1086/282974
- ↵Warren WC, García-Pérez R, Xu S, Lampert KP, Chalopin D, Stöck M, Loewe L, Lu Y, Kuderna L, Minx P, 2018. Clonal polymorphism and high heterozygosity in the celibate genome of the Amazon molly. Nat Ecol Evol 2: 669–679. 10.1038/s41559-018-0473-y
- ↵Xue Y, Prado-Martinez J, Sudmant PH, Narasimhan V, Ayub Q, Szpak M, Frandsen P, Chen Y, Yngvadottir B, Cooper DN, 2015. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 348: 242–245. 10.1126/science.aaa3952