
Abstract
Organs and tissues age at different rates within a single individual. Such asynchrony in aging has been widely observed at multiple levels, from functional hallmarks, such as anatomical structures and physiological processes, to molecular endophenotypes, such as the transcriptome and metabolome. However, we lack a conceptual framework to understand why some components age faster than others. Just as demographic models explain why aging evolves, here we test the hypothesis that demographic differences among cell types, determined by cell-specific differences in turnover rate, can explain why the transcriptome shows signs of aging in some cell types but not others. Through analysis of mouse single-cell transcriptome data across diverse tissues and ages, we find that cellular age explains a large proportion of the variation in the age-related increase in transcriptome variance. We further show that long-lived cells are characterized by relatively high expression of genes associated with proteostasis and that the transcriptome of long-lived cells shows greater evolutionary constraint than short-lived cells. In contrast, in short-lived cell types, the transcriptome is enriched for genes associated with DNA repair. Based on these observations, we develop a novel heuristic model that explains how and why aging rates differ among cell types.
In his poem “The Deacon's Masterpiece,” Oliver Wendell Holmes described a carriage so artfully constructed that it ran flawlessly for 100 years, at which time every single part failed simultaneously (Holmes 1891). The real world of organismal aging is, of course, very different. As animals age, different organs and functions fail at different rates and in different ways (Rando and Wyss-Coray 2021). In humans, hair color, reproductive capacity, and heart function all decline faster than cognitive and gastrointestinal function (Khan et al. 2017). Heterogeneity in aging among organs is observed not only in terms of structural and functional changes but also at the molecular level. For example, in mice, the timing and degree of age-related change in the transcriptome differ among organs (Schaum et al. 2020). Similar among-organ differences are seen in the aging of the proteome (Ori et al. 2015). However, these observations are based on analysis of bulk tissue, which masks variation among cells within tissues (e.g., Ibañez-Solé et al. 2022). Analysis of aging in single-celled organisms has established transcriptome-wide effects of cellular age, including declining coordination among genes expressed within cells, and increased discordance in gene expression profiles among cells (Rando and Wyss-Coray 2021).
Studies using single-cell transcriptomics have made substantial contributions to our understanding of variation in aging among organs and cells. In particular, through analysis of single-cell transcriptome data among cell types within and across organs, researchers have found strong evidence of cell-specific differences in the way that age impacts gene expression levels (Mathys et al. 2019; Menon et al. 2019; Ma et al. 2020; Wang et al. 2020; Yamamoto et al. 2022; Buckley et al. 2023), gene expression variability, and cell-to-cell heterogeneity (Bahar et al. 2006; Enge et al. 2017; Martinez-Jimenez et al. 2017; Salzer et al. 2018; Angelidis et al. 2019; Kimmel et al. 2019; Ximerakis et al. 2019; Zhang et al. 2021; Ibañez-Solé et al. 2022). For example, in mice, age-related changes in gene expression occur in natural killer cells in the lung and spleen but occur to a much lesser extent in B cells from the same organs (Kimmel et al. 2019). Although the relationship between asynchronous variation in the transcriptome and functional decline among organs is speculative, this variation among cell types may, in turn, help us to understand variation in rates of aging among tissues and organs (Cohen et al. 2022).
Despite the potential for single-cell analysis to uncover the asynchrony in tissue and organ aging, we lack a compelling explanation for why transcriptome variation changes with age in some organs but not in others. Prior studies have proposed that transcriptome variation builds from age-driven “noise” in gene expression, which may be caused by somatic mutation (Enge et al. 2017), epigenetic drift (Fraga et al. 2005), or other damage to gene expression programs (Warren et al. 2007; Levy et al. 2020; Schumacher et al. 2021). Others have argued that properly regulated age-dependent gene expression, some of which may prevent or repair cellular damage, is an important component as well (Perez-Gomez et al. 2020; Ibañez-Solé et al. 2022). Some of these potentially causal processes are known to differ among cell types (Enge et al. 2017; Li et al. 2021; Moore et al. 2021). But the root cause of this variation might actually be related to the underlying population dynamics of different cell types. In particular, Warren et al. (2007) suggested that asynchrony in transcriptome variation, rather than being a direct reflection of organ aging, might instead be explained by differences in cell turnover rates.
Cells from slowly-renewing tissues are more likely to have cellular ages similar to the organism's age, whereas fast-turnover cells, being constantly replenished, stay relatively young throughout the life of an organism. If cell age is associated with transcriptome variability, then older cells should accumulate more gene expression noise and show higher transcriptome variability than do short-lived cells (Warren et al. 2007). This explanation is consistent with the observation that age-related increases in transcriptome variability have been detected in low-turnover cardiomyocytes (Bahar et al. 2006) but not in high-turnover granulocytes, naive B and T cells, or endothelial cells of the lung (Warren et al. 2007; Kimmel et al. 2019; Ibañez-Solé et al. 2022).
Cell turnover rate varies by orders of magnitude among cell types, from fast turnover rates in blood or gut epithelial cells, which are renewed every day (Richardson et al. 2014; Sender and Milo 2021), to very slow turnover in neurons, whose renewal during an individual's lifetime might only occur in specific regions of the brain, if at all (Spalding et al. 2013). Such a broad spectrum of cell turnover has led to investigations of gene expression patterns associated with cell longevity (Seim et al. 2016; Castillo-Morales et al. 2019). Here, we took a conceptually different view and investigated the relationship between cell turnover and organismal aging, treating cell turnover as a parameter governing cell population dynamics. We focus specifically on testing the hypothesis that cellular age explains the degree of age-related change among cell types within an individual organism.
The distribution of ages in a population of cells of a given type depends both on cell turnover rate and on the age of the organism. To investigate the relationship between cellular age and transcriptome variability, we first developed a cell demography model to estimate the age distribution of a population of cells of a given cell type within an individual of a particular age. We then examined gene expression variability across cell types with single-cell transcriptome data. To test our hypothesis, we estimated the effect of cellular age on transcriptome variation across cell types at each of three mouse ages. We then tested whether the increased transcriptome variation over cellular age that manifests across tissues at a given mouse age was also true within each cell type. To explore candidate mechanisms underlying the differential aging signals among cell types, we examined the expression pattern of gene groups associated with the hallmarks of aging (López-Otín et al. 2013). We also looked for signs of evolutionary constraint within the transcriptome as a function of cellular longevity. Finally, we have proposed a heuristic model integrating our findings and previous studies on aging, showing the various strategies that short-lived and long-lived cells might use to ensure cell function over the course of an organism's life, and some of the evolutionary implications of variation in cellular longevity and turnover for organismal fitness. These results provide a novel perspective on how “cellular demography” can influence patterns of aging, as well as the mechanisms different cells use to ensure maximal fitness with age.
Results
Cell turnover explains asynchronous trajectories of transcriptome variation with age
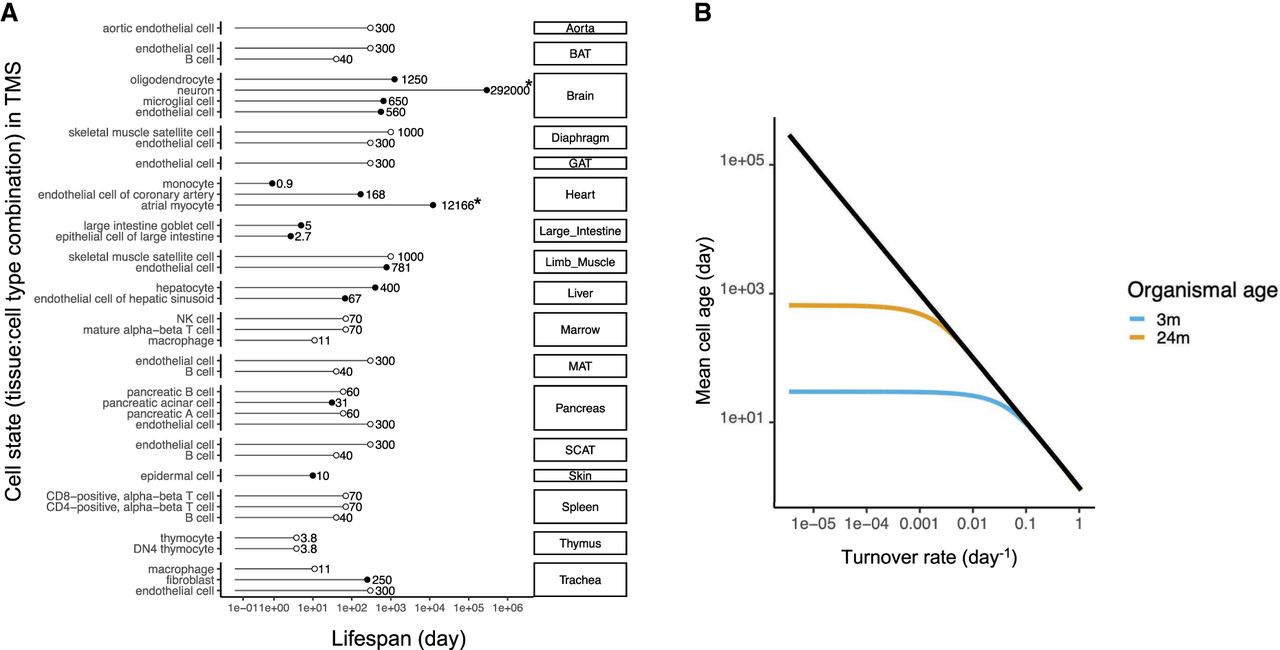
To investigate the relationship between transcriptome variation and cellular age across different cell types, we integrated cell type–specific cellular life span data in mice from Sender and Milo (2021), with single-cell transcriptome data from the mouse aging atlas, Tabula Muris Senis (TMS) (Almanzar et al. 2020; Zhang et al. 2021). Together, this yielded 39 combinations of mostly postmitotic tissue:cell types, within their tissue context, with both life span estimates and age-specific transcriptome profiles (Methods) (Fig. 1A; Supplemental Table S1). We used published measures of cell life span (T) to estimate the mean age of each cell type in mice of different ages. Making the simplifying assumption that cell turnover rates are constant with age, we can estimate the cell turnover rate, b = 1 − e−1/T (Methods) (Supplemental Methods). Given that a cell cannot be older than the organism in which it is found, the distribution of cellular age will be a truncated exponential function determined by cell turnover rate and organismal age. We can then use organismal age to calculate the mean age of a population of cells with turnover b. This cellular demography model illustrates the degree to which cellular age increases with organismal age. For cell types with relatively slow cellular turnover, the mean cellular age increases concomitantly with organismal age. At the extreme, a cell that has no turnover will always be the same age as the organism in which it is found. In contrast, cells with very fast turnover will never be more than a few days old, and barring any changes in turnover rate with host age, the age distribution for that cell type will be constant throughout the life of the organism (Fig. 1B; Supplemental Figs. S1, S2; Supplemental Methods).
The distribution of cell life span data used in this study and of mean cell age distributions during organismal aging. (A) Summary of cellular life span data used in this study. The cellular life span data in rodents (Sender and Milo 2021) were aligned with cell types in the TMS data (see Supplemental Table S1; Almanzar et al. 2020; Zhang et al. 2021). The left-side texts show cell type names; the right-side texts, tissue names: (BAT) brown adipose tissue, (GAT) gonadal adipose tissue, (MAT) mesenteric adipose tissue, and (SCAT) subcutaneous adipose tissue. Filled circles indicate the 15 cell types with tissue-specific life span data; open circles, without tissue-specific life span estimates that correspond to seven distinct cell types. Note that in two cases, the statistical model used by Sender and Milo (2021) gives life span estimates that exceed the maximum life span of laboratory mice, highlighted with an asterisk for neuron and atrial myocytes. However, these values lead to minimal effects on the mean cell age during organismal aging. (B) Mean cell age as a function of cell turnover rate. The black line denotes the theoretical expectation for a cell with an exponential survival function in an infinitely long-lived organism. More realistically, we assume that cell age follows a truncated exponential distribution, such that the mean cell age is constrained by organismal age. We show the analytical solution of the mean cell age as a function of cell turnover rate for a 3-mo-old mouse (blue) and a 24-mo-old mouse (yellow), using the model in Equation 8 (Supplemental Methods).
To quantify cell type–specific transcriptome variability with the TMS data, we initially chose data from the FACS platform from male mice, given its higher sequencing depth and more comprehensive tissue:cell type coverage (Methods) (Supplemental Fig. S3). We measured the variability of individual genes among cells of a given tissue:cell type combination at young and old age separately using the coefficient of variation (CV). To minimize confounding effects of the mean-variance correlation on gene expression (Eling et al. 2018), we limited our analysis to the 16%–83% of genes that are not differentially expressed with age within each of the 39 tissue:cell type combinations (P > 0.1; Methods) (Martinez-Jimenez et al. 2017). Using a mixed model to account for variation owing to gene and to repeated measures within each mouse, we found that gene-level variance increased with age in 22 of 39 tissue:cell type combinations (FDR < 0.05) (Supplemental Fig. S5). Thus, most cell types show at least some increase in transcriptome variability with organismal age, consistent with previous work (Bahar et al. 2006; Enge et al. 2017; Angelidis et al. 2019; Kimmel et al. 2019; Wang et al. 2020; Luo et al. 2022).
Although there was no effect of cellular age on transcriptome variation in young mice (r2 = 0.01, P = 0.610), we found a striking positive association as mice aged, from r2= 0.26 (P = 0.021) in 18-mo-old mice to cell age explaining fully half of the variation in CV in 24-mo-old mice (r2= 0.52, P < 0.001) (Fig. 2A); thus, transcriptome variation accumulated in older mice in a pattern predicted by cellular age. To confirm that this finding did not depend on our metric of transcriptome variability, we applied two alternative metrics (Angelidis et al. 2019; Levy et al. 2020) and found the same pattern (Methods) (Supplemental Fig. S8). Additionally, although data from female mice were limited to mice at ages 3 and 18 mo, the same relationship was true in cells from 18-mo-old female mice (Supplemental Fig. S9). These results support the hypothesis that cellular age is a strong predictor of transcriptome variation among tissues in mice.
Cellular age dynamics predict the increase in transcriptome variability in aging mice. (A) The average log10 coefficient of variation (CV) of each cell type at each of three organismal ages (in months) is plotted over their respective mean cell ages (in days). The colored lines show ordinary least square regressions, with shading indicating 95% confidence intervals, each corresponding to one age group. (B) The mean change in transcriptome variability (βage; Methods) observed in cells over a 21-mo period, from 24-mo-old mice compared with cells from 3-mo-old mice, is correlated with the change in estimated mean cell age for 22 cell types (Pearson's r = 0.702, P = 2.7 × 10−4). The black line shows an ordinary least square regression, with shading indicating 95% confidence intervals. Filled circles indicate the 15 cell types with cell- and tissue-specific life span data; open circles, the seven cell types without tissue-specific life span estimates, and so their βage is the mean from all tissues in which they are detected (see Methods).
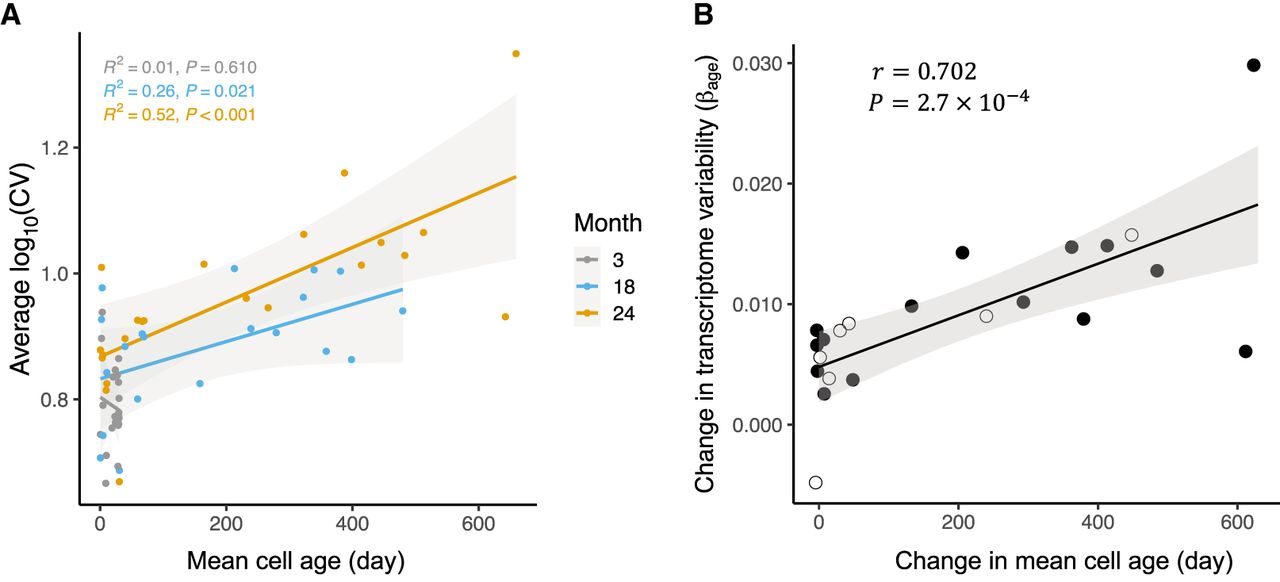
We then asked if the pattern seen across cells at a given mouse age was true as a given cell type aged. More specifically, given the estimates of the age of a cell type in mice of different ages, we can ask if the extent of transcriptome variation across mouse ages is associated with the degree of increase in mean cell age over that time. To estimate the effect of mouse age on transcriptome variability, we fitted a mixed model, treating age as a fixed predictor and both gene and individual mouse as random effects (Methods; Supplemental Fig. S4). After model fitting, the coefficient associated with age was extracted and used as a quantity for the effect of age on transcriptome variation (βage), which was plotted over the age increase for each cell type (Fig. 2B; Supplemental Methods, Equation 9). Here we found a striking positive correlation, with almost half of the increase in transcriptome variation among cells of 3-mo-old mice versus 24-mo-old mice explained by the age increase of each respective cell type (Pearson's r = 0.702; P = 2.7 × 10−4). The increase in cellular age between young and old mice was also a strong predictor of variation measured by two alternative metrics of transcriptome variation, and it held up in females and in males (Fig. 2B; Supplemental Table S2; Supplemental Figs. S8, S9). Taken together, our results show that a substantial proportion of the age-related changes in gene expression variation among cell types can be explained by cellular age.
Cell life span estimates were derived for distinct cell types by Sender and Milo (2021), and in many cases, a given cell type is distributed across different tissues (e.g., we have TMS data for B cells found in brown adipose, mesenteric adipose, and subcutaneous adipose tissue). This allowed us to consider the effect of tissue microenvironment on transcriptome variation. There are tissue-specific turnover estimates for 15 of the 39 tissue:cell type combinations in male mice, whereas the remaining 24 tissue:cell type combinations correspond to seven distinct cell types that lack tissue-specific turnover estimates (Methods) (Fig. 1). To avoid pseudoreplication in our main analysis, we made the simplifying assumption that when tissue-specific turnover was not known, then transcriptome variation βage is best estimated at the cell type level by taking its mean within that cell type across tissues. Tissue microenvironment, however, may affect transcriptome variation, and indeed, when we considered the βage of the same cell type in different tissues, we found that the effect of mouse age on the transcriptome appears to vary by tissue (Supplemental Fig. S10A). However, this variation may be driven by effects of tissue microenvironment on cell turnover, rather than by direct effects of microenvironment on transcriptome variation. In support of this conclusion, among endothelial cells, the only cell type for which we had estimates for both tissue-specific turnover rate and tissue-specific transcriptome variation, βage can be almost entirely explained by tissue-specific age increases of these cells (Supplemental Fig. S10B).
Cell life span associates with signatures of proteostasis and selection
Transcriptome variation is a hallmark of cellular aging and could be an indication of cellular damage and/or dysregulation (Warren et al. 2007; Martinez-Jimenez et al. 2017; Hernando-Herraez et al. 2019). Given the deleterious and cumulative effects of other hallmarks of aging, like somatic mutation, misfolded proteins, and dysfunctional mitochondria, as organisms age (López-Otín et al. 2013), we asked if long-lived cells show increased expression of corresponding repair or maintenance processes in their transcriptome. Using Gene Ontology (GO) terms related to DNA repair, proteostasis, and autophagy, we calculated the expression of each GO term in each cell, as well as the mean of these values for all cells in a cell type (Methods).
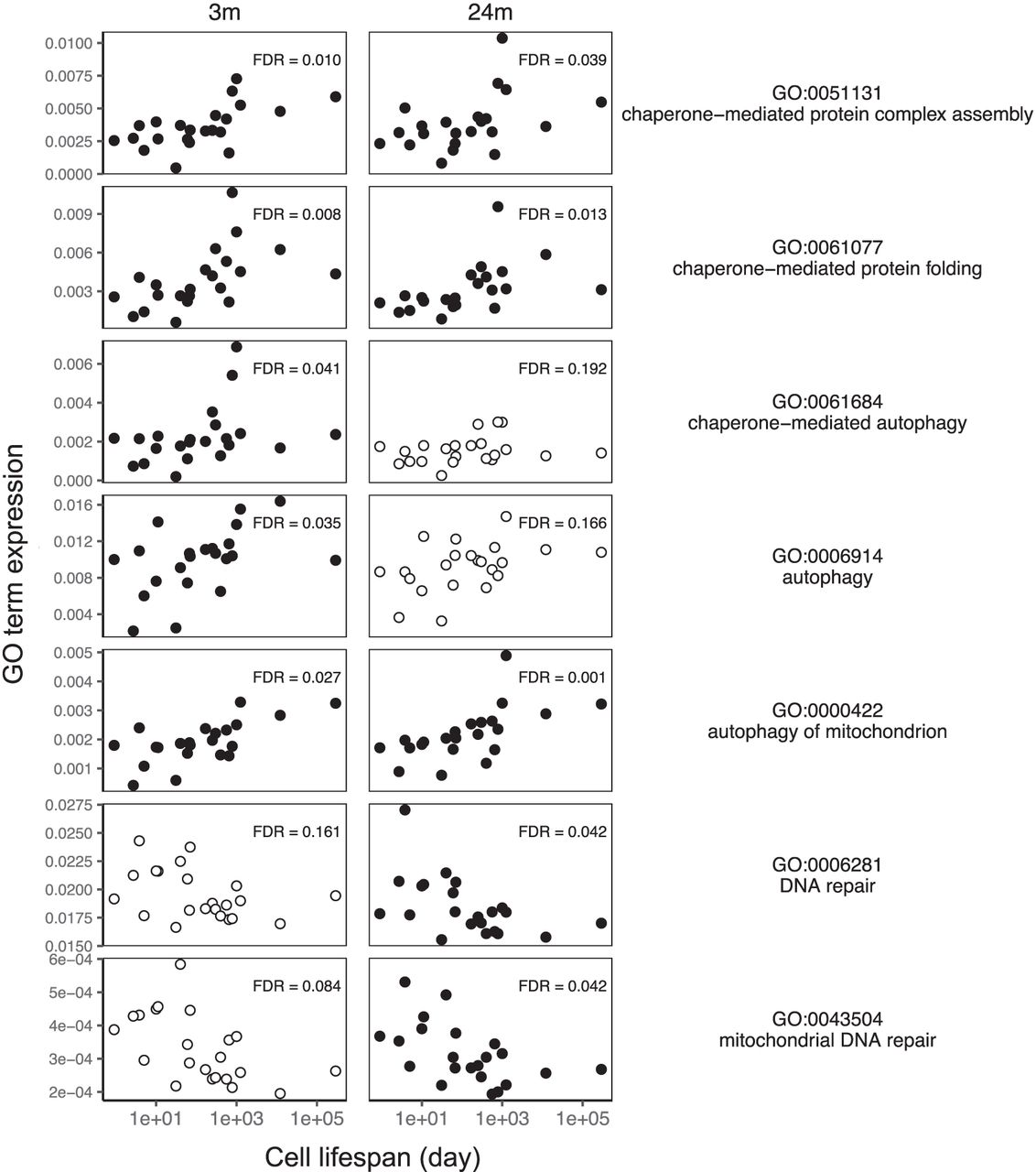
Consistent with our hypothesis, we found that expression of two or three out of the four GO terms associated with chaperone-mediated processes, including protein folding, protein complex assembly, and autophagy, were positively correlated with cell life span in old and young mice, respectively, as was “autophagy of mitochondria” (absolute Kendall's τ > 0.34, FDR < 0.05) (Fig. 3; Supplemental Fig. S11). Counter to our expectation, the expression of two of the six GO terms associated with DNA repair, although not associated with cell life span at young age, was negatively associated with cell life span at old age (absolute Kendall's τ > 0.38, FDR < 0.05) (Fig. 3; Supplemental Fig. S11). That is, in older mice, short-lived cells are more likely than long-lived cells to express genes involved in DNA repair.
Cell life span correlates with chaperone-mediated processes, mitochondrial autophagy, and DNA repair in mice. The mean expression of genes in each GO term in 22 cell types at young age (3 mo), and old age (24 mo) correlates with the life span of those cell types. Panels with filled circles show GO terms with a significant correlation with cell life span (the absolute Kendall's τ > 0.34, FDR < 0.05).
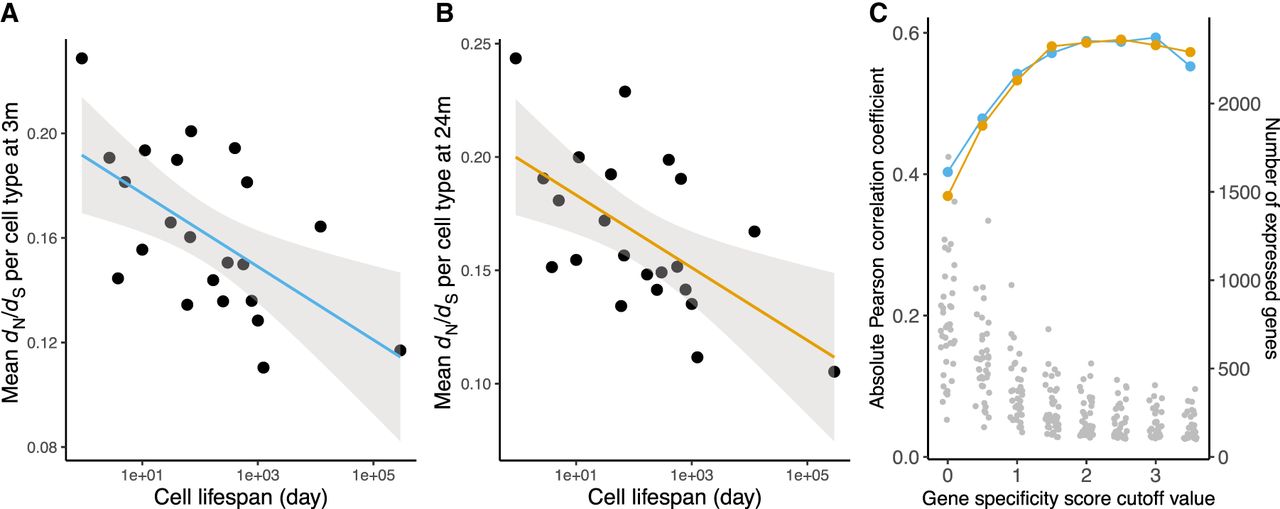
Our results point to the importance in long-lived cells of maintaining proteostasis through chaperone expression (Labbadia and Morimoto 2015) and are consistent with the association between cell longevity and the expression of chaperones in humans (Castillo-Morales et al. 2019). Proteins vary in their dependence on chaperones. As previous work has shown, chaperone clients tend to be less stable than proteins that are not chaperone clients, and they carry higher levels of nonsynonymous codon variation, indicating less evolutionary constraint (Taipale et al. 2012). In light of these findings, we might expect that expression profiles in long-lived cells have evolved to reduce their proteostatic burden by expressing proteins that are, on average, more stable, with relatively low dN/dS ratios (where dN is the nonsynonymous substitution rate, and dS is the synonymous substitution rate) (Drummond and Wilke 2008; Sikosek and Chan 2014). Within each cell, we took the median dN/dS values of cell type–specific genes, with one-to-one rat orthologs, and then used the mean of dN/dS across cells to represent cell type–specific dN/dS (Methods). Consistent with our expectation, we found that dN/dS ratios in the expression profiles of cells from young and old mice were negatively correlated with cell life span (Pearsons's r < −0.58, P < 5.0 × 10−3) (Fig. 4A,B). This result remained unchanged after removing the 76 genes that were more likely to be under recurrent positive selection (dN/dS > 1) (Supplemental Fig. S13; Kryazhimskiy and Plotkin 2008), and was also observed using dN/dS ratios based on the more distant mouse–human orthologs (Pearsons's r < −0.63, P < 1.6 × 10−3) (Supplemental Fig. S14). In addition, we found that the association became stronger as we limited the analysis to genes with increasing cell type–specific expression patterns (Methods) (Fig. 4C).
Cell life span predicts evolutionary constraint among expressed genes. The relaxation of evolutionary constraint (dN/dS) of genes expressed in 22 cell types in young (A) and old (B) mice is negatively correlated with cell life span (Pearson's r = −0.588, P = 3.9 × 10−3 and Pearson's r = −0.586, P = 4.1 × 10−3 for young and old mice, respectively). The median dN/dS is measured per cell, using cell type–specific genes (median dN/dS of genes above specificity score > 2, n = 116 to 971 genes; see Methods). The average dN/dS of a cell type is the mean of dN/dS across cells (n = 56 to 2251 cells; see Methods). The line in each figure shows an ordinary least square regression with shading of 95% confidence intervals. (C) Correlation between dN/dS and cell life span for transcriptomes of increasing cell type specificity (see Methods). The blue or yellow dots corresponding to the left axis show the correlation strength. The gray dots correspond to the right axis and show the number of expressed genes per cell type at each level of specificity.
Discussion
It has long been recognized that the dynamics of aging—its onset, rate, and magnitude—vary across species (Promislow 1991; Gorbunova et al. 2014) and among individuals within species (Kirkwood 2005). But it is also clear that even within individual organisms, different structures and functions age at different rates. Little work has been performed to describe this variation (but see, e.g., Hayward et al. 2015), and we lack a clear framework to explain why some elements age faster than others. A recent evolutionary model posits that those systems under stronger selection—that is, systems that contribute more to organismal fitness—should age faster than others (Moorad and Ravindran 2022), but the theory has yet to be tested, requiring estimates of the age-specific marginal effects on fitness of different components within an organism. Based on observations of cell-specific transcriptome variation with age in just four cell types, Warren et al. (2007) hypothesized that the degree to which the transcriptome shows signs of aging in a particular cell type is positively associated with the life expectancy of that cell type.
Here we provide the first comprehensive test of Warren et al.’s (2007) hypothesis. We show that in mice, the amount of age-related increase in transcriptome variation not only is greater in long-lived cells, such as neurons and muscle cells, relative to short-lived cells like monocytes and gut epithelial cells but also is strongly predicted by the rate at which cellular age increases with increasing organismal age. Although the functional consequences of gene expression variability remain an open question, it appears that differences in aging among cell types, as measured by transcriptome variation among cells within each type, are driven in part by cellular age. Moreover, we show that expression profiles of longer-lived cells are enriched for pathways involved in homeostasis, including chaperone-mediated processes and mitochondrial turnover. We cannot be sure if higher expression of chaperone processes or mitophagy supports long cellular life span or if longer life span leads to greater opportunity for protein damage or mitochondrial malfunction, thereby inducing expression of genes associated with proteostasis or mitophagy. In support of the former interpretation, the proteins expressed in longer-lived cells tend to have evolved under stronger selective constraints.
Our results point to a major role of cell age in influencing age-related changes in cell variability. The underlying mechanisms of this relationship are unknown, however, but likely include both cell autonomous and cell nonautonomous factors, including aging of stem cells (Jones and Rando 2011) and age-related changes in the cellular microenvironment (Brunet et al. 2023), somatic mutation, changes to epigenetic structure, oxidative and other damage, loss of mitochondrial function, diminution of homeostatic mechanisms, and apoptotic cell death (Elliott and Ravichandran 2016; Gladyshev et al. 2021). We consider each factor in light of our results.
First, age-related changes occur in the structure and function of the tissue microenvironment, or cellular niche (Brunet et al. 2023). How the niche affects cellular life span and transcriptome variation across an organism is unknown, but in each instance in which a niche-specific cellular age is available in our study, it is strongly predictive of the effect of age on transcriptome variation (Supplemental Fig. S10B). We speculate that in cells of a given type that occur in diverse organs, variation in rates of aging across different niches is driven by the effect of the niche on rates of cellular turnover rather than directly on rates of increase in transcriptome variation.
Second, there is support for the hypothesis that variation in stem cell dynamics can influence traits associated with aging, especially cancer (Jones and Rando 2011; Tomasetti and Vogelstein 2015). However, our results find little support for a major role for stem cell aging as a driver of transcriptome variation among short- versus long-lived cells. Instead, we see the greatest age-related increase in transcriptome variation among cells with the slowest turnover, in which there is relatively little stem cell activity, contrary to what one might expect if stem cell aging drives transcriptome variation. However, stem cell dynamics could potentially influence patterns that we observe in high-turnover cells. Cell turnover rate should be associated with clonality, owing to clonal expansion among stem cells and their descendants, particularly later in life (Fabre et al. 2022; Mitchell et al. 2022). The prevalence of such clonality across tissues and its effect on transcriptome variation are yet to be systematically assessed, but it is possible that clonality reduces transcriptome variance among high-turnover cells, particularly later in life (Yermanos et al. 2021).
Third, factors intrinsic to the cell, including somatic mutation, cell death, and damage to other biomolecules, to cellular homeostatic mechanisms, and to repair mechanisms, have all been implicated in aging (Gladyshev et al. 2021; Schumacher et al. 2021). Little is known about the degree to which cells differ in their baseline rates of certain kinds of damage, such as free radical damage to biomolecules. However, single-cell genomic analyses have found significant heterogeneity in somatic mutation rates across cell types and organs in both proliferating cells (e.g., Martincorena et al. 2018) and postmitotic cells (Bae et al. 2018; Li et al. 2021; Moore et al. 2021). Somatic mutation, other damage to DNA, and age-related changes to the epigenome and to transcription mechanisms are all associated with transcriptional variation, and so, each could potentially contribute as drivers of the transcriptional variation that accompanies cellular aging (Schumacher et al. 2021; Bozukova et al. 2022; Debès et al. 2023; Gyenis et al. 2023; Ibañez-Solé et al. 2023). We further note that mutation, DNA damage, and epigenetic change need not directly affect the expression of genes in cis, but rather, any indirect effects on gene regulatory mechanisms or cell state may be sufficient to drive genome-wide transcriptional variation within a cell type (Schumacher et al. 2021).
Additionally, programmed cell death (PCD), triggered by cellular damage, directly contributes to cell turnover. This creates opportunities for selection within populations of cells, as we describe below, and should affect transcriptome variation. PCD is critical to maintaining tissue function and preventing cancer and so has received a great deal of attention (Pistritto et al. 2016). And yet, because of the complexity and variety of apoptotic mechanisms and their associated biomarkers, we lack a comprehensive picture of the rates and regulation of apoptotic cell death across cell types (Elliott and Ravichandran 2016).
A heuristic model
We found compelling evidence that cells with relatively slow turnover rates have a much greater age-related increase in transcriptome variance and that this variation is best predicted by the degree to which cell age increases as an organism ages, and we identified additional correlates that point to potential mechanisms that could account for this pattern. In Figure 5, we present a heuristic model to help us integrate our findings and to reconcile observations that might seem, on the face of it, to be contradictory.
Models of cellular aging and cell type–specific transcriptome variation. Here we present a model depicting the accumulation of transcriptome variation with cellular age. (A) As a cell ages, it accumulates genetic damage (light blue) and cellular homeostasis becomes increasingly imbalanced (pink), either of which can directly or indirectly increase transcriptome variation. The rates of accumulation of damage and loss of homeostasis are counteracted by repair and maintenance processes, which in turn slow the rate of increase in transcriptome variation. As cellular transcriptome variation increases, so does the likelihood of triggering cell death (rightward arrow) (Tower 2015). (B) The model of cellular aging when applied to populations of long-lived cells (slow turnover) and short-lived cells (fast turnover) describes the accumulation of transcriptome variation among a cell type (green). Long-lived cells age together with the organism, and transcriptome variation accumulates among these cells as the organism ages. In short-lived cells, damaged cells are frequently replaced by new cells, which prevents accumulation of variation at the cell type level. Cartoons were created with BioRender (https://www.biorender.com).
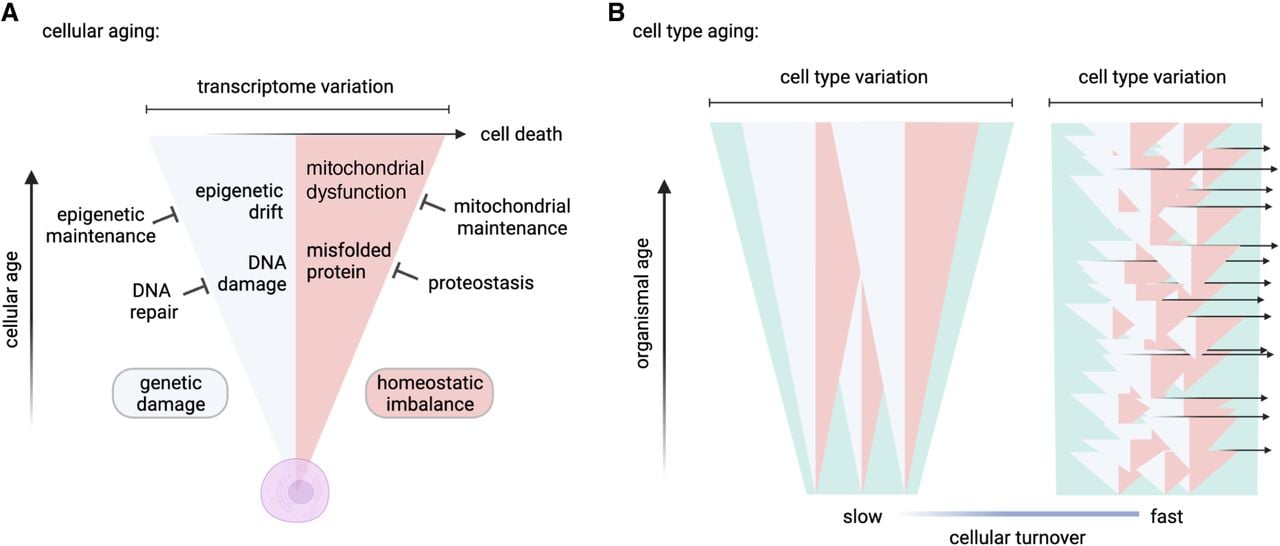
As cells age, we might expect deleterious changes in the transcriptome owing to genetic damage, including somatic mutation and epigenetic drift (Bahar et al. 2006; Schumacher et al. 2021), all of which are counteracted or avoided by repair and maintenance processes (Yousefzadeh et al. 2021; Gyenis et al. 2023). Changes to the transcriptome could also be caused by improper gene regulation because of loss of cellular homeostasis, and cells have evolved elaborate systems to maintain homeostasis through protein chaperones, autophagic recycling of cell components, and so forth (Santra et al. 2019; Lima et al. 2022).
In cells, all of these factors—mutation, oxidative damage, heat stress, etc.—will lead to the gradual accumulation of variation in the transcriptome with age (Fig. 5A). Increased expression of genes associated with proteostasis and mitophagy, as well as expression of proteins with relatively high stability, appear to be ways that long-lived cells mitigate this damage (Castillo-Morales et al. 2019).
Notably, we observe that the expression levels of genes associated with DNA repair are highest not in the longer-lived cells but in short-lived ones. Although this observation might seem to contradict our model, we believe it provides valuable insight. Many different factors lead to DNA mutation and damage, and mitotic division plays a major role (Tomasetti and Vogelstein 2015). Cells with little turnover are protected from this source of damage. Although fully differentiated short-lived cells have little time to accumulate DNA errors, they could inherit DNA damage passed down by the long-lived progenitor stem cells from which they are derived.
When considering these various sources of accumulated error and damage, we are not suggesting that short-lived cells are immune to their effects. In fact, it is possible that short life span goes hand in hand with a higher rate of accumulation of damage and failure, which we depict in our model by the faster rate of accumulation of variation within individual short-lived cells compared with long-lived cells (Fig. 5B). Regardless, rapid turnover and replacement of short-lived cells means that most cells in a population of short-lived cells are very young, with little damage. Thus, short-lived cells can avoid much of the cost of maintaining homeostasis and repairing damage, benefitting from simply being replaced through stem-cell division, with selection removing unfit cells through PCD (Goodell and Rando 2015). This, we suggest, explains the only modest age-related increase in transcriptome variation observed in populations of shorter-lived cells (Fig. 5B).
An evolutionary perspective
How might an individual organism benefit from cells of vastly different life expectancies, like neutrophils, which live for days (McCracken and Allen 2014), versus neurons, which live for decades? There are interesting and potentially important microevolutionary consequences of their differences.
Our results focus primarily on ways that organisms maintain functional long-lived cells, but from an evolutionary perspective, there are several ways that short-lived cells could benefit the organism. For example, organisms might counter the cost of rapid accumulation of damage within short-lived cells with the benefit of limiting major investment in cellular repair and maintenance in those cells (Fig. 5B). Moreover, organisms can facilitate processes that enhance selective removal of these cells (Krakauer and Plotkin 2002). By selectively removing unfit cells, an organism can thus ensure that this population of rapidly aging cells remains healthy, no matter what the age of the organism. Very long-lived cells, like neurons, have little opportunity for such selective removal and replacement, and they instead require investment in intrinsic cellular maintenance. From the organismal perspective, we can think of short cell life span as a strategy that minimizes damage within organs while limiting the burden of having to maintain cell function for more than a few days.
The idea that short cellular life spans lead to faster rates of aging mirrors Medawar's “mutation accumulation” theory of aging, in which older individuals carry late-acting deleterious alleles, albeit not to anyone's benefit (Medawar 1952). Following on from Medawar's model, Williams (1957) argued in his “antagonistic pleiotropy” theory that genes with beneficial effects early in life would be favored by natural selection, even if they had catastrophic effects at late ages. Any cost of these late-acting deleterious effects is deeply discounted owing to the relatively weak force of selection at late age. Although Williams was thinking about fitness effects of genes on individuals within a population, we can also apply this thinking to the evolution of cell types within organisms, which leads to another potential benefit to the organism of producing short-lived cells. If cellular products or functions are beneficial to the organism in the short term but cause problems for the cell in the long run (Labbadia and Morimoto 2015; Santra et al. 2019), a short-lived cell might be able to explore a greater range of the possible phenotypic space, further maximizing tissue function without having to face the long-term costs of those traits. In this sense, short-lived cells might benefit an organism because they are subject to fewer antagonistically pleiotropic effects.
Caveats
There are several limitations to this study worth considering. First, we restricted our analysis to postmitotic cell types to avoid the confounding effect of mitotic division such as cell self-renewal, differentiation, and senescence, as these cellular processes are associated with stem cells or progenitor cells and might be under different regulatory controls or experience different selection pressures (Seim et al. 2016; Derényi and Szöllősi 2017; Brunet et al. 2023). Whether mitotic cells show a similar pattern to the one we observe is an open question. Second, although we did not observe age-related changes in the transcriptomes of short-lived cells when comparing young and old mice, this does not prove that short-lived cells do not experience aging within their short lifetimes. In short-lived cells, the great majority of cells are consistently young, regardless of organismal age (Fig. 1). But given that we cannot distinguish young from old cells whose ages differ by just a matter of days, we lack the resolution to observe patterns of transcriptome variation that might vary with cellular age in those cell types. What is clear is that when comparing the transcriptome in a population of short-lived cells in young versus old mice, these mostly young cells show little difference in transcriptome variation across mouse age. Third, our cell demography model makes simplified assumptions about cellular dynamics, not accounting for more complex tissue architecture (Derényi and Szöllősi 2017), or clonal dynamics, which vary by tissue and might affect the rate of transcriptome variation among cells (Sandén et al. 2020; Sankaran et al. 2022). Fourth, we focused on data from the mouse because of the limited availability of cellular life span estimates and aging atlas-level single-cell transcriptome data in other species. Should data become available, future studies could evaluate the predictions of our theory across more species. Lastly, for simplicity, we assume that the rates of cell turnover do not vary with age, and yet, there is evidence that cell turnover can vary with age and may do so in a cell type–specific manner (for review, see Tower 2015). We lack information on age-related cellular turnover and so examining its effect on transcriptome variation is not currently possible. However, our results indicate that the effect of cell turnover on the aging transcriptome is clearly predicted by estimating cellular age.
Methods
Mouse transcriptome data
We used the TMS data set (accessed from https://figshare.com/articles/dataset/tms_gene_data_rv1/12827615), which provides an integration of transcriptional profiles of multiple tissues at the single-cell level across the life span of mice in both sexes from either a FACS platform (acquired by cell sorting in microtiter well plates followed by Smart-seq2 library preparation) or droplet platform (derived from microfluidic droplets) (Almanzar et al. 2020; Zhang et al. 2021). Our study focused on the TMS FACS data, as they have a higher sequencing depth, which facilitates gene expression variability analysis and has a more comprehensive coverage of tissues and cell types (Supplemental Fig. S3). In our main results, we focused on male mice from 3-mo-old and 24-mo-old age groups as these two age groups have the largest number of animal individuals and the number of cell types (Supplemental Fig. S3). We also analyzed the data from other age groups as well as from female mice to further support our findings.
We obtained fluorescence-activated cell-sorting Smart-seq2 profiles of multiple tissues from young (3-mo-old) and old (24-mo-old) male mice from the TMS database. We selected cell types with at least 40 cells in both young and old mice, which gave 62 cell types among 22 tissues, resulting in transcriptome profiles from 49,819 total cells. Some cell types are detected in multiple organs (e.g., endothelial cells), and so, together the filtered data set contained 84 combinations of tissue:cell type. To account for differences in total captured transcript counts across cells, all cells were down-sampled to have an equal number of transcripts in the two age groups for each tissue:cell type. After removing genes expressed in <10% of cells in each tissue:cell type and cells that express fewer than 100 genes, 49,539 cells remained in our data set.
Cell type–specific life span data
We obtained cell life span data, in which a cell's life span is defined as the number of days since its last division until its death, from fully differentiated postmitotic cell types in the available tissue:cell type contexts in mice (Sender and Milo 2021). Twenty-two cell types with available life span estimates in Sender and Milo 2021’s study corresponded to 39 tissue:cell type combinations in the TMS (Supplemental Table S1). All but one cell type have life span estimates based on mice, whereas an estimate for neurons in the nervous system was only available from human studies (Supplemental Table S1; Spalding et al. 2013; Sender and Milo 2021). Life span estimates in the work of Sender and Milo (2021) involved tracking radiolabeled cells over time, and fitting the data to a model to estimate cellular life span. This statistical framework may give life span estimates that exceed an animal's typical longevity (Sender and Milo 2021). Of the 39 tissue:cell type combinations in the TMS, 15 have life span estimates that were specific to both the cell type and its tissue context, such as endothelial cells in brain, heart, or muscle. The remaining seven cell types lacked context-specific estimates, and so, each shares a life span estimate across the tissues in which they were found. For example, skeletal muscle satellite cells in limbs and in the diaphragm share the same life span as estimated for skeletal muscle satellite cells (Sender and Milo 2021). It is possible that these cells do, in fact, have different life spans. Our simplifying assumption of common life span is a conservative one, as it adds error to our estimates and is likely to reduce the magnitude of any real statistical relationship.
Modeling cell population dynamics
We were able to obtain data on mean cell longevity. However, we recognize that the distribution of cellular ages in an animal of a given age will be a function both of cell turnover rates and of that individual's chronological age. Because the maximum cellular age cannot exceed the organism's age, we used a truncated exponential distribution with an upper bound set by the organismal age. The mean life span of a population of cells can be calculated as the first moment of a truncated exponential distribution.
We first transformed cellular life span of each cell type in days into a finite survival rate per day (Supplemental Methods, Equation 1). We then built a demographic model of cell population dynamics with a two-compartment tissue architecture (Supplemental Methods, Equations 2–4). Assuming constant cell population size, we derived cell type–specific turnover rate (Supplemental Methods, Equation 5), the cellular age distributions (Supplemental Methods, Equations 6, 7), and the analytical solution of the mean cellular age (Supplemental Methods, Equation 8). We plugged in various values of cell type–specific survival rates and the two mouse ages into Equation 8 of the Supplemental Methods to obtain the age increase of each cell type shown in Figure 2 (Supplemental Methods, Equation 9).
Transcriptome variation analysis
We implemented a pipeline to estimate transcriptome variation at a cell type level and to quantify the age effect on transcriptome variation using a linear mixed model framework accounting for individual mouse effect (Supplemental Fig. S4).
In estimating transcriptome variation, we need to control for the confounding effects of the mean-variance relationship on expression abundance (Eling et al. 2018). To address this issue when evaluating the variation of individual genes in young and old animals, we took a conservative strategy and restricted our analysis to genes whose expression did not change significantly with age, an approach used before in studying the effect of age on immune cells (Martinez-Jimenez et al. 2017). We used a linear model to quantify the age effect on gene expression level using the MAST package in R (Finak et al. 2015). We removed genes with a P-value < 0.1 for a given tissue:cell type and then calculated the CV at each age for each of the remaining genes.
Another critical factor to be taken into account when estimating transcriptome variation is the individual mouse effect, as cells from the same individual share common genetic and environmental backgrounds and hence are not statistically independent (Zimmerman et al. 2021). Treating cells of a given type as individual observations irrespective of their animal origin leads to pseudoreplicates in statistical analysis and has been shown to inflate a false-discovery rate and lead to spurious results in differential gene expression analysis (Squair et al. 2021). In TMS FACS data, the 3-mo age group and the 24-mo age group each contain four mouse individuals. To account for the effect of an individual mouse, we calculated CV for each gene in each individual mouse and then tested for effects of age on CV using a linear mixed model with age as a fixed effect and gene and mouse ID as random effects (lmer(log(CV2) age + (1|gene) + (1|mouse)), using the lme4 package in R (Bates et al. 2015). After model fitting, we express the change in transcriptome variability captured by this approach as the coefficient β associated with the effect of age (βage).
To analyze the level of transcriptome variability at the cellular level, we estimated the similarity in gene expression profiles among cells using two metrics: Spearman's rank correlation coefficient (Angelidis et al. 2019) and global coordination level (GCL) (Levy et al. 2020). The Spearman's rank correlation coefficient (ρ) was calculated on the expression matrices in all pairwise cell comparisons using cells from each mouse individual (15 or more cells) per tissue:cell type combination. We then used 1 – ρ as a measure of cell-level transcriptome variation for every cell pair (Supplemental Fig. S6). We took the median of 1 – ρ values within each mouse individual and then used the mean across mice of the same age group to represent cell type–level transcriptome variability measurement. We express the change in transcriptome variability captured by this measure as log2(O/Y), where the transcriptome variability in the young sample is Y and that of the old is O.
The second method, GCL, randomly splits genes into two sets and quantifies their mutual dependence via a distance correlation metric between cell samples evaluated on the two gene sets. This metric was shown to capture both linear and nonlinear gene correlations and is robust to noise in single-cell data (Vaknin et al. 2021). Highly correlated gene expression patterns among cells give a high GCL, whereas a low GCL value indicates low levels of similarity in gene expression profiles. We first measured GCL using cells from each mouse individual (15 or more cells) per tissue:cell type combination and then transformed GCL into a metric describing the lack of gene coordination, or gene discoordination (1 − GCL). Specifically, we randomly split the transcriptomes of cells from each mouse individual per tissue:cell type into halves 100 times and extracted the median value of the resulting 1 − GCL values to represent the mouse individual-specific discoordination value (Supplemental Fig. S7). We used the mean of mouse individual-specific discoordination values of the same age group to represent cell type–level transcriptome variability measurement. We express the change in transcriptome variability captured by this measure as log2(O/Y), where the transcriptome variability in the young sample is Y and that of the old is O.
Cell type–specific GO term expression
We extracted all 12,545 GO terms in the biological process (BP) category in mice via the R package org.Mm.eg.db (version 3.14.0) (https://bioconductor.org/packages/release/data/annotation/html/org.Mm.eg.db.html). Searching all these GO terms using “chaperone,” “DNA repair,” and “autophagy” as the key words, we retained 41 GO terms, 17 of which contained at least six gene members (Supplemental Fig. S11). We calculated a GO term expression for these GO terms for each cell as the proportion of expression abundance among genes in each GO term of the total expression abundance in that cell. Expression levels came from a cell × gene matrix, normalized by the total number of UMIs in each cell and scaled by 106 to counts per million (CPM). In this way, every cell has an expression for each GO term that represents the relative expression of genes in each GO term. We took the mean expressions of cells of a given cell type to represent a cell type–level GO term expression. We calculated the nonparametric correlation between cell life span and GO term expression via Kendall's τ.
Evolutionary constraint analysis
We retrieved dN/dS ratios of 14,185 one-to-one mouse–rat orthologs gene pairs (Supplemental Fig. S12) and 13,532 mouse–human orthologous gene pairs, from the Ensembl BioMart (v.99) (Supplemental Fig. S14; Yates et al. 2020). To evaluate dN/dS among cell type–specific genes, we identified cell type–specific genes using the specificity score approach (Sonawane et al. 2017).
For gene j in cell type k, the specificity score, , is calculated as
To measure dN/dS of the cell type–specific genes in each cell, we took the median dN/dS of the corresponding genes. At each specificity score cutoff, only cells that express at least 100 corresponding cell type–specific genes would be included in the analysis. To represent the evolutionary constraint for each cell type, we took the mean of the dN/dS value of all cells per cell type.
Software availability
Statistical analysis was performed in statistical software R version 4.1.3 (R Core Team 2022). All source code and analyses are available as Supplemental Code and at GitHub (https://github.com/mingwhy/cell.dynamics_aging.asynchrony).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank veterinary pathologist Dr. Kim Waggie (University of Washington) for the curation of the alignment of cells (Sender and Milo 2021) with cells in the TMS (Zhang et al. 2021). This work was supported by the National Institutes of Health (NIH) R01 AG063371 (to D.E.L.P.), NIH R61 AG078428 (to D.E.L.P.), and a Norn Group Impetus grant (to D.E.L.P.). We dedicate our manuscript to the memory of George M. Martin (1927–2022).
Author contributions: M.Y., B.R.H., and D.E.L.P. designed research. M.Y. performed research. M.Y. analyzed data. M.Y., B.R.H., and D.E.L.P. wrote the paper.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278144.123.
References
- ↵Almanzar N, Antony J, Baghel AS, Bakerman I, Bansal I, Barres BA, Beachy PA, Berdnik D, Bilen B, Brownfield D, 2020. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583: 590–595. 10.1038/s41586-020-2496-1
- ↵Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, Tsitsiridis G, Ansari M, Graf E, Strom TM, 2019. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun 10: 963. 10.1038/s41467-019-08831-9
- ↵Bae T, Tomasini L, Mariani J, Zhou B, Roychowdhury T, Franjic D, Pletikos M, Pattni R, Chen BJ, Venturini E, 2018. Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science 359: 550–555. 10.1126/science.aan8690
- ↵Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dollé MET, Calder RB, Chisholm GB, Pollock BH, Klein CA, 2006. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441: 1011–1014. 10.1038/nature04844
- ↵Bates D, Mächler M, Bolker BM, Walker SC. 2015. Fitting linear mixed-effects models using lme4. J Stat Softw 67: 1–48. 10.18637/jss.v067.i01
- ↵Bozukova M, Nikopoulou C, Kleinenkuhnen N, Grbavac D, Goetsch K, Tessarz P. 2022. Aging is associated with increased chromatin accessibility and reduced polymerase pausing in liver. Mol Syst Biol 18: e11002. 10.15252/msb.202211002
- ↵Brunet A, Goodell MA, Rando TA. 2023. Ageing and rejuvenation of tissue stem cells and their niches. Nat Rev Mol Cell Biol 24: 45–62. 10.1038/s41580-022-00510-w
- ↵Buckley MT, Sun ED, George BM, Liu L, Schaum N, Xu L, Reyes JM, Goodell MA, Weissman IL, Wyss-Coray T, 2023. Cell-type-specific aging clocks to quantify aging and rejuvenation in neurogenic regions of the brain. Nat Aging 3: 121–137. 10.1038/s43587-022-00335-4
- ↵Castillo-Morales A, Monzón-Sandoval J, Urrutia AO, Gutiérrez H. 2019. Postmitotic cell longevity-associated genes: a transcriptional signature of postmitotic maintenance in neural tissues. Neurobiol Aging 74: 147–160. 10.1016/j.neurobiolaging.2018.10.015
- ↵Cohen AA, Ferrucci L, Fülöp T, Gravel D, Hao N, Kriete A, Levine ME, Lipsitz LA, Olde Rikkert MGM, Rutenberg A. 2022. A complex systems approach to aging biology. Nat Aging 2: 580–591. 10.1038/s43587-022-00252-6
- ↵Debès C, Papadakis A, Grönke S, Karalay Ö, Tain LS, Mizi A, Nakamura S, Hahn O, Weigelt C, Josipovic N, 2023. Ageing-associated changes in transcriptional elongation influence longevity. Nature 616: 814–821. 10.1038/s41586-023-05922-y
- ↵Derényi I, Szöllősi GJ. 2017. Hierarchical tissue organization as a general mechanism to limit the accumulation of somatic mutations. Nat Commun 8: 14545. 10.1038/ncomms14545
- ↵Drummond DA, Wilke CO. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134: 341–352. 10.1016/j.cell.2008.05.042
- ↵Eling N, Richard AC, Richardson S, Marioni JC, Vallejos CA. 2018. Correcting the mean-variance dependency for differential variability testing using single-cell RNA sequencing data. Cell Syst 7: 284–294.e12. 10.1016/j.cels.2018.06.011
- ↵Elliott MR, Ravichandran KS. 2016. The dynamics of apoptotic cell clearance. Dev Cell 38: 147–160. 10.1016/j.devcel.2016.06.029
- ↵Enge M, Arda HE, Mignardi M, Beausang J, Bottino R, Kim SK, Quake SR. 2017. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell 171: 321–330.e14. 10.1016/j.cell.2017.09.004
- ↵Fabre MA, de Almeida JG, Fiorillo E, Mitchell E, Damaskou A, Rak J, Orrù V, Marongiu M, Chapman MS, Vijayabaskar MS, 2022. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature 606: 335–342. 10.1038/s41586-022-04785-z
- ↵Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, 2015. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16: 278. 10.1186/s13059-015-0844-5
- ↵Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, 2005. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci 102: 10604–10609. 10.1073/pnas.0500398102
- ↵Gladyshev VN, Kritchevsky SB, Clarke SG, Cuervo AM, Fiehn O, de Magalhães JP, Mau T, Maes M, Moritz RL, Niedernhofer LJ, 2021. Molecular damage in aging. Nat Aging 1: 1096–1106. 10.1038/s43587-021-00150-3
- ↵Goodell MA, Rando TA. 2015. Stem cells and healthy aging. Science 350: 1199–1204.
- ↵Gorbunova V, Seluanov A, Zhang Z, Gladyshev VN, Vijg J. 2014. Comparative genetics of longevity and cancer: insights from long-lived rodents. Nat Rev Genet 15: 531–540. 10.1038/nrg3728
- ↵Gyenis A, Chang J, Demmers JJPG, Bruens ST, Barnhoorn S, Brandt RMC, Baar MP, Raseta M, Derks KWJ, Hoeijmakers JHJ, 2023. Genome-wide RNA polymerase stalling shapes the transcriptome during aging. Nat Genet 55: 268–279. 10.1038/s41588-022-01279-6
- ↵Hayward AD, Moorad J, Regan CE, Berenos C, Pilkington JG, Pemberton JM, Nussey DH. 2015. Asynchrony of senescence among phenotypic traits in a wild mammal population. Exp Gerontol 71: 56–68. 10.1016/j.exger.2015.08.003
- ↵Hernando-Herraez I, Evano B, Stubbs T, Commere PH, Jan Bonder M, Clark S, Andrews S, Tajbakhsh S, Reik W. 2019. Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nat Commun 10: 4361. 10.1038/s41467-019-12293-4
- ↵Holmes OW. 1891. The deacon's masterpiece or, the wonderful ‘one-hoss shay’: a logical story. Houghton, Mifflin, Boston.
- ↵Ibañez-Solé O, Ascensión AM, Araúzo-Bravo MJ, Izeta A. 2022. Lack of evidence for increased transcriptional noise in aged tissues. eLife 11: e80380. 10.7554/eLife.80380
- ↵Ibañez-Solé O, Barrio I, Izeta A. 2023. Age or lifestyle-induced accumulation of genotoxicity is associated with a length-dependent decrease in gene expression. iScience 26: 106368. 10.1016/j.isci.2023.106368
- ↵Jones DL, Rando TA. 2011. Emerging models and paradigms for stem cell ageing. Nat Cell Biol 13: 506–512. 10.1038/ncb0511-506
- ↵Khan SS, Singer BD, Vaughan DE. 2017. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell 16: 624–633. 10.1111/acel.12601
- ↵Kimmel JC, Penland L, Rubinstein ND, Hendrickson DG, Kelley DR, Rosenthal AZ. 2019. Murine single-cell RNA-seq reveals cell-identity- and tissue-specific trajectories of aging. Genome Res 29: 2088–2103. 10.1101/gr.253880.119
- ↵Kirkwood TBL. 2005. Understanding the odd science of aging. Cell 120: 437–447. 10.1016/j.cell.2005.01.027
- ↵Krakauer DC, Plotkin JB. 2002. Redundancy, antiredundancy, and the robustness of genomes. Proc Natl Acad Sci 99: 1405–1409. 10.1073/pnas.032668599
- ↵Kryazhimskiy S, Plotkin JB. 2008. The population genetics of dN/dS. PLoS Genet 4: e1000304. 10.1371/journal.pgen.1000304
- ↵Labbadia J, Morimoto RI. 2015. The biology of proteostasis in aging and disease. Annu Rev Biochem 84: 435–464. 10.1146/annurev-biochem-060614-033955
- ↵Levy O, Amit G, Vaknin D, Snir T, Efroni S, Castaldi P, Liu YY, Cohen HY, Bashan A. 2020. Age-related loss of gene-to-gene transcriptional coordination among single cells. Nat Metab 2: 1305–1315. 10.1038/s42255-020-00304-4
- ↵Li R, Di L, Li J, Fan W, Liu Y, Guo W, Liu W, Liu L, Li Q, Chen L, 2021. A body map of somatic mutagenesis in morphologically normal human tissues. Nature 597: 398–403. 10.1038/s41586-021-03836-1
- ↵Lima T, Li TY, Mottis A, Auwerx J. 2022. Pleiotropic effects of mitochondria in aging. Nat Aging 2: 199–213. 10.1038/s43587-022-00191-2
- ↵López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. 2013. The hallmarks of aging. Cell 153: 1194–1217. 10.1016/j.cell.2013.05.039
- ↵Luo OJ, Lei W, Zhu G, Ren Z, Xu Y, Xiao C, Zhang H, Cai J, Luo Z, Gao L, 2022. Multidimensional single-cell analysis of human peripheral blood reveals characteristic features of the immune system landscape in aging and frailty. Nat Aging 2: 348–364. 10.1038/s43587-022-00198-9
- ↵Ma S, Sun S, Geng L, Song M, Wang W, Ye Y, Ji Q, Zou Z, Wang S, He X, 2020. Caloric restriction reprograms the single-cell transcriptional landscape of Rattus norvegicus aging. Cell 180: 984–1001.e22. 10.1016/j.cell.2020.02.008
- ↵Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, 2018. Somatic mutant clones colonize the human esophagus with age. Science 362: 911–917. 10.1126/science.aau3879
- ↵Martinez-Jimenez CP, Eling N, Chen HC, Vallejos CA, Kolodziejczyk AA, Connor F, Stojic L, Rayner TF, Stubbington MJT, Teichmann SA, 2017. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 355: 1433–1436. 10.1126/science.aah4115
- ↵Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, 2019. Single-cell transcriptomic analysis of Alzheimer's disease. Nature 570: 332–337. 10.1038/s41586-019-1195-2
- ↵McCracken JM, Allen LAH. 2014. Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death 7: 15–23. 10.4137/JCD.S11038
- ↵Medawar PB. 1952. An unsolved problem of biology. HK Lewis & Co, London.
- ↵Menon M, Mohammadi S, Davila-Velderrain J, Goods BA, Cadwell TD, Xing Y, Stemmer-Rachamimov A, Shalek AK, Love JC, Kellis M, 2019. Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat Commun 10: 4902. 10.1038/s41467-019-12780-8
- ↵Mitchell E, Spencer Chapman M, Williams N, Dawson KJ, Mende N, Calderbank EF, Jung H, Mitchell T, Coorens THH, Spencer DH, 2022. Clonal dynamics of haematopoiesis across the human lifespan. Nature 606: 343–350. 10.1038/s41586-022-04786-y
- ↵Moorad JA, Ravindran S. 2022. Natural selection and the evolution of asynchronous aging. American Naturalist 199: 551–563. 10.1086/718589
- ↵Moore L, Cagan A, Coorens THH, Neville MDC, Sanghvi R, Sanders MA, Oliver TRW, Leongamornlert D, Ellis P, Noorani A, 2021. The mutational landscape of human somatic and germline cells. Nature 597: 381–386. 10.1038/s41586-021-03822-7
- ↵Ori A, Toyama BH, Harris MS, Bock T, Iskar M, Bork P, Ingolia NT, Hetzer MW, Beck M. 2015. Integrated transcriptome and proteome analyses reveal organ-specific proteome deterioration in old rats. Cell Syst 1: 224–237. 10.1016/j.cels.2015.08.012
- ↵Perez-Gomez A, Buxbaum JN, Petrascheck M. 2020. The aging transcriptome: read between the lines. Curr Opin Neurobiol 63: 170–175. 10.1016/j.conb.2020.05.001
- ↵Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D'Orazi G. 2016. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging 8: 603–619. 10.18632/aging.100934
- ↵Promislow DEL. 1991. Senescence in natural populations of mammals: a comparative study. Evolution 45: 1869–1887. 10.2307/2409837
- ↵Rando TA, Wyss-Coray T. 2021. Asynchronous, contagious and digital aging. Nat Aging 1: 29–35. 10.1038/s43587-020-00015-1
- ↵R Core Team. 2022. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/.
- ↵Richardson RB, Allan DS, Le Y. 2014. Greater organ involution in highly proliferative tissues associated with the early onset and acceleration of ageing in humans. Exp Gerontol 55: 80–91. 10.1016/j.exger.2014.03.015
- ↵Salzer MC, Lafzi A, Berenguer-Llergo A, Youssif C, Castellanos A, Solanas G, Peixoto FO, Stephan-Otto Attolini C, Prats N, Aguilera M, 2018. Identity noise and adipogenic traits characterize dermal fibroblast aging. Cell 175: 1575–1590.e22. 10.1016/j.cell.2018.10.012
- ↵Sandén C, Lilljebjörn H, Orsmark Pietras C, Henningsson R, Saba KH, Landberg N, Thorsson H, von Palffy S, Peña-Martinez P, Högberg C, 2020. Clonal competition within complex evolutionary hierarchies shapes AML over time. Nat Commun 11: 579. 10.1038/s41467-019-14106-0
- ↵Sankaran VG, Weissman JS, Zon LI. 2022. Cellular barcoding to decipher clonal dynamics in disease. Science 378: eabm5874. 10.1126/science.abm5874
- ↵Santra M, Dill KA, De Graff AMR. 2019. Proteostasis collapse is a driver of cell aging and death. Proc Natl Acad Sci 116: 22173–22178. 10.1073/pnas.1906592116
- ↵Schaum N, Lehallier B, Hahn O, Pálovics R, Hosseinzadeh S, Lee SE, Sit R, Lee DP, Losada PM, Zardeneta ME, 2020. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583: 596–602. 10.1038/s41586-020-2499-y
- ↵Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ. 2021. The central role of DNA damage in the ageing process. Nature 592: 695–703. 10.1038/s41586-021-03307-7
- ↵Seim I, Ma S, Gladyshev VN. 2016. Gene expression signatures of human cell and tissue longevity. NPJ Aging Mech Dis 2: 16014. 10.1038/npjamd.2016.14
- ↵Sender R, Milo R. 2021. The distribution of cellular turnover in the human body. Nat Med 27: 45–48. 10.1038/s41591-020-01182-9
- ↵Sikosek T, Chan HS. 2014. Biophysics of protein evolution and evolutionary protein biophysics. J R Soc Interface 11: 20140419. 10.1098/rsif.2014.0419
- ↵Sonawane AR, Platig J, Fagny M, Chen CY, Paulson JN, Lopes-Ramos CM, DeMeo DL, Quackenbush J, Glass K, Kuijjer ML. 2017. Understanding tissue-specific gene regulation. Cell Rep 21: 1077–1088. 10.1016/j.celrep.2017.10.001
- ↵Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Boström E, Westerlund I, Vial C, Buchholz BA, 2013. Dynamics of hippocampal neurogenesis in adult humans. Cell 153: 1219–1227. 10.1016/j.cell.2013.05.002
- ↵Squair JW, Gautier M, Kathe C, Anderson MA, James ND, Hutson TH, Hudelle R, Qaiser T, Matson KJE, Barraud Q, 2021. Confronting false discoveries in single-cell differential expression. Nat Commun 12: 5692. 10.1038/s41467-021-25960-2
- ↵Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. 2012. Quantitative analysis of Hsp90-client interactions reveals principles of substrate recognition. Cell 150: 987–1001. 10.1016/j.cell.2012.06.047
- ↵Tomasetti C, Vogelstein B. 2015. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347: 78–81. 10.1126/science.1260825
- ↵Tower J. 2015. Programmed cell death in aging. Ageing Res Rev 23: 90–100. 10.1016/j.arr.2015.04.002
- ↵Vaknin D, Amit G, Bashan A. 2021. A top-down measure of gene-to-gene coordination for analyzing cell-to-cell variability. Sci Rep 11: 11075–11075. 10.1038/s41598-021-90353-w
- ↵Wang S, Zheng Y, Li J, Yu Y, Zhang W, Song M, Liu Z, Min Z, Hu H, Jing Y, 2020. Single-cell transcriptomic atlas of primate ovarian aging. Cell 180: 585–600.e19. 10.1016/j.cell.2020.01.009
- ↵Warren LA, Rossi DJ, Schiebinger GR, Weissman IL, Kim SK, Quake SR. 2007. Transcriptional instability is not a universal attribute of aging. Aging Cell 6: 775–782. 10.1111/j.1474-9726.2007.00337.x
- ↵Williams GC. 1957. Pleiotropy, natural selection, and the evolution of senescence. Evolution 11: 398–411. 10.2307/2406060
- ↵Ximerakis M, Lipnick SL, Innes BT, Simmons SK, Adiconis X, Dionne D, Mayweather BA, Nguyen L, Niziolek Z, Ozek C, 2019. Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci 22: 1696–1708. 10.1038/s41593-019-0491-3
- ↵Yamamoto R, Chung R, Vazquez JM, Sheng H, Steinberg PL, Ioannidis NM, Sudmant PH. 2022. Tissue-specific impacts of aging and genetics on gene expression patterns in humans. Nat Commun 13: 5803. 10.1038/s41467-022-33509-0
- ↵Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, 2020. Ensembl 2020. Nucleic Acids Res 48: 682–688. 10.1093/nar/gkz966
- ↵Yermanos A, Neumeier D, Sandu I, Borsa M, Waindok AC, Merkler D, Oxenius A, Reddy ST. 2021. Single-cell immune repertoire and transcriptome sequencing reveals that clonally expanded and transcriptionally distinct lymphocytes populate the aged central nervous system in mice. Proc Biol Sci 288: 20202793. 10.1098/rspb.2020.2793
- ↵Yousefzadeh M, Henpita C, Vyas R, Soto-Palma C, Robbins P, Niedernhofer L. 2021. DNA damage—how and why we age? eLife 10: e62852. 10.7554/eLife.62852
- ↵Zhang MJ, Pisco AO, Darmanis S, Zou J. 2021. Mouse aging cell atlas analysis reveals global and cell type-specific aging signatures. eLife 10: e62293. 10.7554/eLife.62293
- ↵Zimmerman KD, Espeland MA, Langefeld CD. 2021. A practical solution to pseudoreplication bias in single-cell studies. Nat Commun 12: 738. 10.1038/s41467-021-21038-1