
Abstract
The successful discovery of novel biological therapeutics by selection requires highly diverse libraries of candidate sequences that contain a high proportion of desirable candidates. Here we propose the use of computationally designed factorizable libraries made of concatenated segment libraries as a method of creating large libraries that meet an objective function at low cost. We show that factorizable libraries can be designed efficiently by representing objective functions that describe sequence optimality as an inner product of feature vectors, which we use to design an optimization method we call stochastically annealed product spaces (SAPS). We then use this approach to design diverse and efficient libraries of antibody CDR-H3 sequences with various optimized characteristics.
Biologics, such as monoclonal antibody therapeutics, are commonly discovered by expressing diverse candidate libraries in phage or yeast display systems followed by multiple rounds of affinity selection against a biological target of interest (Lu et al. 2020). Many other protein engineering tasks, including the discovery of adeno-associated vectors (AAV) for gene therapy (Wang et al. 2019; Bryant et al. 2021), T cell receptor (TCR) design (Holler et al. 2000; Li et al. 2005; Smith et al. 2015), and aptamer screening (Keefe et al. 2010; Maier and Levy 2016), can also be framed as selection from a library of candidates.
Therapeutic discovery by selection requires candidate libraries that are both highly diverse and enriched in desirable candidates in order to isolate even a single lead for further preclinical development. We define library diversity as the number of sequences in a library that are sufficiently different from each other to produce different therapeutics. We define library efficiency as the proportion of library sequences with favorable therapeutic, delivery, and manufacturing properties (Ponsel et al. 2011). State-of-the-art antibody candidate libraries are typically random libraries that are produced via mutagenesis or sequential synthesis using trinucleotide (codon) mixes (Shim 2015). Random libraries are highly diverse and thus prioritize exploration of the possible sequence space. However, random libraries can be inefficient and contain sequences with undesirable qualities such as polyspecificity, hydrophobicity, and instability. Such properties have negative consequences that range from manufacturing difficulty to dangerous clinical side-effects (Raybould et al. 2019). In recent years, rationally designed libraries have been proposed in which each library sequence is individually specified and synthesized, which we refer to here as enumerated libraries. Sequences in enumerated libraries can have superior developability profiles, resulting in efficient libraries (Liu et al. 2020; Shin et al. 2021). However, enumerated libraries can be both computationally intensive to design and costly to manufacture. At present, the cost of enumerated libraries is prohibitive for library complexities above ∼106 sequences, and thus, enumerated libraries are typically not sufficiently diverse for de novo therapeutic discovery (Hughes and Ellington 2017).
Here, we will introduce factorizable libraries in which each library member is a combination of designed segments in which each segment library is much less complex than a resulting factorizable library. To create a factorizable library, segment libraries are combined, inspired in part by the natural use of recombination to create highly diverse natural libraries of antibodies and T cell receptors. Importantly, this factorization allows for the synthesis of segment libraries at a low cost that when combined result in a high-complexity library with desirable properties. We develop a method for designing factorizable libraries efficiently, which we call stochastically annealed product spaces (SAPS). SAPS iteratively improves segment libraries with respect to an objective function that evaluates the full-length factorizable library that results from the concatenation of the segment libraries. After the synthesis of segment library DNA oligonucleotides, segment libraries can be joined with a combination of overhang and blunt end ligation similar to Golden Gate assembly to create a factorizable library (Engler et al. 2009; Chockalingam et al. 2020).
In this work, we aim to formalize the problem of designing a factorizable library, develop a computational method for the problem, establish theoretical and empirical properties pertaining to the problem and method, and show the utility of our method by designing and analyzing factorizable libraries that randomize the third complementarity determining region of antibody heavy chains (CDR-H3s).
Methods
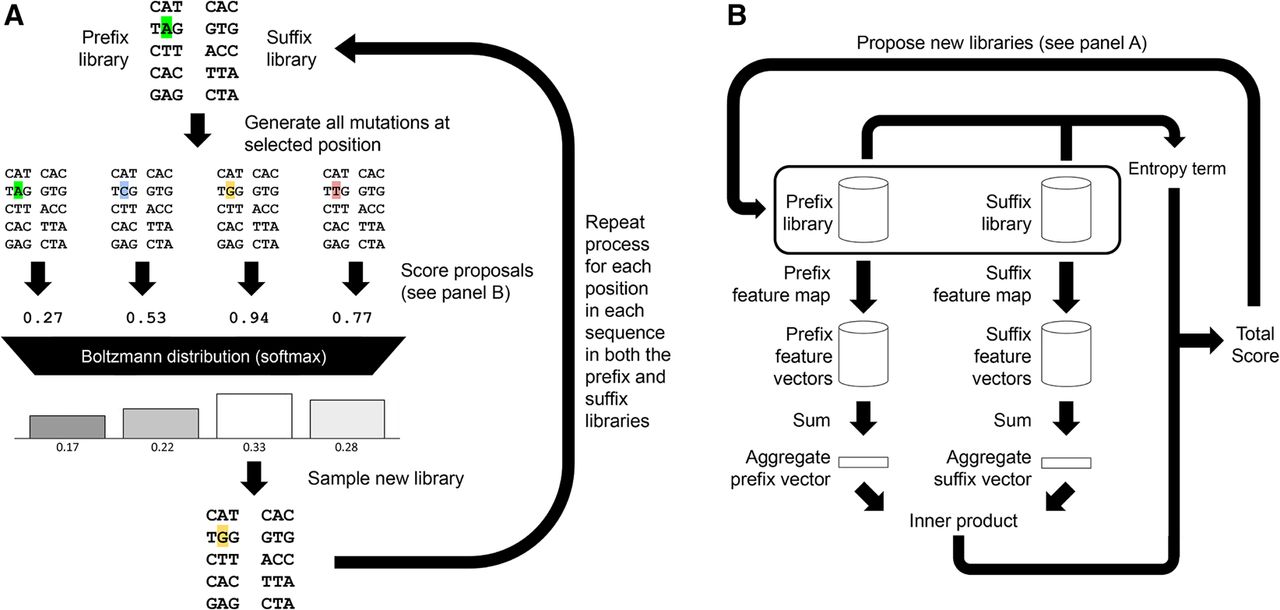
We introduce a method for designing segment libraries that when joined create a factorizable library that is optimized for a specific performance objective. The performance objective consists of an efficiency term and a diversity (entropy) term. Here we focus on factorizable libraries that consist of two segments: a prefix segment and a suffix segment. However, this work generalizes to factorizable libraries with an arbitrary number of segments. We first formalize the task of designing a factorizable library and show that this task is computationally intractable. We then propose the use of simulated annealing as a heuristic meta-algorithm (Fig. 1A). Simulated annealing requires multiple evaluations of the objective function to generate distributions of proposed updates, which is prohibitively expensive if computing the objective function requires scoring every sequence in the product space. We speed up this evaluation by expressing our objective function as the inner product of features of the prefix segment and features of the suffix segment. This objective formulation allows us to rapidly evaluate the objective by keeping a running sum of these features. For added sequence diversity, we also include an entropy term in the optimization objective (Fig. 1B). The result of library design is the set of sequences in the prefix library and the set of sequences in the suffix library. Joining all prefix segments with all suffix segments combinatorially yields a factorizable library that realizes the provided objective.
Factorizable library optimization and evaluation. (A) Optimization is achieved through iterative stochastic updates. An update step is performed by selecting a position in a sequence in one of the libraries and generating all possible mutations for that position. The mutated libraries are then scored, and then a Boltzmann distribution over the libraries is generated using the negated scores as energy values. The update is then sampled from the distribution. A full update sweep performs this for all positions in all sequences in both segment libraries. Multiple sweeps are performed, and the temperature of the Boltzmann distribution is lowered over time. For simplicity, the figure depicts this optimization on small DNA libraries. In our application to antibody CDR-H3 library design, we operate on longer length protein sequences composed of amino acids. (B) Evaluation of the objective function of a factorizable library is performed by mapping all the sequences in its prefix and suffix libraries to feature spaces. The feature vectors are then aggregated, and an inner product is taken between them, which by the distributive property produces the total score for the whole factorizable library. A position-based entropy term is evaluated to quantify the diversity of sequences in the library, and a weighted sum of the two is then used to guide optimization.
Preliminaries
Fix Σ to be a finite alphabet, and fix L to be a positive integer. Let ΣL denote the set of strings of length L whose symbols are in Σ. We also fix two positive integers, Lp and Ls, such that Lp + Ls = L. We will call ΣL the sequence space, ΣLp the prefix space, and ΣLs the suffix space.
For a pair of strings x and y, we use x⨁y to denote their concatenation. If X and Y are instead sets of strings, then their concatenation X⨁Y is defined to be {x⨁y | x ∈ X, y ∈ Y}.
We use ⟨x, y⟩ to denote the inner product of x and y, and we will use x·y to explicitly denote a dot product if x and y belong to some Euclidean space. For any set X, we will use 2X to denote the power set of X.
We will state various theorems as we explain our method. The proofs are omitted from the main text for improved flow and can be found in section B of the Supplemental Methods.
Designing a factorizable library is computationally difficult
Our goal is to design a factorizable library of sequences that is efficient. To formalize this, suppose we are given some scoring function f : ΣL → ℝ that characterizes the utility of a single sequence, and we are given a pair of positive integers, np and ns. Then the goal is to find a set S⊆ΣL such that the total score ∑s∈S f(s) is maximized, subject to the constraint that S is the concatenation of Sp⊆ΣLp and Ss⊆ΣLs such that |Sp| = np and |Ss| = ns.
The factorizable constraint adds an additional layer of complexity to the problem, making it much more difficult than library design without the constraint. Normally, if the objective is reasonably easy to compute, then we can always find an optimal library by enumerating and scoring all possible sequences and picking the best ones. However, once factorizability is enforced, then under standard complexity assumptions, it is impossible to reliably design a library that is appreciably better than a randomly selected library, even when sufficient resources to evaluate the entire sequence landscape are provided. A precise statement of this result is provided in Theorem 3 located in section A of the Supplemental Methods.
SAPS generates factorizable libraries
We introduce SAPS for designing segment libraries that when combinatorially joined optimize a provided objective for the resulting factorizable library. Because producing an efficient factorizable library is intractable, we rely on heuristic methods that employ Gibbs sampling and simulated annealing.
We initially start with randomly generated prefix and suffix libraries with sizes that are chosen to achieve a desired complexity for the factorizable library. We then perform a Gibbs sampling sweep over all positions in all sequences in both libraries. At each position, we generate all possible substitutions at that position and stochastically accept one of them such that the probability of selecting that update is proportional to the exponential of the score of the concatenated library resulting from that update divided by a temperature parameter. The temperature parameter is then lowered over time according to some schedule so that the stationary distribution approaches an indicator function over the optimal library. This process is illustrated in Figure 1.
The runtime of this procedure given a library scoring oracle scales linearly with the number of positions we need to sweep over, which scales linearly with the lengths of the sequences. Because of the factorizable nature of the library, the number of positions we need to sweep over scales with O(np + ns) as opposed to the number of sequences in the factorizable library (O(npns)), which makes designing large libraries tractable when np + ns, is significantly smaller than npns.
The reverse kernel trick allows for efficient library score evaluation
A major bottleneck of the simulated annealing approach is evaluating the total score of the factorizable library. Modifying a single sequence in the prefix or suffix libraries can affect the scores of O(max(np,ns)) sequences in the factorizable library, so potentially that many reevaluations of f(.) would be needed.
Suppose we find a pair of functions φp : ΣLp → V and φs : ΣLs → V for some inner product space V such that f(x⨁y) = ⟨φp(x), φs(y)⟩. Then by the distributive property of inner products,
For any function f(.), we can find a pair of prefix and suffix feature maps that map prefix and suffix sequences to finite dimensional Euclidean spaces and have the desired properties. Theorem 1 shows that the loss of accuracy through computing f(.) using the dot product of prefix and suffix feature maps is bounded by the dimensionality of the Euclidean spaces employed for V.
Theorem 1. Let m ≤ |Σmin(Lp,Ls)| be a positive integer. Then it is possible to find for every f : ΣL → ℝ a φp : ΣLp → ℝm′ and a φs : ΣLs → ℝm′ such that
Although Theorem 1 does guarantee the existence of feature maps φp and φs, it also implies that the dimension of those feature spaces can get very large for certain functions. The reverse kernel trick is unhelpful if the dimension becomes so large that adding vectors and evaluating dot products are inefficient. It is therefore essential to find feature maps with codomains where sums and inner products can be efficiently evaluated. To be more explicit, we must be able to find compact representations of sequences for our optimization to be useful.
A special case in which small feature spaces can be found is when our scoring function can be described with an Ising model or, more generally, a Potts model. Because the only interactions modeled are between pairs of sequence positions, an encoding of size O(|Σ|L) will suffice express all the interactions. Additional details can be found in section C.1 of the Supplemental Methods.
More generally, we will rely on deep learning to produce these feature maps. We can parametrize φp(.) and φs(.) with a pair of neural networks, and we can make a f(.) predictor by taking the dot product of the outputs of these networks. We can then train this predictor using standard deep learning methodology. Specific details on the architecture and training can be found in sections C.3 and C.4 of the Supplemental Methods. Although there is no mathematical guarantee that a compact representation can be learned using this approach and, in the worst case, it is possible for feature vectors to scale exponentially with sequence length, we show empirically that this can be performed (see the section Inner Products of Small Feature Vectors Produce CDR-H3 Enrichment Predictions That Are Comparable to State of the Art). Further, deep neural networks are widely used for learning lower-dimensional representations of sequential data in natural language processing.
Sequences of different lengths can be represented using padding
To allow for sequences of differing lengths in the factorizable library, we introduce a padding character in Σ. Note that the padding character is not a gap character, so it should not appear in the middle of a sequence. We avoid malformed sequences by explicitly specifying where padding characters occur and leaving these positions static throughout the optimization procedure. We can then ensure that the padding characters only occur at the beginning of sequences (for prefixes) or at the end of sequences (for suffixes), so sequences will always be well formed. A desirable outcome of this approach is that the factorizable library will contain a diversity of sequence lengths. If sequences are instead sampled without prespecified lengths, for instance, by allowing the padding characters to be proposed and assigning libraries with malformed strings to have a score of −∞, the tendency will be to sample longer sequences because the number of longer sequences vastly outnumbers the number of shorter sequences.
If sequences have fixed lengths, duplicates can be eliminated if we enforce that each sequence in the prefix library is unique and each sequence in the suffix library is unique. This is insufficient if sequences can vary in length. We may, for instance, propose a prefix library that contains “AC” and “*A” and a suffix library that contains “D*” and “CD,” where “*” is the padding symbol. Concatenating the two libraries then generates “ACD” twice.
Note that there is no sequence we can remove from the prefix or suffix library without reducing the number of unique sequences in the concatenated library, so it is unclear whether preventing such proposals is desirable. Therefore, we choose to ignore this case and treat the differently padded duplicates in the concatenated library as distinct sequences for library generation purposes. The impact of such duplicates is low: If Δ is the difference between the longest and the shortest sequence in the prefix or suffix library, then the number of truly unique sequences in the library can drop by no more than a factor of Δ because each sequence can be shifted at most Δ times.
Sequence diversity can be explicitly enforced with an entropy term
Beyond attaining efficiency, we would also like a factorizable library to explore a diverse range of sequences. This is partially achieved through the sheer number of unique sequences that factorizable libraries can contain, but this does not necessarily preclude excessive exploitation of certain parts of the sequence space. For example, given a seed peptide of length 20, it is possible to generate a library of size 109 consisting of nothing but mutants that have mutated at most five residues away from the initial seed sequence.
To ensure library diversity, we add an entropy term to our optimization objective. Let Sp[i]⊆Sp be the subset of the prefix library of sequences of length i, and let Ss[i]⊆Ss be the subset of the suffix library of sequences of length i. Let 1sj= c be one if sj = c and zero otherwise, where sj denotes the jth letter of s. The entropy objective H can then be given by the following formula, where for simplicity we define an empty summation, 0/0, and 0ln(0) to all evaluate to zero for the purposes of this formula:
Theorem 2. Let d∈ΣL, and let m < L. Let Sp⨁Ss⊆ΣL be a library in which every sequence can be obtained through at most m substitutions of d. Then
The parameter λ trades off efficiency versus diversity in library design
We introduce the hyperparameter λ that controls the trade-off between the entropy term and the objective function score term that represents efficiency. Using λ, we can write down the SAPS objective function:
Changes in this quantity induced by changing a single symbol in a single sequence in the prefix library roughly scales with |Ss|. Similarly, changes induced by changing a single symbol in a single sequence in the suffix library roughly scales with |Sp|. An informal derivation can be found in section C.2 of the Supplemental Methods.
Therefore, to ensure that proposal distributions remain diverse even for large libraries, we divide the score by those quantities before generating the proposal distributions.
Results
Evaluation of SAPS library design performance on simulated data sets
As a benchmark for the ability of SAPS to produce high-scoring factorizable libraries, we first chose a simple design domain. We randomly generate nonlattice Ising models in which coupling energies between any two spins at any two positions are drawn independently and uniformly at random from {−1,0,1}, and in which the spins at each position has an independent energy also drawn independently and uniformly at random from {−1,0,1} (for additional details, see section D.1 of the Supplemental Methods). The Ising models we generate operate on sequences of length 14, 16, 18, and 20, and we generate 100 models for each length. From a biological perspective, these Ising models can be viewed as analogous to modeling the energy of a peptide threaded through some designed structure under a two-residue hydrophobic-polar scheme evaluated with something akin to a pairwise distance-based potential.
We then generate factorizable libraries of sizes 400, 1600, 3600, 6400, 10,000, and 14,400 for each model that optimize for the highest average energy using our proposed approach, in which the length of the prefixes and suffixes are exactly half the length of the total sequence. We drop the entropy term for evaluating these factorizable libraries to focus on how the optimization performs.
We benchmarked SAPS against five other approaches, which we sketch out here. Additional details may be found in section D.2 of the Supplemental Methods. The first approach is the greedy approach, in which, instead of stochastically sampling changes to the libraries, we deterministically pick the optimal change, and iterate until convergence. The second approach is to use an expectation heuristic, in which we determine the average value of a prefix or suffix sequence by averaging over all sequences with that prefix or suffix and then select the top prefixes and suffixes. The third approach is to use a max heuristic, in which we take the prefixes and suffixes of the top scoring sequences. The fourth and fifth approaches are to take the proposals generated by the second and third approaches, respectively, as a seed proposal and then apply a greedy refinement to it using the greedy approach. Save for the greedy approach, all the approaches we benchmark against would produce the optimal result if there were no couplings between positions in the prefix or suffix.
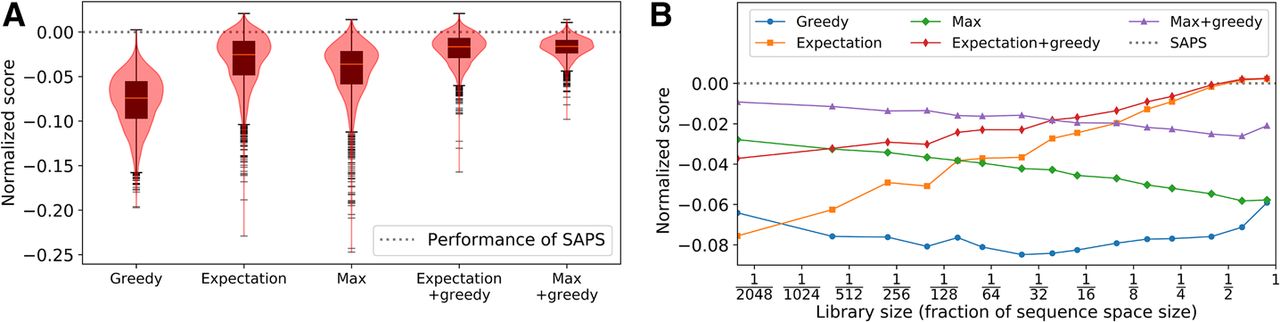
We find SAPS tends to outperform all other methods. Out of 2400 trials over varying conditions, SAPS achieves the best scoring sequence 2099 times (87%). The greedy approach was able to outperform SAPS 1 of 2400 times, the expectation heuristic 199 of 2400 times (231 times after applying greedy refinement), and the max heuristic 30 of 2400 times (76 times after applying greedy refinement). The scores achieved by different methods in relation to SAPS are given in Figure 2A. When the library size occupies a significant fraction of the sequence space (around one-fourth to one-half), we find that the expectation heuristic tends to outperform SAPS (Fig. 2B). This is likely owing to how the prefix heuristic becomes a better approximation to the true utility when the suffix library covers a significant chunk of the suffix space and vice versa. In practice, there is rarely a reason to design a library in this regime. If we are allowed such library sizes, it would be more sensible to simply use the entire sequence space as a library.
Simulated annealing outperforms other approaches on random Ising models. We evaluate our method against five benchmarks on 400 randomly generated Ising models that operate on varying sequence sizes. For each model, six libraries of varying sizes are generated for a total of 2400 experimental conditions. To normalize over the varying conditions, we scale the scores such that the expected score of a library of the desired size generated uniformly at random is one unit apart from the maximum possible score of any arbitrary library of the desired size. The scores are then shifted such that SAPS achieves a score of zero, which is indicated by the dotted gray line in the figures. We do this because the variability of the optimums between different Ising models is much larger than the difference between the approaches, making it difficult to see that SAPS outperforms the other methods in most instances. (A) Distribution of normalized scores for the approaches we benchmark against using a box plot in conjunction with a violin plot. (B) Mean of the normalized score for each approach as a function of library size.
Inner products of small feature vectors produce CDR-H3 enrichment predictions that are comparable to the state of the art
We next sought to show that we can learn prefix and suffix feature maps that map to small spaces and can successfully predict affinity to biological targets. We use high-throughput sequencing data from three rounds of affinity selection on a random synthetic antibody library that uses position-specific codon frequencies to improve library efficiency against multiple specific targets, including the antibodies ranibizumab, omalizumab, trastuzumab, and etanercept (for additional details, see section E.1 of the Supplemental Methods). We also select against a baculovirus extract (BV), which is a mixture of viral DNA, proteins, and lipids commonly used to assay polyspecificity of antibody therapeutics in late-stage preclinical development with a smaller set of candidates (Jain et al. 2017). After each round of selection, antibodies expressed via phage display are isolated and sequenced, hence providing per-round read counts for unique CDR-H3 sequences, which we use to generate training and held-out test sets. For comparison to experimentally generated random synthetic antibody libraries, we collected high-throughput sequencing data using the same random synthetic antibody library panned against no target for a single round (FW_kappa), which is the also the same seed library used for previously published phage panning experiments (Liu et al. 2020). The majority of sequenced CDR-H3s (∼99%) in this library range in length from eight to 20 residues, so we filter out sequences outside this range. We use the log10-enrichment from round 2 (R2) to round 3 (R3) (log10(R3/R2)) as a measure of affinity and regression label for this sequence domained prediction task. log10(R3/R2) enrichment was found to have a better signal-to-noise ratio compared with the inclusion of round 1 (R1) reads, and previous work has shown that this measure correlates well with ground-truth CDRH3 affinity measured by individual binding assays, such as enzyme-linked immunosorbent assay (ELISA) (Liu et al. 2020).
We use deep learning to find prefix and suffix feature maps that map to low-dimensional feature spaces (see sections C.3 and C.4 of the Supplemental Methods). Specifically, we use a deep convolutional neural network with residual connections (i.e., a ResNet) to map prefixes and suffixes to 16-dimensional vectors. We chose this architecture as it is commonly used and attains performance comparable to prior work (see the end of this section), and additional details on the model architecture may be found in section C.3 of the Supplemental Methods. The inner product between a prefix and suffix feature vector then gives the predicted log10(R3/R2) enrichment. We will refer to this entire pipeline as a reverse kernel model.
It is possible for the reverse kernel model to appear to perform well even if it fails to capture the nonlinear interactions between the prefix and the suffix positions of a sequence if those interactions are sufficiently negligible. To control for this, we train a pair of ResNets, where one predicts a score on the prefix and one predicts a score on the suffix. The scores are then added to produce the overall prediction. We will refer to this pipeline as the independent model. We use this as a baseline to evaluate how well the reverse kernel model captures the nonlinear interactions.
Finally, we also trained a ResNet with no restrictions on its functional form, which we will refer to as the unrestricted model. Unlike the reverse kernel model, it is able to share information between the prefix and suffix in any of its layers, which allows it to capture a much richer set of interactions between the prefix and suffix. As a consequence, using the unrestricted model for factorizable library design would be intractable. However, it provides a point of comparison for how much the constraint of expressing the reverse kernel model as an inner product of feature vectors impacts performance. Furthermore, because we expect this model to produce the most accurate approximation of the true underlying sequence landscape, we will use this model to evaluate the libraries we produce.
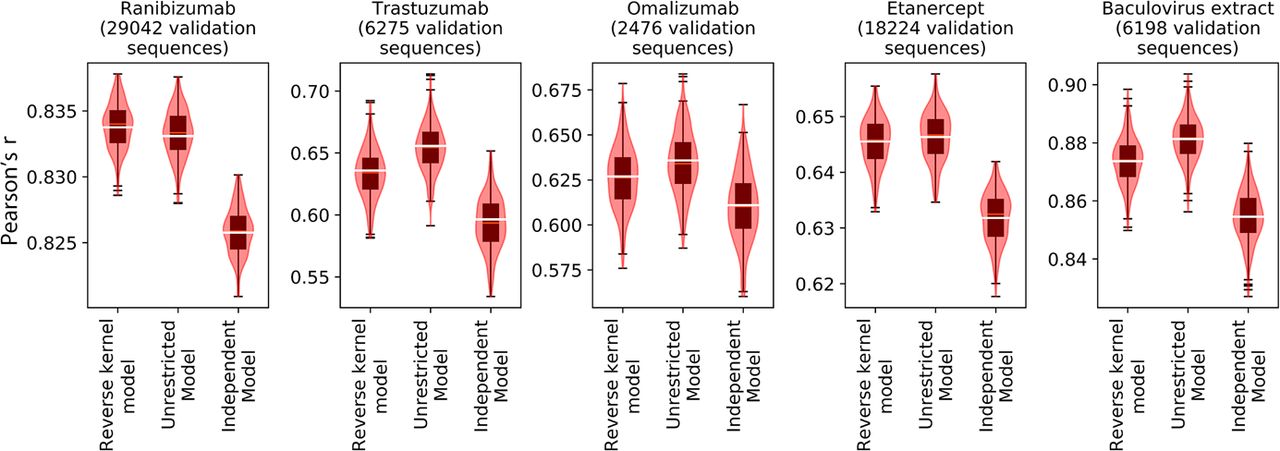
We compare the performance of our models by computing the Pearson's r correlation between the predicted and observed log10(R3/R2) enrichment on held out validation sets. The results are presented in Figure 3, where we see that generally the unrestricted model does indeed perform the best (with the one exception of ranibizumab), suggesting that it does provides the best approximation of the sequence landscape. We also see that the reverse kernel model outperforms the independent model, which shows that it is able to combine prefix and suffix properties in a nonlinear way to produce better generalizations. Our reverse kernel models attain Pearson's r values of 0.83, 0.64, 0.63, 0.65, and 0.87 on validation sets for ranibizumab, trastuzumab, omalizumab, etanercept, and BV, respectively, which is comparable to the values that were reported in prior work, which were 0.79, 0.65, and 0.64 for ranibizumab, trastuzumab, and etanercept, respectively (Liu et al. 2020).
Reverse kernel models outperform independent models and approach unrestricted models for predicting antibody enrichment. We compare the performance of our reverse kernel models with that of our unrestricted models and independent models on validation antibody enrichment data. The Pearson's r values on the validation set are indicated with a white bar in the above plots, and we use a box plot in conjunction with a violin plot to show the uncertainty as measured using 250 bootstrap samples of the validation data set.
SAPS produces diverse factorizable libraries with optimized affinity for specific targets
To generate the factorizable libraries, we take the reverse kernel models we trained and performed the SAPS procedure for 500 sweeps, decreasing the temperature by a factor of 1.1 every five rounds. For the purposes of SAPS, the values output by the reverse kernel models were divided by their standard deviation, which was estimated with 100,000 randomly generated sequences following the same length distribution as the factorizable library, with residues generated uniformly and independently.
We generated pairs of prefix and suffix libraries for each target that each contain 35,000 sequences of length four to 10. When combined, they produce factorizable libraries containing over 109 designed sequences that are optimized for binding to ranibizumab, omalizumab, trastuzumab, and etanercept (for additional details, see section E.2 of the Supplemental Methods). We will refer to these libraries as ranibizumab(+), omalizumab(+), trastuzumab(+), and etanercept(+), respectively. The diversity hyperparameter λ used for designing these libraries was chosen by observing its effects on smaller libraries (see section E.3 and Supplemental Figure S1 in the Supplemental Methods). We report chosen hyperparameters for each generated library in Supplemental Table S1 in the Supplemental Methods. Generally, we recommend using hyperparameters between 0.1 and 0.3 depending on whether a user's priority is exploration or exploitation of the sequence space.
We find that our libraries are highly diverse in comparison to FW_kappa, the experimentally randomized synthetic antibody library created with per-position frequencies used in the previously described phage display and affinity selection experiments. The expected Levenshtein distances between a pair of randomly selected sequences of length 12 within our libraries are around two edits further than the expected Levenshtein distances between a pair of randomly selected sequences of length 12 within FW_kappa and are around only one edit closer than the expected Levenshtein distances between a pair of uniformly random sequences of length 12. Although FW_kappa serves as an approximation of the diversity of a random library, we note that it may contain a biased sequence distribution owing to the round of no-target panning and other experimental constraints before high-throughput sequencing, similar to other proposed antibody CDR-H3 libraries (Kelly et al. 2018). The full distributions are presented in Figure 4A. Next, we show that diversity is similar in the prefix and suffix region for each factorizable library and is not derived from one side of the CDR-H3 sequence alone in Supplemental Figure S2 of the Supplemental Methods.
Factorizable library optimization and evaluation. (A) Histograms of the pairwise Levenshtein distance between pairs of length 12 sequences in each library and a uniformly random library. The mean distance, μ, is reported. (B) The sequence optimality (efficiency) of generated libraries compared with that of FW_kappa by scoring the sequences of FW_kappa and 1 million uniformly random samples from the designed libraries with the corresponding unrestricted model. Score distributions are reported as boxplots laid over violin plots for FW_kappa (blue) against each designed library (red). (C) Joint plot shows predicted enrichment of FW_kappa (red), equally weighted libraries (blue), ranibizumab weighted libraries (green), and BV weighted libraries (yellow) by the ranibizumab unrestricted model on the x-axis and the BV unrestricted model on the y-axis. The mean scores for FW_kappa are indicated by the dotted lines. Panels D–F show sequence logos (Schneider and Stephens 1990) for enriched nonspecific motifs in the ranibizumab weighted library over the BV weighted library discovered by STREME. (D) RRY motif (P-value = 4.8 × 10−2315). (E) VVT motif (P-value = 4.2 × 10−38). (F) WWH motif (P-value = 9.8 × 10−21).
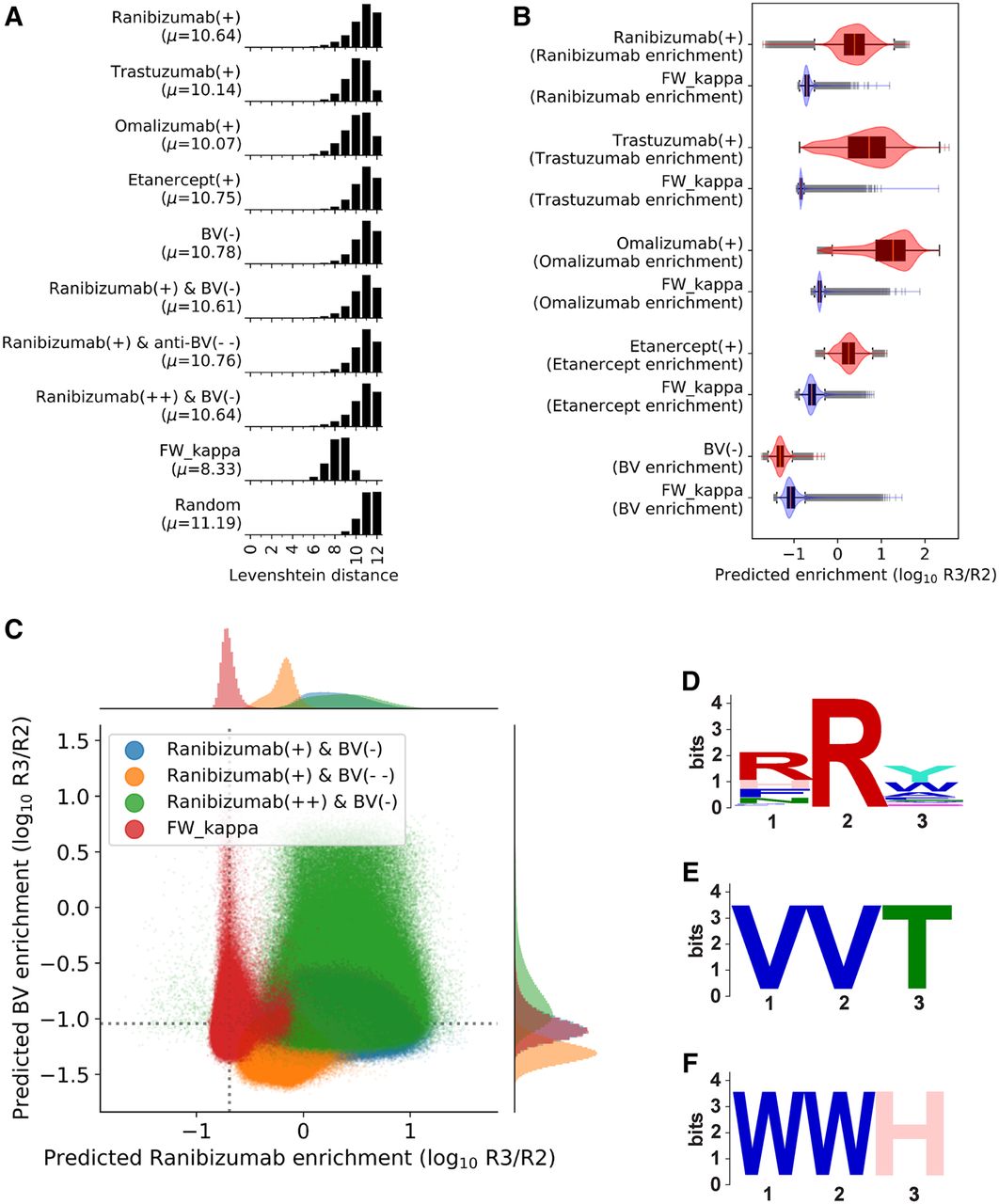
We also find that our libraries score significantly better on enrichment for the targets they were optimized for in comparison to FW_kappa. The target-specific enrichments were estimated by running the corresponding unrestricted model. The distributions of the enrichment scores are presented in Figure 4B. Together, these results suggest that SAPS is able to generate factorizable libraries that are both diverse and efficient. Details on pairwise distance computation and model scoring are provided in section F.1 of the Supplemental Methods.
Flexible SAPS parameters allow for the design of limited polyspecificity factorizable libraries
Next, we show that SAPS can produce factorizable libraries with specified diversity and sequence optimality constraints. In this task, we focus on designing a library with limited polyspecificity using the aforementioned BV target. Generally, if an antibody binds a target like BV, it is likely a polyspecific sequence that will bind many different targets in the body, leading to diminished efficacy by fast immune clearance or even clinically dangerous off-target effects (Hötzel et al. 2012).
We used SAPS to design a factorizable library with low affinity for BV, which we will refer to as BV(−). This is performed by negating the output of the reverse kernel model and is intended to reduce the number of polyspecific members included in the libraries. We show that this library has high diversity but lower affinity for BV than the FW_kappa library, indicating a better polyspecificity profile in Figure 4, A and B. Further, we conduct basic motif enrichment analysis and show that hypothesized nonspecific motifs, as theorized by Kelly et al. (2018), are less prominent in the designed factorizable library (see section F.3 and Supplemental Figure S3 in the Supplemental Methods). Further, we conduct STREME motif enrichment analysis of FW_kappa over BV(−) and find that known nonspecific motifs are significantly enriched (see Supplemental Table S2 in the Supplemental Methods).
Next, we used SAPS to design factorizable libraries that were optimized for both increased affinity to ranibizumab and for decreased affinity to BV. To achieve this, we use a weighted sum of the ranibizumab reverse kernel model output and the negated BV reverse kernel model output to score sequences for our optimization objective. We generate three factorizable libraries: one in which both outputs are unscaled (“ranibizumab(+) & BV(−)” or “equally weighted”), one in which ranibizumab is scaled by 0.1 whereas BV is unscaled (“ranibizumab(+) & BV(− −)” or “BV weighted”), and one in which BV is scaled by 0.1 and ranibizumab is unscaled (“ranibizumab(++) & BV(−)” or “ranibizumab weighted”). We evaluate efficiency by scoring each library with unrestricted models predicting ranibizumab affinity and BV affinity, showing that the libraries designed have the intended score distribution (Fig. 4C). Further, we conducted motif enrichment in the ranibizumab weighted library (ranibizumab(++) & BV(−)) over the BV weighted library (ranibizumab(+) & BV(− −)) using STREME (for details, see section F.3 of the Supplemental Methods; Bailey 2021) and observed significant enrichment of known nonspecific motifs such as valine (VV), tryptophan (WW), and arginine (RR) pair enrichment. These results are presented in Figure 4, D through F and show that SAPS is flexible for highly specific design parameters. This also illustrates a functional use case for rationally designed factorizable libraries, as it is common in antibody discovery to spend significant resources on both finding a sequence with optimized affinity for a target and rejecting sequences with high polyspecificity (Ponsel et al. 2011).
Discussion
We introduce SAPS, a computational method to design factorizable libraries, a library synthesis strategy that enables the rational design of highly diverse libraries with optimized properties at moderate cost. As a result of their skewed focus on exploration and exploitation of the sequence space, respectively, random libraries and enumerated libraries are not ideal for the discovery of novel therapeutics, especially for difficult targets. Further, it is currently not feasible to create libraries with 109 enumerated members by direct synthesis. With rationally designed factorizable libraries, smaller segment libraries are synthesized at low cost and combined to produce a full-length library that is combinatorially larger. By guiding the design of the segment libraries, factorizable giga-libraries can contain a higher proportion of optimized sequences for use in therapeutic selection experiments, increasing the probability of discovering novel therapeutics targeting difficult biological and disease targets. We show SAPS by designing factorizable antibody CDR-H3 sequence libraries against various targets. We note that SAPS-designed factorizable libraries can be used for any discovery task that can benefit from the direct synthesis of diverse and functionally efficient sequencing libraries, such as TCR libraries (Holler et al. 2000; Li et al. 2005; Smith et al. 2015), AAV capsid libraries (Wang et al. 2019; Bryant et al. 2021), DNA/RNA libraries such as aptamers (Keefe et al. 2010; Maier and Levy 2016), and protein design to an objective, among other examples.
We also note that independently designed factorizable libraries can be synthesized, ligated, and subsequently mixed to form a single integrated factorizable library that integrates the objective functions of each of the underlying factorizable libraries. This method allows dependencies between segments to be captured in each component factorizable library.
We have shown that reverse kernel models can reliably recapitulate sequence–function relationships as measured by experimental affinity selection. We show that these models can be used as scoring functions for SAPS to generate factorizable libraries that, upon combination, contain 109 or more members. We show that these factorizable libraries explore the sequence space by computing the pairwise edit distances between sequences in these giga-libraries, showing their superior diversity compared with an experimentally generated random library. By scoring generated sequences using validated unrestricted models, we show that designed factorizable libraries are more efficient than random libraries and reflect intended objectives for given tasks and exploit the sequence landscape. Finally, we show that our method flexibly allows for fine-tuning of design parameters such as the overall edit distance between sequences and trade-offs between multiple desired sequence properties.
Software availability
Custom scripts and training data are provided in the Supplemental Code and Supplemental Data, respectively. They are also available at GitHub (https://github.com/gifford-lab/FactorizableLibrary).
Competing interest statement
G.H., C.B., and S.E. are employees of Novartis. The remaining authors declare no competing interests.
Acknowledgments
This work was funded by National Institutes of Health grant R01 CA218094 and a gift from Schmidt Futures to D.K.G., and the experimental work was funded by Novartis.
Author contributions: Conceptualization, methodology, software, data curation, formal analysis, visualization, writing the original draft, and review editing were by Z.D. and S.D.S. Investigation, resources, and data curation were by C.B. and G.H. Investigation, resources, data curation, review editing, supervision, and project administration were by S.E. Conceptualization, methodology, validation, writing the original draft, review editing, supervision, project administration, and funding acquisition were by D.K.G.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276593.122.
References
- ↵Bailey TL. 2021. STREME: accurate and versatile sequence motif discovery. Bioinformatics 37: 2834–2840. 10.1093/bioinformatics/btab203
- ↵Bryant DH, Bashir A, Sinai S, Jain NK, Ogden PJ, Riley PF, Church GM, Colwell LJ, Kelsic ED. 2021. Deep diversification of an AAV capsid protein by machine learning. Nat Biotechnol 39: 691–696. 10.1038/s41587-020-00793-4
- ↵Chockalingam K, Peng Z, Vuong CN, Berghman LR, Chen Z. 2020. Golden gate assembly with a bi-directional promoter (GBid): a simple, scalable method for phage display Fab library creation. Sci Rep 10: 2888. 10.1038/s41598-020-59745-2
- ↵Engler C, Gruetzner R, Kandzia R, Marillonnet S. 2009. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One 4: e5553. 10.1371/journal.pone.0005553
- ↵Holler PD, Holman PO, Shusta EV, O'Herrin S, Wittrup KD, Kranz DM. 2000. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc Natl Acad Sci 97: 5387–5392. 10.1073/pnas.080078297
- ↵Hötzel I, Theil F-P, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, 2012. A strategy for risk mitigation of antibodies with fast clearance. MAbs 4: 753–760. 10.4161/mabs.22189
- ↵Hughes RA, Ellington AD. 2017. Synthetic DNA synthesis and assembly: putting the synthetic in synthetic biology. Cold Spring Harb Perspect Biol 9: a023812. 10.1101/cshperspect.a023812
- ↵Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, 2017. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci 114: 944–949. 10.1073/pnas.1616408114
- ↵Keefe AD, Pai S, Ellington A. 2010. Aptamers as therapeutics. Nat Rev Drug Discov 9: 537–550. 10.1038/nrd3141
- ↵Kelly RL, Le D, Zhao J, Wittrup KD. 2018. Reduction of nonspecificity motifs in synthetic antibody libraries. J Mol Biol 430: 119–130. 10.1016/j.jmb.2017.11.008
- ↵Li Y, Moysey R, Molloy PE, Vuidepot A-L, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, 2005. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol 23: 349–354. 10.1038/nbt1070
- ↵Liu G, Zeng H, Mueller J, Carter B, Wang Z, Schilz J, Horny G, Birnbaum ME, Ewert S, Gifford DK, 2020. Antibody complementarity determining region design using high-capacity machine learning. Bioinformatics 36: 2126–2133. 10.1093/bioinformatics/btz895
- ↵Lu R-M, Hwang Y-C, Liu I-J, Lee C-C, Tsai H-Z, Li H-J, Wu H-C. 2020. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci 27: 1. 10.1186/s12929-019-0592-z
- ↵Maier KE, Levy M. 2016. From selection hits to clinical leads: progress in aptamer discovery. Mol Ther Methods Clin Dev 3: 16014. 10.1038/mtm.2016.14
- ↵Ponsel D, Neugebauer J, Ladetzki-Baehs K, Tissot K. 2011. High affinity, developability and functional size: the holy grail of combinatorial antibody library generation. Molecules 16: 3675–3700. 10.3390/molecules16053675
- ↵Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, Bujotzek A, Shi J, Deane CM. 2019. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci 116: 4025–4030. 10.1073/pnas.1810576116
- ↵Schneider TD, Stephens RM. 1990. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18: 6097–6100. 10.1093/nar/18.20.6097
- ↵Shim H. 2015. Synthetic approach to the generation of antibody diversity. BMB Rep 48: 489–494. 10.5483/BMBRep.2015.48.9.120
- ↵Shin J-E, Riesselman AJ, Kollasch AW, McMahon C, Simon E, Sander C, Manglik A, Kruse AC, Marks DS. 2021. Protein design and variant prediction using autoregressive generative models. Nat Commun 12: 2403. 10.1038/s41467-021-22732-w
- ↵Smith SN, Harris DT, Kranz DM. 2015. T cell receptor engineering and analysis using the yeast display platform. Methods Mol Biol 1319: 95–141. 10.1007/978-1-4939-2748-7_6
- ↵Wang D, Tai PWL, Gao G. 2019. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov 18: 358–378. 10.1038/s41573-019-0012-9