
Native RNA sequencing in fission yeast reveals frequent alternative splicing isoforms
- José Carlos Montañés1,
- Marta Huertas1,
- Simone G. Moro1,
- William R. Blevins1,4,
- Mercè Carmona2,
- José Ayté2,
- Elena Hidalgo2 and
- M. Mar Albà1,3
- 1Evolutionary Genomics Group, Research Program on Biomedical Informatics, Hospital del Mar Medical Research Institute (IMIM) and Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain;
- 2Oxidative Stress and Cell Cycle Group, Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain;
- 3Catalan Institution for Research and Advanced Studies (ICREA), 08010 Barcelona, Spain
Abstract
The unicellular yeast Schizosaccharomyces pombe (fission yeast) retains many of the splicing features observed in humans and is thus an excellent model to study the basic mechanisms of splicing. Nearly half the genes contain introns, but the impact of alternative splicing in gene regulation and proteome diversification remains largely unexplored. Here we leverage Oxford Nanopore Technologies native RNA sequencing (dRNA), as well as ribosome profiling data, to uncover the full range of polyadenylated transcripts and translated open reading frames. We identify 332 alternative isoforms affecting the coding sequences of 262 different genes, 97 of which occur at frequencies >20%, indicating that functional alternative splicing in S. pombe is more prevalent than previously suspected. Intron retention events make ∼80% of the cases; these events may be involved in the regulation of gene expression and, in some cases, generate novel protein isoforms, as supported by ribosome profiling data in 18 of the intron retention isoforms. One example is the rpl22 gene, in which intron retention is associated with the translation of a protein of only 13 amino acids. We also find that lowly expressed transcripts tend to have longer poly(A) tails than highly expressed transcripts, highlighting an interdependence between poly(A) tail length and transcript expression level. Finally, we discover 214 novel transcripts that are not annotated, including 158 antisense transcripts, some of which also show translation evidence. The methodologies described in this work open new opportunities to study the regulation of splicing in a simple eukaryotic model.
The unicellular eukaryote Schizosaccharomyces pombe, with around 7000 genes, is an ideal model to study cellular processes that are conserved across eukaryotes (Wood et al. 2002; Kim et al. 2010). About 43% of the genes contain introns, often multiple ones. Thus, in contrast to other unicellular yeast species such as Saccharomyces cerevisiae, which has a very limited number of introns, S. pombe can also be used to study the molecular basis of splicing. Previous studies using intron lariat sequencing, short-read RNA sequencing (RNA-seq), and Iso-Seq have uncovered many low-frequency alternative isoforms (Bitton et al. 2015a; Stepankiw et al. 2015; Kuang et al. 2017), suggesting that splicing fidelity in the species is relatively low.
Little is known about the impact of alternative splicing (AS) in generating functional isoforms and expanding the proteome of S. pombe. One of the few well-studied cases is rem1, encoding a cyclin required for meiosis. The expression of the Rem1 protein is regulated at the level of splicing; the retention of an intron ensures that no protein is produced before the start of meiosis (Malapeira et al. 2005; Moldón et al. 2008). At the same time, the intron retention (IR) isoform results in a 17-kDa protein with a role in recombination in the premeiotic S phase. Other possible examples of functional AS events are three exon skipping (ES) transcripts that have been reported to be conserved between S. pombe and humans (Awan et al. 2013). A complete catalog of AS isoforms occurring at high frequencies, together with the putative encoded proteins, is still missing.
Here we use native RNA-seq (Garalde et al. 2018; Workman et al. 2019), in combination with ribosome profiling (Ingolia et al. 2009; Brar and Weissman 2015), to uncover the complete transcriptome and translatome of S. pombe. Oxford Nanopore Technologies (ONT) direct RNA (dRNA) sequencing (dRNA-seq) offers several important advantages over previous RNA-seq approaches: (1) It provides an unbiased snapshot of the native polyadenylated RNAs in the cell; (2) there is no need to assemble the transcripts using reads that are much shorter than the RNA molecule; (3) it is highly quantitative, as each sequence corresponds to a single RNA molecule; and (4) it is very sensitive because millions of reads can be generated per experiment. dRNA-seq has been successfully used to discover new gene isoforms in Homo sapiens (Workman et al. 2019), Arabidopsis thaliana (Zhang et al. 2020), and Caenorhabditis (Li et al. 2020; Roach et al. 2020). Additionally, unlike Nanopore cDNA sequencing, dRNA provides information on the orientation of the transcript, which is essential to be able to detect new antisense transcripts. As we have recently shown in S. cerevisiae, antisense transcripts can originate rapidly during evolution, providing new functionalities (Blevins et al. 2021). No dRNA-seq of S. pombe has yet been produced, limiting our knowledge on the complexity of the transcriptome of this model eukaryotic species.
Nanopore dRNA-seq starts from the 3′-end of the molecule, capturing the full-length poly(A) tail of each RNA. This enables the investigation of poly(A) tail variation among different transcripts and individual mRNA molecules (Workman et al. 2019). Poly(A) tail length is the result of polyadenylation and deadenylation processes and has been related to transcript stability and translatability (Dreyfus and Régnier 2002). Poly(A) tail shortening can initiate mRNA degradation in the cytoplasm (Parker and Song 2004). In humans, it has been shown that poly(A) polymerase activity can result in the decay of nuclear noncoding RNAs (ncRNAs) or mRNAs with retained introns (Bresson et al. 2015). By using dRNA, it is possible to study both AS and alterations in the poly(A) length, obtaining new clues about the possible regulatory functions of poly(A) length.
AS isoforms can encode proteins that are different from the canonical ones. These proteins remain poorly annotated because they are frequently short and partially overlap the annotated protein. A high-throughput method to test for translation activity in putative open reading frames (ORFs) is ribosome profiling (Ribo-seq) (Ingolia et al. 2009). This technique is based on the sequencing of ribosome protected RNA fragments and has single-nucleotide resolution. The 3-nucleotide (nt) periodicity of the reads has been used to discover novel translated ORFs in long ncRNA and 5′ untranslated regions (5′ UTRs) (Ingolia et al. 2009; Duncan and Mata 2014; Ji et al. 2015; Ruiz-Orera and Albà 2019b), as well as in alternative transcript isoforms (Reixachs-Solé et al. 2019). Here we use Ribo-seq data to investigate the hallmarks of translation of alternative protein isoforms, as well as to identify translated ncRNAs and novel transcripts. Our aim is to exploit Nanopore dRNA data in conjunction with Ribo-seq to uncover parts of the transcriptome and translatome that might have remained hidden owing to previous technical limitations.
Results
Native sequencing of poly(A)+ RNAs in S. pombe
We extracted total RNA from S. pombe cells growing at log-phase and subsequently performed poly(A)+ selection. Then we performed dRNA-seq of the polyadenylated RNA using an ONT Gridion instrument (Garalde et al. 2018). We obtained a total of 7,297,642 dRNA-seq reads from four sequencing runs. Each of these reads corresponds to a single native poly(A)+ RNA. The average read length was ∼650 nt (for more details, see Supplemental Table S1).
Nanopore reads are remarkably long compared with other short-read sequencing technologies, and they contain more errors, which need to be corrected (Amarasinghe et al. 2020). One commonly used approach to try to decrease the proportion of errors is to select reads that pass a certain quality score (typically Q ≥ 7). However, we found that eliminating reads with Q < 7 had nearly no effect on the error rate (Fig. 1A), and thus, we did not apply this filter. Instead, we performed a correction based on Illumina reads with the program fmlrc (Wang et al. 2018), taking advantage of a previous S. pombe Illumina RNA-seq experiment performed in the same growth conditions as here (Blevins et al. 2019). The error rate decreased to about half its original values, but some regions, such as the 3′-end of transcripts, still remained largely uncorrected. For this reason, we subsequently applied TranscriptClean, which uses the genome sequence as reference to correct ONT reads (Wyman and Mortazavi 2019). The final “clean” set had an average error rate of only 1.24%, which basically corresponded to short indels.
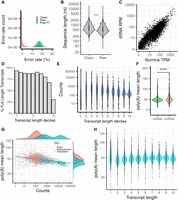
dRNA sequencing (dRNA-seq) of S. pombe. (A) Error rate distribution of raw and clean reads. Error rate is the percentage of aligned positions that contain a mismatch or indel. (Raw) The original reads, (raw P7) original reads with quality score Q ≥ 7, and (clean) corrected reads used in this work. (B) Sequence length distribution of raw and clean reads. Number of raw reads, 7,097,130; number of clean reads, 5,054,233; median value raw reads, 500; and median value clean reads, 620. Differences in the distribution are significant by a Wilcoxon test (P-value < 2.2 × 10−16). (C) Correlation transcript abundance ONT dRNA versus Illumina. The reads were mapped to the PomBase transcriptome. In the case of dRNA reads, we simply divided the number of reads by the number of million reads (reads per million [RPM]). For Illumina reads, we calculated transcripts per million (TMP), normalizing by transcript length as well as number of million reads. We selected transcripts expressed in at least one of the two data sets; transcripts with multimapping Illumina reads were removed. Number of transcripts analyzed was 4999. (D) Estimated number of transcripts with at least one full-length read with respect to transcript length. The data are shown for different transcript length deciles: (71.0–633.2), (633.2–923.4), (923.4–1175.0), (1175.0–1395.0), (1395.0–1637.0), (1637.0–1911.2), (1911.2–2227.4), (2227.4–2695.0), (2695.0–3444.6), (3444.6–15,022.0]. Number of transcripts was 6453. (E) dRNA counts with respect to transcript length. Bins are the same as in D. (F) Poly(A) tail distribution in mRNAs and ncRNAs. Poly(A) tail is estimated as the mean of the poly(A) tail length of all the reads that map to each transcript. Differences are significant according to a Wilcoxon test (P-value < 10−5). (G) Relationship between poly(A) tail length and transcript abundance. For each transcript, the average poly(A) tail length of all the reads mapping to the transcript is taken. Only mRNAs are taken into account for this calculation (n = 4995). Genes related with reproduction (GO:0000003) and translation (GO:0006412) are highlighted. Highly expressed transcripts tend to have shorter poly(A) tails. The correlation is significant (Spearman's ρ = −0.376; P-value = 9.3 × 10−168) (H) Distribution of poly(A) tail length with respect to transcript length. Bins are the same as in D. Poly(A) tail length is homogeneously distributed across different transcript length classes.
The reads were mapped to the PomBase gene annotations with minimap2 (Li 2018). The total number of mapped reads was 5,054,233. The longest mapped read was 13,899 nt long. The mapped reads had an average length of 756 nt and were significantly longer than the raw reads (Fig. 1B). We could see expression of the vast majority of the protein-coding transcripts (97.8%), as well as of a very large percentage of ncRNAs (87.8%) and smaller amounts of other RNA classes (Supplemental Fig. S1). In general, ncRNAs were expressed at much lower levels than mRNAs (average 70 dRNA reads vs. 1130 dRNA reads).
We inspected the correlation between dRNA and Illumina derived transcript abundances. Whereas each dRNA read corresponds to one native molecule, Illumina sequencing involves cDNA synthesis and PCR amplification, and the number of mapped reads needs to be normalized by length. In addition, we found that 13.41% of the Illumina reads were multimapping, increasing the uncertainty in the transcript abundance estimates. There was a high positive correlation between the abundance estimates obtained with the two technologies (Spearman's ρ = 0.849, P < 10−12) (Fig. 1C), after excluding transcripts with multimapping reads. Inclusion of the 1800 transcripts with Illumina multimapping reads caused an overestimation of transcript expression levels for some of the transcripts (Supplemental Fig. S2).
Nanopore mRNA sequencing starts from the 3′-end of the transcript and proceeds toward the 5′-end. Some of the mRNAs are sequenced to completion (full-length reads), whereas others are truncated at their 5′-end. We estimated the number of full-length reads by mapping the reads to the gene annotations and then comparing the length of each mapped read with the length of the corresponding annotated transcript. To be considered full length, the read had to be equal or longer than the annotated transcript, or in case it was shorter, the difference should be <50 nt. This accounted for the fact that the first 10–15 nt of the 5′ UTR are systematically missed with Nanopore and that the real 3′-end might also show some variation with respect to the annotated transcript. We estimated that the total number of full-length reads was 1,013,789 (20.06%). Perhaps more importantly, the vast majority of the transcripts with expression evidence had at least one full-length read (5165 out of 6453, 80.04%). As expected, the fraction of transcripts recovered as full-length reads decreased with transcript length, with the strongest effect being observed in transcripts >3.45 kb (Fig. 1D, last decile; Supplemental Fig. S3). We observed that this subset of very long transcripts also tended to be expressed at lower levels than transcripts of intermediate length (Fig. 1E; Supplemental Fig. S4). Transcripts in the first decile (<633 nt) were expressed at even lower values, but because of their short size, they were normally recovered as full-length reads.
Poly(A) tail length depends on expression level but not transcript length
Extended poly(A) tail lengths have been previously associated with increased transcript's stability and translatability (Dreyfus and Régnier 2002). We used nanopolish to measure poly(A) tail length directly from the dRNA data. The average poly(A) length was ∼50 nt, similar to that observed in humans (Workman et al. 2019). Poly(A) tails tended to be slightly shorter in mRNAs (median, 48.9 nt) than ncRNAs (median, 51 nt) (Fig. 1F). We found that poly(A) length and transcript abundance were negatively correlated (Spearman's ρ = −0.376 and P-value < 10−5) (Fig. 1G). The median number of counts for the top 10% transcripts with the shortest poly(A) tail (length <40) was 581.5, whereas the 10% of genes with the longest poly(A) tail (length >58) had a median of 131 counts. Gene Ontology (GO) term enrichment analysis indicated that genes with the shortest poly(A) tail were significantly enriched in translation-related functions, whereas those with the longest poly(A) tail were in sexual reproduction and meiosis-related functions (Supplemental Fig. S5). Consistently, these two groups also showed clear differences in their expression levels, with the translation genes being expressed at very high levels and the meiosis genes at much lower levels (Fig. 1G). No major differences in poly(A) length were observed in relation to transcript length (Fig. 1H).
Identification of hundreds of alternative transcript isoforms
We used StringTie2 to identify possible transcript isoforms supported by the dRNA reads (Kovaka et al. 2019). This program has the advantage that it does not require that the reads are full length, something which a priori cannot be determined for dRNA reads. StringTie2 yielded 5799 transcripts that showed a length distribution similar to that of annotated transcripts (Supplemental Table S2; Supplemental Fig. S6).
We identified a total of 332 alternative isoforms, in 262 different genes, that had an effect on the coding sequence. These events were novel and not annotated in PomBase. Because not all reads corresponded to full-length transcripts, a small proportion of the reads, 3.7%, mapped to different gene isoforms (7271 multimapping reads out of 189,281). The formation of alternative isoforms decreased the relative amount of the reference protein and, in some cases, could potentially lead to different protein products. We could distinguish between four types of events: intron retention (IR), intron inclusion (II), use of alternative splicing (AS) sites, and exon skipping (ES) (Fig. 2A). IR events were denoted by dRNA sequences in which the intron was not spliced out. In the case of II, a nonannotated intron was observed in a subset of the reads. AS sites implied the use of different splice site donor or acceptor signals in a subset of the mRNA molecules. Finally, ES was represented by sequences that lacked a complete exon. The most common event was IR, which represented ∼80% of all events, followed by II in ∼12% of the cases (Fig. 2B).
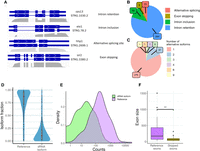
Identification of alternative isoforms using dRNA-seq. (A) Alternative splicing (AS) isoform classes. We built a transcriptome using the dRNA reads with StringTie2. We identified 332 alternative isoforms in 263 different genes. The plot shows one example of each of the four main classes of alternative isoforms detected. Diagrams of the exons of the reference and the alternative isoform as shown, together with the dRNA coverage along the gene. (B) Number of different types of splicing isoforms. Intron retention (IR) represents ∼80% of the events. (C) Number of isoforms per gene. In most cases, only one alternative isoform was detected. The most extreme case corresponds to wtf19, with two annotated isoforms and nine additional alternative isoforms detected here. (D) Relative abundance of reference and alternative isoforms for each gene. Data are for the genes containing at least one alternative isoform. The abundance is computed using the number of mapped dRNA reads; the fraction is then calculated over all isoforms containing mapped reads. Number of reference isoforms is 263 (two for wtf19); number of new alternative isoforms detected here, 332; median fraction reference isoforms, 0.873; and median fraction alternative isoforms, 0.105. (E) Abundance of reference and alternative isoforms. Number of dRNA reads mapped to reference and alternative isoforms. Numbers of isoforms as in D. (F) Skipped exons tend to be smaller than the complete set of exons in the reference annotations. Median length reference exons is 170; median length skipped exons, 63. P-value = 0.00752 Wilcoxon test.
In general, the number of alternative isoforms observed was one or at most two, although in some cases, a larger number of isoforms could be observed (Fig. 2C). The latter cases corresponded to genes of the killer meiotic drive system, a rapidly evolving family of parasitic and antidote genes (Eickbush et al. 2019). The maximum number of alternative isoforms recovered by StringTie2 was nine in wtf19. These isoforms apparently originated from different types of exon/intron inclusion and exclusion events, as well as by the use of alternative splice sites. The capacity of this gene to generate many alternative isoforms might be important for the ongoing arms race that characterizes the gene family.
To quantify the isoform expression levels, we mapped the dRNA reads to the transcriptome and used only uniquely mapped reads. This allowed us to unambiguously distinguish between the reference and alternative isoform transcripts. As expected, alternative isoforms were, in general, found at lower frequencies than the annotated isoform (Fig. 2D). Nevertheless, some nonannotated isoforms were found at very high frequencies, and there was a clear overlap between the expression levels of alternative isoforms and already annotated transcripts (Fig. 2E). As many as 92 alternative isoforms had a frequency >20%; 172 cases, >10%. For example, retention of the first intron in rpl22, a gene encoding 60S ribosomal protein 22, showed a frequency of 30% (12,003 dRNA reads vs. 28,910 for the reference mRNA). In gdt2, coding for a Golgi calcium ion transporter, the IR isoform was supported by 40% of the dRNA reads (565 vs. 858). In the case of elo1, encoding an enzyme involved in fatty acid elongation, an isoform in which the third intron was included represented 42% of all transcripts (201 dRNA reads vs. 276 for the reference mRNA). An extreme case was etp1, a gene involved in the adaptation to high concentrations of ethanol (Snowdon et al. 2009). In this case, the transcript containing the intron was the predominant one (85% of the dRNA reads, 182 out of 212). These examples were further validated by RT-PCR (Supplemental Fig. S7; Supplemental Table S7).
We observed a moderate but significant tendency for the first intron to be retained. In genes with two introns and for which only one of the introns was retained, we found 64 cases in which the first intron was retained and 36 in which the second intron was retained (P-value = 0.007 compared with 50/50, proportion test). We identified nine ES events, mostly affecting very small exons (Fig. 2F). One example was sir2, encoding a histone deacetylase. An isoform in which the fourth exon was skipped represented 17.2% of the transcripts (37 dRNA reads vs. 178 for the reference isoform).
Virtual translation of the sequences of the alternative isoforms indicated that, except in 10 cases, they resulted in proteins that were shorter than the annotated one (Fig. 3A). We sought evidence of protein translation using previously published Ribo-seq data (Duncan and Mata 2017). We focused on IR isoforms, which are the easiest to analyze, because we do not expect to have Ribo-seq reads mapping to the intron except if the intron is retained and translated. In 18 cases, we found a minimum of five Ribo-seq reads supporting the alternative protein. One remarkable example was the translation of an ORF coding for a protein of only 13 amino acids (aa) in the IR isoform of rpl22 (Fig. 3B). The 13-aa protein was supported by 311 isoform-specific Ribo-seq reads. The number of Ribo-seq reads that map to a sequence can be used as a proxy of translation level, because each mapped Ribo-seq read potentially corresponds to a translating ribosome (Ingolia et al. 2009). Examination of the Ribo-seq coverage in the isoform-specific intronic region indicated that, although the 13-aa isoform was translated at levels estimated to be around 1/10 of the canonical 117-aa-long protein, it was still among the top 10% most expressed proteins in the cell.
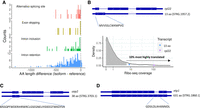
Proteome expansion by AS. (A) Difference in size between the putative alternative protein and the reference protein. Data are shown for the four main classes of AS events. In the vast majority of cases, the alternative protein would be smaller than the canonical protein. (B) Alternative 13-aa protein isoform in the rpl22 gene. The diagram shows the putative coding sequence in the alternative IR isoform. A stop codon in frame in the intronic region results in the translation of a 13-aa protein. We obtained Ribo-seq support for the 13-aa alternative protein. We also estimated the Ribo-seq coverage of the Rpl22 canonical protein (SPAC11E3.15.1) and the 13-aa alternative isoform (STRG.1957.2) using isoform-specific coding sequences. The values are compared with those for the coding sequences of all transcripts with five or more Ribo-seq reads mapped to the P-site (n = 5669). The gray area covers 90% of cases. (C) Alternative 38-aa protein isoform in the uap2 gene. IR in uap2 generates a shorter coding sequence, encoding a putative 38 aa protein. (D) Alternative 361-aa protein isoform in the etp1 gene. IR in etp1 results in a protein that is 15 aa longer than the reference one.
Another example of IR supported by ribosome profiling data was uap2, encoding the U2 snRNP-associated protein Uap2. About one-fourth of the transcripts corresponded to retention of the first intron, resulting in a putative protein of 38 aa instead of the standard 367-aa protein (Fig. 3C). Other genes displaying similar patterns were mal3, encoding a microtubule protein (des Georges et al. 2008); rpb4, encoding a RNA polymerase II complex subunit (Sakurai et al. 1999); and not11, encoding a CCR4-NOT complex subunit involved in the shortening of the poly(A) length and initiation of cytoplasmic mRNA decay (Ukleja et al. 2016). In mal3, IR was found at a frequency of 38% and resulted in a putative protein of 26 aa; in rpb4, 26.8% and a protein of 20 aa; and in not11, 60% and a protein of 55 aa. A very different case was etp1; the intron contained no stop codon in frame, and for this reason, the resulting protein was predicted to be 15 aa longer than the reference one (Fig. 3D).
In other genes, different isoforms were generated by the use of AS sites, ES, or II. One example was pat10 (SPAC18B11.08c), a gene that encodes an endoplasmic reticulum protein that is part of a chaperone complex involved in the biogenesis of proteins with multiple transmembrane domains (Chitwood and Hegde 2020). The reference transcript is composed of five exons and encodes a protein that is 95 aa long. The dRNA reads provided direct evidence of an alternative isoform arising from a downstream alternative splice site in intron 3 and skipping of exon 4. The alternative isoform had a frequency of 27% (161 dRNA reads alternative isoform vs. 431 for the reference) and resulted in a putative protein of 74 aa (Supplemental Fig. S8).
IR is associated with extended poly(A) tails
We next investigated poly(A) tail length with respect to AS events. First, for each type of event, we compared the poly(A) length of the dRNA reads in the alternative isoform and the reference isoform. Collectively, we could observe significant differences for IR events and AS site events (Fig. 4A). In both cases, poly(A) tails tended to be longer in the alternative isoform. However, when we compared the differences in poly(A) length for each gene, taking the average value for all reads mapping to the same isoform, a consistent difference was only observed for IR events (Fig. 4B).
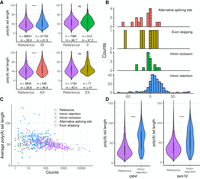
Poly(A) length in alternative transcript isoforms. (A) Distribution of poly(A) length by isoform type. We computed poly(A) length for all dRNA reads with nanopolish. Only reads with the label PASS were considered. (n) Number of reads with poly(A) length information; (m) median poly(A) length. (IR) Intron retention; (II) intron inclusion; (AS) alternative splicing site; and (ES) exon skipping. Significant differences were identified for isoform retention and AS site events compared with the corresponding reference isoforms. (***) P-value < 10−5, Wilcoxon test. (B) Difference in the average poly(A) length between the alternative and reference isoforms. Longer poly(A) lengths were consistently observed for IR isoforms. (C) Negative correlation between average poly(A) length and expression level for reference and alternative transcript isoforms. The data are only for genes in which we detected alternative isoforms. Reference refers to the annotated isoform. Spearman's ρ = −0.41, P = 3.14 × 10−23. (D) Examples poly(A) length differences between the reference and the IR isoforms. We computed the poly(A) length for the dRNA reads that correspond to each of the isoforms. The first example corresponds to RNA polymerase II subunit 4 (rpb4, SPBC337.14), with ∼27% of the reads corresponding to the IR isoform. The second example corresponds to a nucleolar RNA-binding protein also implicated in mRNA processing (tam10, SPBC14C8.19), with ∼18% of the reads mapping to the IR isoform. In both cases poly(A) length showed a significant tendency to be longer in the IR isoform. (***) P-value < 10−5; (ns) nonsignificant, Wilcoxon test.
In a recent study in S. cerevisiae, the investigators concluded that the poly(A) tail of newly transcribed transcripts is 50 adenosines long on average and is shortened in the cytoplasm to 40 adenosines on average (Tudek et al. 2021). We thus considered the possibility that longer poly(A) tails could be indicative of not fully processed transcripts still retained in the nucleus. However, isoforms with translation evidence, and thus presumably located in the cytoplasm, also had longer poly(A) tails than the reference isoforms (Supplemental Fig. S9). Thus, the data fit quite well the previously observed negative correlation between expression level and poly(A) length (Fig. 4C). Another possible explanation was that only a fraction of the molecules was being translated, whereas the rest was retained in the nucleus, resulting in overall longer poly(A) tails. Because the latter possibility cannot be tested with the current data, the question remains open.
In general, IR isoforms tended to be less abundant than the reference isoform and also tended to have longer poly(A) tails, as shown in the examples in Figure 4D. When we examined cases in which the alternative and reference isoforms had relatively similar abundances, the results varied depending on the gene. In some cases, such as not11, the poly(A) tail length of the alternative and reference isoform was not significantly different. In other cases, including mal3, rps13, and vps38, the differences were significant, although relatively small (Supplemental Fig. S10). In contrast, slm3 and red1 showed very strong and significant differences in poly(A) tail length between the alternative and reference isoforms (approximately 80 vs. 50 nt, respectively) (Supplemental Fig. S11).
Discovery of new transcribed loci
The reconstruction of the transcriptome using the dRNA reads also resulted in the discovery of 214 completely novel transcripts, whose coordinates on the genome did not show any overlap to annotated genes on the same strand. Mapping the Illumina reads to these transcripts confirmed the expression of the majority of them (168 out of 214). We found that about three-fourths of them, 158 (74%), overlapped other genes on the opposite orientation and were classified as antisense. The remaining 56 transcripts were located in regions with no other annotated features and were classified as intergenic.
The novel transcripts tended to be shorter than the annotated ones, especially the intergenic ones (Fig. 5A; Supplemental Table S2). They also tended to be expressed at lower levels than annotated transcripts, although in this case, there were no significant differences between antisense and intergenic transcripts. Because novel transcripts are lowly expressed, their detection might largely depend on the sequencing coverage. To explore this, we generated saturation curves by subsampling the number of original mapped dRNA reads. Whereas the number of known genes that could be detected reached a plateau at approximately 1.5 million reads, the number of novel transcripts showed an approximately linear relationship with the number of sequencing reads (Supplemental Fig. S12). We also investigated the fitness associated with the novel transcripts by using data from a previous saturating transposon mutagenesis experiment (Grech et al. 2019). We found that the level of constraints in the set of novel transcripts showed no significant differences to that of annotated ncRNAs and was clearly weaker than in coding sequences (Supplemental Fig. S13).
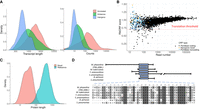
New transcripts and peptides. (A) Transcript length and gene expression for novel antisense and intergenic transcripts. Novel antisense transcripts tend to be longer than intergenic transcripts, and both classes of transcripts are shorter than already characterized ones (Wilcoxon test, P-value < 10−5). Gene expression levels are lower in novel transcripts compared with not novel ones (Wilcoxon test, P-value < 10−5). Novel antisense and intergenic transcripts show a similar expression level distribution. (TPM) Transcripts per million, quantification provided by StringTie. (B) Prediction of translated ORFs. The plot shows the RibORF score versus the number of Ribo-seq mapped reads for different classes of transcripts. ORFs with at least 10 mapped reads and a RibORF score higher than 0.7 were selected as translated. The score is based on the Ribo-seq read 3-nt periodicity and homogeneity. Nanopore novel indicates transcripts that did not overlap any annotated transcript, 12 of these transcripts contained ORFs with translation signatures. PomBase noncoding indicates annotated ncRNAs that also had translation signatures. (C) Novel translated ORFs are shorter than annotated ones. Comparison of aa length for ORFs with evidence of translation in novel transcripts and annotated coding sequences. Differences are statistically significant (Wilcoxon test, P-value < 10−5). (D) New protein identified in S. pombe. The protein labeled as STRG.2094.1 showed significant similarity to other bacteria proteins (BLASTP e-values between 10−2 and 10−7) and, in eukaryotes, only to a protein from the fungus Macrophomina phaseolina (e-value <10−9). The blue box represents the homologous region; lines at the side represent additional protein sequence.
We next used the ribosome profiling data to investigate the translation patterns in the novel transcripts. We predicted putatively translated ORFs using RibORF (Ji et al. 2015). This program produces a score based on 3-nt periodicity and homogeneity of the Ribo-seq reads along the ORF. In previous studies, we established that a RibORF score >0.7 was associated with significant translation activity (Blevins et al. 2021; Moro et al. 2021). As expected, the vast majority of the annotated coding sequences in mRNAs were classified correctly by the program (4514 out of 4560, 98.99%) (Fig. 5B). In addition, 16% of the annotated ncRNAs also contained ORFs with evidence of translation (Supplemental Table S3; Supplemental Fig. S14). These findings are in line with the translation signatures observed in a large fraction of the long ncRNAs in other biological systems (Ruiz-Orera et al. 2014; Ji et al. 2015; Chen et al. 2020).
Among the newly discovered transcripts we identified 12 cases with evidence of translation: eight antisense and four intergenic. One of the intergenic transcripts contained two putatively translated ORFs. The encoded proteins were small, with a median length of 44.5 aa for antisense transcripts and 25 aa for intergenic transcripts compared with 393 aa for canonical ORFs (Fig. 5C). The orientation of four of these novel transcripts, as well as the distance to the nearest transcription start site (<400 bp), suggested divergent transcription from a bidirectional promoter (Supplemental Table S4). One of the newly identified proteins showed significant homology with several prokaryotic proteins as well as to an uncharacterized protein from the fungus Macrophomina phaseolina (Fig. 5D). Given the sparse species distribution, it seems likely that this protein has originated by horizontal gene transfer, probably from bacteria. The rest of genes did not have homology with any other annotated protein or to a set of novel translated ORFs recently discovered in S. cerevisiae (Blevins et al. 2021). Therefore, these genes might have originated de novo in the S. pombe lineage.
Finally, we also used the dRNA-seq data to annotate 5′ and 3′ UTRs, focusing on those that were not yet annotated in PomBase (266 mRNA without a 5′ UTR and 337 mRNAs without a 3′ UTR). Nanopore data are expected to be very accurate for the 3′-end but less so for the 5′-end, as the first 10–15 nt of the mRNA are normally not recovered. Using the dRNA-based transcriptome, we annotated 105 5′ UTRs and 75 3′ UTRs that were previously missing. The median size of these sequences was 217 and 256, respectively, which was comparable to the length of the annotated ones (median 168 for 5′ UTR and 259 for 3′ UTR, respectively). Thus, dRNA provides an effective way to annotate 3′ UTRs and, to some extent, also 5′ UTRs.
Discussion
Native or direct RNA sequencing (dRNA) provides unprecedented resolution to study the transcriptome. The technique has provided new insights into the features of the transcripts expressed in several eukaryotic species, including human, C. elegans, and Arabidopsis (Workman et al. 2019; Li et al. 2020; Roach et al. 2020; Zhang et al. 2020). Here we applied dRNA to the fission yeast S. pombe, an intron-rich unicellular eukaryote that has become a very useful model to study splicing (Yan et al. 2015; Fair and Pleiss 2017). Our strategy was based on obtaining a very high coverage of the transcriptome to uncover alternative splice forms and lowly expressed transcripts. We obtained RNA sequences for 97% of the annotated mRNAs and 87% of the ncRNAs. Additionally, we characterized 332 nonannotated alternative isoforms and 214 completely new transcripts, about three-fourths of which overlapped other genes in antisense orientation. The work presents a new view of the S. pombe transcriptome because a substantial number of the newly identified AS isoforms occur at high frequencies, and some are likely to translate alternative proteins, indicating that the transcriptome is more complex and functionally diverse than previously thought.
By using dRNA-seq, it is possible to recover poly(A) tail length information from the sequencing reads. In eukaryotes, poly(A) tail lengthening is associated with increased mRNA stability and poly(A) tail shortening with mRNA degradation (Richter 2000; Dreyfus and Régnier 2002). Here we characterized poly(A) length in S. pombe and investigated if transcripts that showed diverse splicing patterns presented alterations in poly(A) length. For the complete S. pombe poly(A)+ transcriptome, we found that the average poly(A) tail is ∼50 nt, very similar to humans (Workman et al. 2019) and C. elegans (Roach et al. 2020), highlighting the high evolutionary conservation of this trend. We also found that poly(A) length tends to be shorter in mRNAs encoding highly expressed proteins, such as translation-related proteins, than in mRNAs that are expressed at low levels during exponential growth conditions, such as many meiosis-specific proteins. Similar results were recently observed using TAIL-seq data in C. elegans (Lima et al. 2017). These results are unexpected given previous experimental evidence that poly(A) tail elongation promotes transcript stability and translatability (Preiss et al. 1998; Eichhorn et al. 2016), and point to yet poorly understood mechanisms controlling poly(A) tail dynamics in different kinds of transcripts.
Upon synthesis, transcripts are polyadenylated and later exported to the cytoplasm, where they eventually decay, a process that involves poly(A) tail deadenylation (Tudek et al. 2021). We found that the poly(A) tail of the alternative isoforms was generally longer than that of the reference transcripts, especially in the case of IR events. This could be explained by the negative relationship between expression level and poly(A) tail length, but it could also be that some IR isoforms were retained in the nucleus, whereas others, including those for which we found translation evidence, are exported to the cytoplasm. To eliminate the influence of expression level, we examined the differences in poly(A) length for cases in which the reference and alternative isoform were expressed at similar levels. We found two different scenarios. In the first one, the alternative isoform had a similar poly(A) tail length to the reference isoform. One example was not11, with a median poly(A) length of 51.2 for the reference isoform and 50.1 for the IR isoform. In the case of rps13, poly(A) length was 34 for the reference isoform and 37.6 for the IR isoform, in line with the high expression of this gene. In the second scenario, there was a very clear difference in poly(A) length between the two isoforms. This was the case with slm3 and red1, with a median poly(A) tail of around 80 for the IR isoform and 50 for the fully spliced form. These results pointed to the existence of two classes of isoforms: the first class representing possibly functional alternative proteins and the second class transcripts retained in the nucleus. Nuclear retention of incompletely processed mRNAs might be an additional layer of gene expression control. For example, in mouse cells, Gabbr1 RNA remains incompletely spliced on the chromatin in embryonic stem cells, being only fully processed and exported for translation upon neuronal differentiation (Yeom et al. 2021).
Nanopore native mRNA sequencing is a powerful technique to uncover the full set of transcripts generated by different combinations of exons and introns, which cannot be accurately solved by Illumina reads. At the same time, the cost and scalability it offers is comparable to that of Illumina sequencing. We generated around 7 million dRNA reads in an organism with about 7000 genes. This high coverage and the lack of amplification biases allowed us to perform a very precise estimation of the abundance of AS transcripts. We found that about one-third of the events occurred at a very high frequency (>20%), which suggests that many of the events are functional. IR events were the most common ones, as also observed in other fungi and plants (Gonzalez-Hilarion et al. 2016; Ullah et al. 2018). IR often results in premature termination codons, which could potentially trigger nonsense mediated decay (NMD). However, studies in Cryptococcus neoformans have shown that IR is largely independent of NMD because mutants that do not express Upf proteins, which are the proteins that mediate NMD (Kervestin and Jacobson 2012), do not show IR up-regulation (Gonzalez-Hilarion et al. 2016). By using ribosome profiling data, we obtained evidence that some IR isoforms are likely to translate alternative proteins. Thus, in some cases, the same gene may be used to express multiple proteins. A previously described example is cardiolipin synthase, which is specifically produced by the intron IV retention isoform of SPAC22A12.08c mRNA (Virčíková et al. 2018). Here we found several possible examples of novel proteins generated by IR isoforms, which will need to be inspected in more detail. In addition to IR, other not yet characterized proteins can be formed by II, ES, or the use of AS sites. Taken together, the results show that AS in fission yeast is likely to play a more important role in proteome diversification than previously anticipated.
ORFs encoding proteins smaller than 100 aa are difficult to annotate because they cannot be distinguished from randomly occurring ORFs using computational means. The emergence of ribosome profiling has changed this situation because it enables the identification of ORFs with significant translation signatures regardless of the size of the ORF (Ingolia et al. 2009; Ruiz-Orera and Albà 2019b). The technique has revealed that the number of small proteins in the cells is probably much higher than previously suspected, including many micropeptides resulting from the translation of small ORFs in transcripts currently annotated as long ncRNAs (Ji et al. 2015; Calviello et al. 2016; Ruiz-Orera and Albà 2019a; Chen et al. 2020; Douka et al. 2021). When we examined the Ribo-seq data in S. pombe for the complete transcriptome, we found very similar results to those previously described in mammals. The genome contains a relatively large number of annotated ncRNAs, 1527, many of which are antisense to protein-coding genes. Here we identified 214 additional ones. In line with previous findings in S. pombe (Duncan and Mata 2014), a sizable fraction of these ncRNAs (16%) showed translation evidence. Some of the translated ORFs in ncRNAs might encode functional micropeptides, whereas others, especially for very lowly abundant ncRNAs, could represent pervasive nonfunctional translation activities.
In summary, deep native RNA sequencing using Nanopore has uncovered an unexpectedly large number of high-frequency AS isoforms in S. pombe. Many of these isoforms could encode alternative, generally smaller, proteins, of as-yet-unknown functions. We have also identified a group of IR RNAs that show abnormally long poly(A) tails and that could potentially be regulating gene expression. The work provides new resources and methodologies for researchers investigating differential splicing in the fission yeast model.
Methods
S. pombe cultures
S. pombe (strain CBS5682) was grown in a rich medium at 30°C and harvested during log-phased growth (OD600 ∼ 0.5). The medium was identical to the one previously used to perform RNA sequencing of the same S. pombe isolate using Illumina Technology (Blevins et al. 2019). The composition of the medium, defined by Tsankov et al. (2010), can be found in Supplemental Tables S5 and S6.
RNA extraction
We extracted total RNA from S. pombe using the phenol chloroform extraction method (Castillo et al. 2003). Briefly, cells were grown to a final OD600 of 0.5. Yeast cultures (25–50 mL) were then centrifuged at 1500 rpm for 3 min and washed with H2O, and cell pellets were immediately kept on ice. Each sample was then resuspended in 0.4 mL of AE buffer (50 mM sodium acetate at pH 5.3, 10 mM EDTA at pH 8.0). Sodium dodecyl sulfate was then added to a final concentration of 1%, and proteins and DNA were extracted by adding 0.6 mL of acidic phenol/chloroform (V/V), followed by incubation for 5 min at 65°C. The aqueous phase was separated by centrifugation at 14,000 rpm for 2 min at 4°C and washed with a volume of chloroform and separated by centrifugation at 14,000 rpm for 2 min at 4°C. RNA was precipitated from the aqueous phase with ethanol. RIN quality scores were in the range of 9.6–10. We subsequently performed poly(A)+ RNA purification using the NEBNext Poly(A) magnetic isolation module and concentration with the Monarch RNA cleanup kit. The poly(A)+ purification steps were performed at the Genomics Core Facility of the Universitat Pompeu Fabra.
Direct RNA sequencing
The poly(A)+ RNA was used for dRNA-seq in an ONT Gridion X4. dRNA-seq offers the advantage over cDNA sequencing in that strand orientation information is maintained. The protocol involves adaptor ligation, and the molecules pass through an ionic current, adaptors and poly(A)+ tail first and then the rest of the molecule. The S. pombe samples were run in four flowcells. For each run, we used ∼600 ng of poly(A)+ RNA in 10 µL of volume. The dRNA-seq kit SQK-RNA002 was used. The base-calling was performed on live mode (during the sequencing) through the Guppy v.4.0.11 integrated on minKNOW v.4.0.5, using the HAC model. Nanopore dRNA-seq and base-calling was performed by the Centro Nacional de Análisis Genómico (CNAG).
We pulled together the output of the four runs, obtaining a total of 7,297,641 reads. We discarded any reads smaller than 150 bases and longer than 15,000 bases (likely artifacts) and removed any possible adapters with Porechop (https://github.com/rrwick/Porechop).
Read mapping and correction
To decrease the error rate of the reads and facilitate the subsequent de novo transcript assembly, we decided to correct the dRNA reads with Illumina RNA-seq reads from yeast grown in the same conditions using the software fmlrc with default parameters (Wang et al. 2018). Subsequently, we used TranscriptClean to correct the remaining errors (Wyman and Mortazavi 2019). The set of Illumina reads comprised 22,389,887 strand-specific 50-bp reads (Blevins et al. 2019). To run fmlrc, we first had to transform all uracil (U) bases in the dRNA reads to thymine (T) bases and, once the reads had been corrected, transform them back to U's. The reads were mapped to the genome using minimap2 (Li 2018) with the following options: minimap2 -t 6 -ax splice -uf -k14 ‐‐secondary = no -G 260. The Nanopore/Illumina hybrid reads showed an error rate of 7.2%, mainly because of poor coverage of the mRNA 3′-ends by Illumina reads. Individual read error rate was calculated using the CIGAR values of the reads aligned to the reference genome. Average error rate was calculated using SAMtools stats (Li et al. 2009). Reads that had a mapping quality of less than five were eliminated. The final set of “clean” reads comprised 5,054,233 reads.
To estimate the number of reads that were full length, we first obtained the length of the mapped reads with bam_alignment_length.py from the wub package. Reads with a length equal or longer than the transcript in which they are aligned minus 50 nt were estimated to be full-length reads.
Transcript assembly
We used StringTie2 to obtain a S. pombe transcriptome directly from the set of dRNA clean reads, as previously described (Kovaka et al. 2019). The parameters were as follows: –l – conservative –G -t -c 1.5 -f 0.05. The program uses the reference genome and the mapped reads for the assembly and, optionally, a gene annotation file. We chose to use the gene annotations to guide the assembly because this option provided a direct mapping to already known genes, facilitating the assembly. The reference genome and gene annotation files were downloaded from the PomBase database on February 1, 2021 (Lock et al. 2019). For the assembly, we considered all mapped reads except those that mapped to multiple sites (those with a MAPQ score greater than five and that had alignments with the flags 0 and 16 in the SAM file). The number of reads used for the assembly was 5,054,233 reads (“clean” reads). We eliminated any assembled transcripts <150 nt. The resulting annotation file was named “StringTie transcriptome.” It contained 5799 different transcripts.
The identification of alternative isoforms was based on the StringTie transcriptome. StringTie2 recovered all isoforms with an estimated frequency >5% with respect to the most common isoform. StringTie transcripts that corresponded to annotated genes but were different from the reference transcript were classified into one of the following transcript alternative isoform types: IR, II, ES, and AS site. The classification was based on the number and genomic position of the exons represented in the dRNA reads. We focused on events affecting the coding sequence as they were the ones most likely to have functional consequences. These selection steps resulted in 262 genes with alternative isoforms. The total number of events was 332 because some genes had more than one possible event.
The identification of novel transcribed loci was also based on the StringTie transcriptome. Novel transcripts were defined as those not overlapping any annotated gene. We obtained 214 novel transcripts, 158 of which overlapped another gene in antisense orientation. Saturation curves were produced by subsampling the mapped reads with SAMtools -s (0.1 to 0.9 of the total number of reads) and running StringTie2 again. The assembled transcripts were then compared with the set of novel or annotated transcripts using the intersect function from BEDTools (Quinlan and Hall 2010).
Transcript expression quantification with Nanopore reads
The number of mapped dRNA transcripts was calculated first mapping the transcripts to the transcriptome with minimap2 (arguments were -t 6 -ax map-ont -K 10G ‐‐for-only ‐‐no-long-join -r 10,10 ‐‐secondary = no) and then using the script bam_counts_reads.py from the nanoporetech package with the -a argument set to five to consider only uniquely mapped reads. To quantify the expression of already annotated transcripts, we used the PomBase transcriptome. We mapped reads to 5026 annotated mRNAs (97.8%); the average number of mapped reads per mRNA was 1130 and the median 1259. As each read corresponds to a native mRNA molecule, we consider the number of mapped reads to be equivalent to coverage. To quantify the expression of different isoforms and novel transcripts we used the StringTie transcriptome. The data allowed to compare the relative abundance of different isoforms with high accuracy. For information on gene expression data, see Supplemental Table S8.
Transcript expression quantification with Illumina reads
Illumina reads from S. pombe grown in rich medium were obtained from a previous study (Blevins et al. 2019). The Illumina reads were 50 bp long and strand specific. They were mapped to the reference transcriptome with HISAT2 (Kim et al. 2019) with the ‐‐rna-strandness “RF” option. Reads that were not aligned in the expected direction (tag XS:A:-) were discarded from the resulting BAM file. The reads mapping to each transcript were quantified with bam_ counts_reads.py from the ONT nanoporetech package (https://github.com/nanoporetech/wub). The counts were normalized to fragments per kilobase per million mapped reads (FPKM).
Poly(A) tail quantification
Poly(A) tail lengths were estimated at the read level using the nanopolish (v 0.13.3; Loman et al. 2015) polya script. As input to the command nanopolish polya, we used the raw FAST5 and FASTQ files in addition to the corrected reads mapped to the genome of S. pombe. Finally, only those reads with the quality control provided by nanopolish with the tag “PASS” were considered. For information on poly(A) length, see Supplemental Table S8.
GO enrichment
GO term enrichment was performed using the web application AnGeLi (Bitton et al. 2015b). We identified overrepresented or underrepresented GO terms for genes in the top 10% or bottom 10% regarding average poly(A) tail length. As a predefined background, we used all genes. GO term information about individual genes was extracted from AnGeLi too.
Analysis of ribosome profiling data
To study the translatability of the transcripts, we used previously published ribosome profiling data (Duncan and Mata 2017). The data were from untreated cells (ArrayExpress [https://www.ebi.ac.uk/arrayexpress/] under accession numbers ERR1994961 and ERR1994962). We filtered out ribosomal RNAs and mapped the reads to the S. pombe genome with TopHat (v 2.1.1) (Kim et al. 2013) with default options. We used the script offsetCorrect.pl from RibORF to identify the P-site of each read (Ji et al. 2015). The number of mapped P-sites was used to evaluate the level of translation of the intronic regions in IR isoforms. Ribo-seq coverage was calculated in all the transcripts with CDS in PomBase using htseq-count (-m intersection-strict) with the genome and P-sites obtained before. Ribo-seq coverage of the alternative isoform of SPAC11E3.15.1 was calculated using only the intronic region until the stop codon included in the intron. RibORF was also used to predict translated ORFs in novel transcripts and ncRNAs. We required at least 10 mapped reads and a ribORF score >0.7. When two or more ORFs showed overlapped on the same transcript, we kept the longest ORF. For information on isoforms with translation evidence (five or more mapped Ribo-seq reads), novel nonannotated transcripts, and UTRs, see Supplemental Table S8. For information on annotated lncRNAs with evidence of translation (RibORF score >0.7), see Supplemental Table S9.
Genomic DNA preparation
Genomic DNA was prepared from 10 mL of yeast cultures grown to saturation. Cells were pelleted at 1500 rpm for 3 min and washed with H2O; pellets were immediately frozen in liquid nitrogen. Samples were resuspended in 0.2 mL of genomic DNA preparation buffer (10 mM Tris-HCl at pH 8.0, 100 mM NaCl, 2% Triton X-100, 1% SDS, 1 mM EDTA), 0.1 mL neutral phenol, and 0.1 mL chloroform. Glass beads were added, and cells were lysed in a Vortex Genie 2 (Scientific Industries). After removal of glass beads, homogenates were centrifuged at 20,000 g for 5 min (4°C), supernatants were collected, and 0.2 mL of chloroform was added. Following centrifugation, supernatants were collected, and DNA was precipitated with 1/10 volume of 3 M sodium acetate (pH 5.2) and 2.5 volumes of EtOH, followed by incubation 30 min at −80°C. Following centrifugation, pellets were washed with 1 mL EtOH (70%), air-dried, and resuspended in 40 µL of TE-buffer (10 mM Tris-HCl at pH 8.0, 1 mM EDTA) containing 1 µL RNase A. RNA was digested for 30 min at 37°C, and DNA was stored at −20°C.
cDNA preparation
RNA (25 µg) was treated with 0.5 µL DNase I for 30 min at 37°C and then inactivated for 10 min at 75°C. Reverse transcriptase (RT) reactions were performed with 8 µg of DNase I-digested RNA, following the manufacturer's instructions (high-capacity RT kit, Thermo Fisher Scientific; 10 min at 25°C, 120 min at 37°C, and 5 min at 85°C) in the presence or absence of RT.
Polymerase chain reactions
Polymerase chain reactions (PCRs) were performed in a total volume of 20 µL using 0.5 µL of the cDNA reactions with primers listed in Supplemental Table S7. Fifty nanograms of genomic DNA was used as reference for the unspliced transcript. PCR products were separated on 2% agarose TBE gels. Digital images were acquired with Bio-Rad software.
Statistical tests and plots
The generation of plots and statistical tests was performed using the R package (R Core Team 2020). Figures were made with ggplot2 (Wickham 2016).
Data access
The ONT dRNA raw sequences generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA791394. Additional Supplemental Data can be found at Figshare (https://doi.org/10.6084/m9.figshare.19368146). This comprises the set of clean reads used for transcript reconstruction and quantification, the dRNA-based StringTie transcriptome (noncurated), the dRNA-based S. pombe transcriptome for genes with alternative isoforms (curated), 5′ UTR and 3′ UTR annotation files, and Supplemental Tables S8 and S9. Supplemental Table S8 contains information on gene expression values, alternative gene isoforms, and transcript poly(A) tail length. Supplemental Table S9 contains information on ncRNAs containing ORFs with evidence of translation.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work benefited from preliminary Nanopore RNA-seq data analyses performed by Bea Calvo and Audald Lloret-Villas, as well as discussions with Eduardo Eyras and Ivan de la Rubia. We acknowledge funding from Ministerio de Ciencia e Innovación (MCI), Agencia Estatal de Investigación (AEI) grant PGC2018–094091-B-I00, cofunded by Fondo Europeo de Desarrollo Regional (FEDER), and from Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR), Generalitat de Catalunya, grant 2017SGR01020.
Author contributions: W.R.B., J.C.M., E.H., J.A., and M.M.A. contributed to the conceptualization of the study and design of experiments. W.R.B., J.C.M., and M.C. performed yeast growth cultures and RNA extraction. M.C. performed the RT-PCR experiments. J.C.M. developed most pipelines and performed the majority of analyses. M.H. and S.G.M. analyzed the data related to novel genes and the annotation of UTRs. J.C.M. and M.M.A. wrote the manuscript with input from all authors.
Footnotes
-
↵4 Present address: CNAG-CRG, Centre for Genomic Regulation (CRG), 08028 Barcelona, Spain
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276516.121.
- Received December 20, 2021.
- Accepted May 9, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








