
A general framework for identifying oligogenic combinations of rare variants in complex disorders
- 1Department of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, Pennsylvania 16802, USA;
- 2Bioinformatics and Genomics Graduate Program, Huck Institutes of the Life Sciences, University Park, Pennsylvania 16802, USA;
- 3Department of Anthropology, Pennsylvania State University, University Park, Pennsylvania 16802, USA
Abstract
Genetic studies of complex disorders such as autism and intellectual disability (ID) are often based on enrichment of individual rare variants or their aggregate burden in affected individuals compared to controls. However, these studies overlook the influence of combinations of rare variants that may not be deleterious on their own due to statistical challenges resulting from rarity and combinatorial explosion when enumerating variant combinations, limiting our ability to study oligogenic basis for these disorders. Here, we present RareComb, a framework that combines the Apriori algorithm and statistical inference to identify specific combinations of mutated genes associated with complex phenotypes. RareComb overcomes computational barriers and exhaustively evaluates variant combinations to identify nonadditive relationships between simultaneously mutated genes. Using RareComb, we analyzed 6189 individuals with autism and identified 718 combinations significantly associated with ID, and carriers of these combinations showed lower IQ than expected in an independent cohort of 1878 individuals. These combinations were enriched for nervous system genes such as NIN and NGF, showed complex inheritance patterns, and were depleted in unaffected siblings. We found that an affected individual can carry many oligogenic combinations, each contributing to the same phenotype or distinct phenotypes at varying effect sizes. We also used this framework to identify combinations associated with multiple comorbid phenotypes, including mutations of COL28A1 and MFSD2B for ID and schizophrenia and ABCA4, DNAH10 and MC1R for ID and anxiety/depression. Our framework identifies a key component of missing heritability and provides a novel paradigm to untangle the genetic architecture of complex disorders.
Recent human population growth has led to a rapid increase in the load of rare variants affecting functionally important regions of the genome (Coventry et al. 2010; Keinan and Clark 2012; Tennessen et al. 2012). Thus, rare variants are collectively more abundant in the population compared to common variants, many of which confer significant risk for neurodevelopmental disorders such as autism and intellectual disability (McClellan and King 2010; The 1000 Genomes Project Consortium 2015; Taliun et al. 2021; Backman et al. 2021). In fact, recent studies have directly implicated rare damaging mutations that are very recent or de novo in >100 genes toward neurodevelopmental disorders (Sebat et al. 2007; Iossifov et al. 2014; Wilfert et al. 2021). The ability to establish robust associations between rare variants of high effect size and complex disease has made this class of variants the primary focus of recent studies. However, a much larger class of rare and variably expressive variants that are individually less deleterious but, in combination, exert large effects towards disease is often overlooked. Variants in this category are often transmitted across generations without adverse effects on their carriers until they encounter other similar variants that, when combined, lead to genetic interactions conferring a higher risk for disease than their individual risks (Badano and Katsanis 2002; Gifford et al. 2019). Whereas this phenomenon underpins oligogenic models proposed over the years, studies so far have not focused on detecting combinatorial effects of specific sets of rare variants toward disease phenotypes (Badano et al. 2006; Girirajan et al. 2010; Leblond et al. 2012; Pizzo et al. 2019).
Identifying the effects of specific combinations of rare variants toward disease etiology has been challenging for many reasons. First, combinations of rare variants are rarer, and extremely large cohorts are required to observe even a few recurrent instances of specific variant combinations (Uricchio et al. 2016). Prior studies of oligogenic models for rare variants evaded this problem by aggregating variant information at the sample level and comparing the overall burden of rare variants between groups of individuals (such as cases and controls) (Sebat et al. 2007; Iossifov et al. 2014; Krumm et al. 2015; Halvorsen et al. 2020). Second, the combinatorial explosion resulting from even a small set of rare variants makes it difficult to exhaustively evaluate all combinations. Whereas sophisticated frameworks such as network analysis and machine learning provide powerful tools to model the composite effects of thousands of variables on a complex system and predict emergent behaviors and quantitative outcomes, adapting them to exhaustively search and delineate the effects of specific combinations of variables is daunting (Murdoch et al. 2019; Molnar et al. 2020). Furthermore, incorporating an efficient search tool into these frameworks and extending them to detect higher-order combinatorial effects would be nearly impossible. Third, even when all combinations of rare variants could be exhaustively evaluated within a large cohort, there is a lack of methods that are sensitive enough to detect small differences between comparison groups to establish statistical significance. Therefore, an alternate approach that is highly flexible, scalable, and sensitive is necessary to address computational and statistical challenges associated with assessing rare variant combinations.
Here, we present a combinatorial framework called RareComb that couples the Apriori algorithm with binomial tests to overcome the limitations of data sparsity and high dimensionality and systematically analyzes patterns of rare variants between groups of interest to identify specific combinations that are significantly associated with phenotypes (Agrawal and Ramakrishnan 1994). We demonstrate the utility and adaptability of our framework by identifying mutated gene combinations significantly associated with one or more phenotypes among children with autism. Our generalizable and modular framework does not depend on a priori knowledge and can detect rare patterns from high-dimensional genetic data to generate interpretable results, making it readily applicable for analyzing cohorts of all size ranges to dissect the genetic basis of complex disorders.
Results
We hypothesized that two or more genes disrupted simultaneously by rare deleterious mutations contribute to a highly penetrant phenotype, as in an oligogenic model, or lead to a more severe phenotype than when each of the same genes are disrupted individually. We developed RareComb as a framework that combines data mining and statistical analysis to identify specific combinations (such as pairs, triplets, etc.) of rare variants that show significant associations with one or more phenotypes. RareComb analyzes an “n × p” sparse Boolean matrix with “p” genes in “n” individuals in two discrete steps (Fig. 1). First, it applies the Apriori algorithm independently in cases and controls to enumerate the frequency of all simultaneously mutated combinations that meet a preset minimum frequency threshold (Supplemental Fig. S1). Second, for each qualifying combination of variants, the method derives the expected frequency of simultaneously observing mutations in the constituent genes under the assumption of independence. It then independently quantifies the magnitude of deviation of the observed from the expected frequencies using binomial tests in cases and controls and uses multiple-testing adjusted P-values to identify combinations that are statistically enriched in cases but not in controls. Finally, the method calculates effect sizes using Cohen's d and statistical power at 1% and 5% significance thresholds to enable prioritization of a high-confidence set of combinations that contribute to the phenotype in an oligogenic manner.
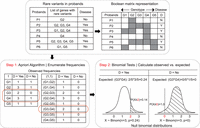
Conceptual overview of combinatorial analyses using RareComb. A Boolean representation of genotype (mutated genes, G1, G2, etc.) and disease status for probands (P1, P2, etc.) is shown. In step 1, the Apriori algorithm is applied to the Boolean input matrix to calculate the frequencies of individual (e.g., G1) and simultaneous occurrences of events (G1 and G2) that meet the user-specified criteria, including the size of combinations (pairs, triplets, etc.) and minimum frequency threshold of simultaneous occurrences. In step 2, independently in case and control groups, for each combination, the binomial test is applied to compare the observed frequency of simultaneous occurrence of events with its corresponding null binomial distribution of the expected frequencies calculated under the assumption of independence. Binomial test for gene pair G3 and G4 is shown as an example.
RareComb identifies oligogenic combinations associated with ID and autism
We sought to identify pairs and triplets of mutated genes that are significantly associated with intellectual disability/cognitive impairment (ID) phenotypes by analyzing 6189 affected males from the Simons Foundation Powering Autism Research or SPARK (The SPARK Consortium 2018) cohort for discovery and 1878 affected males from the Simons Simplex Collection or SSC (Fischbach and Lord 2010) cohort for validation. To facilitate cross-cohort comparison, we identified 10,217 rare variants (MAF ≤ 1%) that were predicted to be deleterious by multiple methods and observed in both cohorts and aggregated these variants to genes for the analysis (see Methods). For this study, we first categorized 1215 probands from the SPARK cohort diagnosed with ID/cognitive impairment as “cases” and 4974 probands without ID as “controls” (Fig. 2A). We then applied RareComb to cases after constraining it to only evaluate those gene combinations in which simultaneous mutations are observed in at least five probands (i.e., minimum frequency threshold). We identified 25,602 pairs involving 1956 mutated genes in cases that were observed at a higher frequency than expected under the assumption of independence.
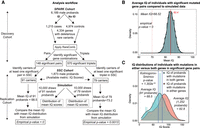
Combinations of rare variants contributing to intellectual disability (ID) phenotype. (A) An outline of the approach used to identify and validate mutated gene pairs and triplets enriched in probands with ID is shown. We tested whether mutated gene pairs identified as significant in one cohort (SPARK) are also associated with severe phenotypes in an independent cohort (SSC). To test this, we obtained the mean IQ score of individuals from the SSC cohort carrying significant combinations identified from the SPARK cohort. Empirical P-values were then calculated based on the deviation of the mean IQ from the distribution of mean IQ scores obtained from 10,000 random draws in the simulation. (B) The mean IQ of individuals with mutated gene pairs in the SSC cohort was significantly lower (empirical P-value = 0) when compared to the distribution of mean IQ scores obtained from the simulation. (C) Histogram shows the distributions of IQ scores of SSC probands who carried mutations in either of the genes versus both constituent genes of the significant gene pairs. The distributions were significantly different from each other (P-value = 1.302 × 10−16, Kolmogorov–Smirnov test).
Similarly, analyzing the controls using only the 1956 genes mutated in cases, RareComb identified 148 pairs of mutated genes that were significantly enriched in cases but not in controls (Supplemental Table S1), with moderate to high effect sizes (Cohen's d, 0.08–0.15) and adequate statistical power (70%–100% at 5% significance threshold) (Supplemental Fig. S2). These 148 gene pairs belonged to 142 probands, with 74% (105/142) of them carrying more than one significant pair. These observations suggest that an individual can carry multiple combinations, each contributing to the same phenotype at varying effect sizes (Supplemental Fig. S3). To identify enrichment for specific variant types within combinations, we examined the 148 significant gene pairs by mutation type, including missense, stop-loss, and stop-gain mutations. We identified 871 instances of variant pairs, of which 95.64% (833/871) contained a missense mutation in both genes, 4.36% (38/871) contained a missense in one of the genes and a stop-gain in the other. We found no instances of pairs of genes with missense/stop-loss, stop-loss/stop-loss, stop-gain/stop-loss, or stop-gain/stop-gain mutations. To evaluate the statistical significance of these observed proportions, we generated all possible variant pairs from all male probands and calculated the expected proportion for each possible pair of variant types (Supplemental Table S2). Out of all possible variant pairs, 93.9% were missense/missense variant pairs and 5.89% were missense/stop-gain pairs. One-tailed binomial tests showed that the 95.64% observed in our data for missense/missense pairs was higher than the expected 93.9% (P-value = 0.015), but the 4.36% observed for missense/stop-gain pairs was not significantly lower than the expected 5.89% (P-value = 0.58). These results suggest that there is a higher propensity for missense mutations to form combinations than the relatively higher impact stop-gain or stop-loss mutations.
We next sought to validate the association of these 148 mutated gene pairs toward intellectual disability. We hypothesized that if the association of the gene pairs with ID in the SPARK cohort were truly significant, carriers of mutations in those gene pairs would tend to have lower than average IQ scores in the independent SSC cohort. We found that 90 of the 148 significant pairs identified in the SPARK cohort were observed in at least one proband in the SSC cohort. These 90 mutated gene pairs were carried by 91 unique probands, whose average full-scale IQ scores (average IQ = 68.52) were lower than those of all ascertained probands in the SSC cohort (average IQ = 86). To assess the significance of this result, we performed 10,000 random draws of 91 probands from the SSC cohort to generate a simulated distribution of their average IQ scores. The average IQ of carriers of mutated gene pairs (average IQ = 68.52) was significantly lower than the overall distribution of average IQ derived from simulations (average IQ ranged from 73 to 92; empirical P = 0) (Fig. 2B). Furthermore, the average IQ of the 91 SSC probands with both mutated genes was significantly lower than the average IQ of 1252 carriers of mutations in only one of the two genes (68.5 vs. 82.8; Kolmogorov–Smirnov P = 1.302 × 10−16) (Fig. 2C). When each of the 90 combinations was evaluated individually, carriers of mutations in both genes for 73% (66/90) of the combinations showed lower IQ than individuals with mutations in individual genes of the same combination, with 39/90 remaining significant after multiple testing correction (Supplemental Table S3; Supplemental Fig. S4). These results provide evidence for synergistic effects of deleterious mutations within specific pairs of genes towards ID phenotypes. We note that this analysis only considered combinations that were enriched in cases but not in controls for multiple testing. Therefore, we repeated the analysis using a more conservative approach that considered all combinations that met the frequency threshold in cases for multiple-testing correction (see Methods) and obtained 115 significant pairs belonging to 79 probands (Supplemental Table S4). The average IQ of carriers of mutated gene pairs (average IQ = 69.11) remained significantly lower than the overall distribution of average IQ derived from simulations (average IQ ranged from 73 to 92; empirical P = 0) (Supplemental Fig. S5). We also conducted the analysis on the entire cohort by combining 6189 male and 1528 female probands together and identified 199 gene pairs belonging to 82 males and 14 female probands (Supplemental Table S5). Our results held true even when both male and female probands were considered together, with the average IQ of these 96 probands (average IQ = 69.46) being significantly lower than the simulated distribution of average IQ (average IQ ranged from 71 to 92; empirical P-value = 0) (Supplemental Fig. S6).
Next, we applied RareComb to identify gene triplets associated with intellectual disability using the two cohorts of affected males and repeated the simulations to identify 1593 significant combinations in the SPARK cohort. We selected 570 high-confidence triplets (with ≥90% statistical power at 5% significance threshold) (Supplemental Table S6) and found that 79 probands in the SSC cohort carried at least one of these deleterious triplets. The average IQ score of individuals carrying significant gene triplets (average IQ score = 73) was significantly lower than a distribution of average IQ scores from 10,000 draws of 79 SSC probands (average IQ ranged from 72 to 94; empirical P = 0.0011) (see Supplemental Fig. S7). This result reiterated that carriers of mutations in the significant gene combinations have lower IQ than a random group of probands. Our results also demonstrate the ability of the framework to identify higher-order combinations of mutations that are significantly associated with specific phenotypes in individuals with complex disorders.
Oligogenic combinations are enriched for specific inheritance patterns
As individual variants can arise de novo or be inherited maternally or paternally, variants in pairs of genes can have six possible patterns of transmission (Supplemental Fig. S8A). We identified a total of 926 occurrences of the 148 pairs of mutated genes enriched among SPARK probands with ID (n = 142 probands), of which inheritance could be determined without ambiguity for 887 instances. We found that one variant occurred de novo and the other variant was inherited from the mother in 244/887 instances (27.5%). Similarly, both mutated genes were inherited from the mother in 226/887 instances (25.4%) or occurred de novo in 221/887 instances (24.9%), and the remaining fraction (∼22%) of variant pairs were either inherited from both parents, inherited from the father, or transmitted de novo and paternally. To assess the significance of our observations, we performed simulations to establish a baseline expectation of proportions for each category of parental inheritance pattern. We selected 926 pairs of genes in 1000 random draws of all possible mutated gene pairs among SPARK probands and calculated the fraction of instances that fell into each of the six transmission categories. Unaffected siblings were not considered for this simulation. The observed proportion was higher than the simulated proportions for instances when both variants occurred de novo (24.9% vs. 17%, empirical P = 0) and when one variant was de novo and the other was inherited maternally (27.5% vs. 25%, P = 0.028) (Fig. 3A). We note that the depletions observed in categories “Maternal + Paternal” and “Both Paternal” could simply be due to the numerical offset resulting from enrichment of other categories. We repeated this analysis for 7596 children affected with autism in the SPARK cohort compared to 11,740 unaffected parents and identified 110 gene pairs significantly associated with autism (Supplemental Table S7). Similar to the results obtained for the ID phenotype, we found that both variants of a gene pair were more likely to occur de novo (24% vs. 18%, empirical P = 0) or one variant occurring de novo and the other inherited maternally (33% vs. 26%, P = 0) than expected based on simulation studies (Supplemental Fig. S9). The enrichment of de novo or maternally inherited variants for significant gene pairs aligns with published reports that severely affected children tend to carry multiple de novo mutations or inherit pathogenic rare variants from mildly affected or unaffected carrier mothers (Girirajan et al. 2012; Krumm et al. 2015; Turner et al. 2017).
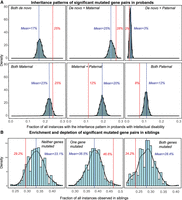
Analysis of parental and sibling inheritance patterns of significant gene pairs associated with ID. (A) Fraction of all instances of significant gene pairs observed within each of the six possible parental inheritance patterns (red) compared against 1000 simulations is shown (blue). During each simulation, random mutated gene pairs from the SSC cohort were selected, the inheritance status of the mutations was identified, and the fraction of those instances belonging to one of the six predefined categories was calculated. Comparing the observed fractions with the simulated fractions indicates statistical enrichment for two specific inheritance patterns based on empirical P-values: both variants being de novo, and one variant being de novo and the other transmitted from the mother. (B) Histograms show the carrier status of significant gene pairs in siblings of carrier probands (red) compared against 1000 simulations (blue). Among significant pairs, both genes were mutated in only 24.2% of all siblings (compared to 28.4% in simulations), whereas one of the two genes was mutated in 46.6% of all siblings (compared to 38.5% in simulations). These results show that mutations are more likely to be observed in just one of the two genes within the gene pairs and are less likely to be observed simultaneously in siblings of carrier probands.
We then assessed whether the mutated gene pairs associated with ID were also found in siblings of carrier probands. Restricting our analysis to families with unaffected siblings whose probands had mutations in ID-enriched gene pairs, we found that both variants were present in the corresponding sibling for only 53/219 (24.2%) instances of gene pairs, whereas 102/219 (46.6%) had variants in only one of the two genes and 64/219 (29.2%) instances had no variants in either of the genes in the siblings (Supplemental Fig. S8B). Using simulations, we found a significantly higher proportion of instances with only one of the two variants present in siblings compared to the expected values (46.6% vs. 38.5%, P = 0.007). Furthermore, the proportion of observed instances with neither of the variants present in siblings (29.2% vs. 33.1%, empirical P = 0.098) or both variants present in siblings (24.2% vs. 28.4%, P = 0.079) was lower than expected (Fig. 3B). The observation that only a small fraction of unaffected siblings carried both mutated gene pairs suggests a strong association of these gene pairs with ID phenotypes. These results suggest that mutations in pairs of genes significantly associated with a severe phenotype in probands are more likely to occur individually than simultaneously in unaffected siblings of the same family.
Genes forming oligogenic combinations are distinct from canonical autism genes
We expanded our analysis to include all 16,556 mutated genes in the SPARK male cohort, as opposed to genes with mutations present in both the SPARK and SSC male cohorts, and identified 52 significant gene pairs (Supplemental Table S8) and 230 triplets associated with the ID phenotype (with ≥90% statistical power at 1% significance threshold) (Supplemental Table S9). Due to the expanded search space, the 52 mutated gene pairs showed more significant P-values from the binomial tests when compared to those obtained from the more restricted set of variants overlapping both SPARK and SSC cohorts (Supplemental Fig. S10). Mutated genes within the 52 combinations included several genes related to nervous system development, such as NIN, HDC, NGF, and BRD8. Furthermore, 5/52 pairs and 59/230 triplets contained at least one gene associated with autism in the SFARI database—including FGFR1, associated with multiple disorders including Kallmann syndrome (Dodé et al. 2003) and Pfeiffer syndrome (Schell et al. 1995); RELN, associated with temporal lobe epilepsy (Dazzo et al. 2015); SYNE1, associated with spinocerebellar ataxia (Synofzik et al. 2016; Yoshinaga et al. 2017); and PNPLA7, associated with autism and ID (Prasad et al. 2012). Thus, most genes forming combinations are not involved in canonical autism or ID disorders, suggesting synergistic effects of these genes without prior association to disease. We also analyzed 14,708 variants from 1528 female probands (375 probands with ID and 1153 without ID) and identified 19 significant pairs associated with ID, indicating that significant combinations can be identified even when the sample sizes are small (Supplemental Table S10).
We performed Gene Ontology (GO) enrichment analysis for genes within the 52 combinations and identified seven out of nine significantly enriched GO terms to be exclusively associated with nervous system-related functions, including synthesis and metabolism of catecholamines, axon/neuron regeneration, and neuron generation and differentiation (Supplemental Fig. S11; Mi et al. 2019). Enrichment of several annotations related to growth and maintenance of brain cells such as “axon development,” “neurogenesis,” “axon regeneration,” “neuron differentiation,” “neuron projection regeneration,” and “response to axon injury” indicate the physiological relevance of the genes identified by our method. Furthermore, the differences in the type and specificity of GO terms enriched for significant pairs versus triplets were apparent, with genes forming pairs involved in nervous system function and genes forming triplets associated with both nervous system as well as other biological processes. We next assessed the enrichment and depletion of Human Phenotype Ontology (HPO) terms for genes forming significant pairs towards ID phenotypes (Köhler et al. 2021). First, we calculated the fraction of all 4484 genes within the HPO database associated with each HPO term. For example, 30% (1366/4484) of all genes in HPO were associated with ID. We compared these expected values calculated for each HPO term with the corresponding fractions observed within the 95 genes forming 52 ID-associated pairs using binomial tests. Genes associated with HPO terms related to neurodevelopmental phenotypes, such as ID, global developmental delay, seizure, and microcephaly, were significantly depleted within the set of 95 genes forming gene pairs (Supplemental Table S11). Next, we evaluated whether genes within each of the 52 significant pairs shared one or more common HPO phenotype or disease. Of the 52 pairs, only one pair (DNASE1 and MTR) shared an HPO phenotype (“epilepsy”). This was significantly lower than the expected value obtained from the distribution of the number of shared HPO phenotypes between all possible pairs of genes in the HPO database (1/52, 1.9% ID gene pairs compared to 31.5% of all HPO gene pairs shared one HPO phenotype, P = 2.2 × 10−16; one-sided binomial test) (Supplemental Fig. S12; Supplemental Table S12). We note that the 4484 genes within HPO are potentially biased toward well-studied disorders, making pairs of genes drawn from HPO more likely to share phenotypes than random pairs of genes from the genome. Overall, GO and HPO analyses show that genes forming oligogenic combinations are involved in neuronal processes but have not been previously connected to neurodevelopmental phenotypes, indicating the novelty of the associations between these genes and ID phenotypes.
Identifying variant combinations toward specific patterns of comorbid phenotypes
We adapted our framework to identify significant associations of two or more genotypes with multiple comorbid phenotypes. To identify novel comorbid associations, we eliminated phenotypes that were highly correlated with each other, such as ADHD and reading disorder (Gilger et al. 1992). We analyzed variant profiles of 6189 autism probands from the SPARK cohort with records of comorbid features, including 1215 individuals with ID, 1825 with anxiety and depression, and 332 with schizophrenia features. We assessed for significant co-occurrences of two or more mutated genes with two or more of the above phenotypes (Fig. 4). Using one-tailed binomial tests to compare the observed frequency of combinations of genotypes and phenotypes to the expected frequency, we first identified 169 significant associations between pairs of mutated genes and two comorbid phenotypes as well as 82 combinations of three mutated genes and two comorbid phenotypes (Supplemental Tables S13, S14). As some of these significant genotype-phenotype combinations can be confounded by a high degree of co-occurrence of mutated genes, we next calculated genotype-only P-values using binomial tests for all significant genotype-phenotype associations. For 32/169 combinations of two mutated genes and two comorbid phenotypes and 5/82 combinations of three mutated genes and two comorbid phenotypes, the composite genotype-phenotype P-values were significant, whereas genotype-only P-values were not significant, suggesting stronger associations between these variant combinations and phenotypes. For example, even when variants in genes COL28A1 and MFSD2B did not co-occur more frequently than expected under the assumption of independence, these mutated genes co-occurred more frequently than expected among probands with ID and schizophrenia phenotypes. Loss-of-function and rare missense mutations in COL28A1 have been reported in individuals with autism (Krumm et al. 2013; Guo et al. 2017), and MFSD2A, a paralog of MFSD2B, has been directly implicated in an autosomal recessive disorder associated with progressive microcephaly, spasticity, and brain imaging abnormalities (Guemez-Gamboa et al. 2015). Similarly, we found ARVCF and FAT1 to be significantly associated with ID and schizophrenia, with ARVCF mapping within the 22q11.2 DiGeorge syndrome region (Sanders et al. 2005), whereas rare de novo mutations in FAT1 being associated with autism and schizophrenia (Iossifov et al. 2014; Kenny et al. 2014). Finally, we found that the mutations in genes ABCA4, DNAH10, and MC1R significantly co-occurred in individuals with ID and anxiety/depression phenotypes. These results demonstrate the utility of identifying higher-order associations between genotypes and phenotypes in complex disorders such as autism.
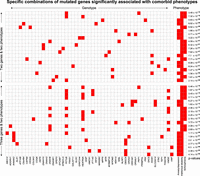
Analysis of comorbid phenotypes using RareComb. We analyzed the genotypes of probands with anxiety/depression, ID, or schizophrenia. The heat map shows combinations of two or three mutated genes that were significantly enriched in individuals with specific patterns of comorbid phenotypes compared to the expected frequency under the assumption of independence.
Discussion
Current rare variant analysis strategies are geared toward either searching for individual variants of high effect size whose influence on the phenotype is evident, such as de novo gene-disruptive mutations, or comparing rare variant burden to explain collective effects on phenotypes (Sebat et al. 2007; Girirajan et al. 2011; Zheng et al. 2016). The wider space between these two extremes of the analysis spectrum that involves combinations of rare variants has largely remained understudied. Although digenic diseases and multihit models of complex diseases have been used to provide post hoc explanations for an observed phenomenon, they are not equipped to serve as a framework to actively search for and identify rare variant combinations that fit oligogenic models for specific phenotypes (Badano et al. 2006; Leblond et al. 2012; Gifford et al. 2019). Although machine learning has become the de facto approach for disease outcome predictions, the lack of holy-grail predictors and reduced interpretability due to data sparsity makes it less fit to detect combinatorial effects (Murdoch et al. 2019). In addition, the common practice of evaluating feature importance metrics of machine learning classifiers falls short of the objective to identify combinations of features that exert higher effect on the phenotype than evident from their independent effects (Murdoch et al. 2019; Molnar et al. 2020). Even if this black-box nature of machine learning could be overcome, identifying even a handful of truly oligogenic variants to use as “ground truth” for training a classifier can be challenging. Whereas a few studies managed to use a small number of examples as training sets effectively, those approaches were limited to digenic models (Papadimitriou et al. 2019). Furthermore, prior studies to assess combinatorial effects have been inherently biased due to their need to minimize the search space by restricting the analysis to only a subset of genes chosen based on a priori knowledge (Schaaf et al. 2011; Papadimitriou et al. 2019; Kerner et al. 2020). Here, we provide a proof-of-concept analytical framework that remains agnostic to prior evidence and performs exhaustive searches to identify combinatorial effects among rare variants while retaining high granularity of data and interpretability of results.
Here, we use our framework to identify gene pairs and triplets significantly associated with intellectual disability and show that several constituent genes are associated with nervous system processes. These mutated gene combinations are more likely to be inherited maternally or occur de novo, are depleted in unaffected siblings from the same family, and are less likely to involve canonical autism or ID genes, suggesting that genes forming significant combinations are less deleterious on their own but manifest effects only when combined with other similar genes carrying rare mutations. Whereas previous studies have linked aggregate rare variant burden toward intellectual disability (Fitzgerald et al. 2015; Singh et al. 2017), our results fine-map the association to specific combinations of constituent genes contributing to the burden. Based on these observations, we propose a novel paradigm for dissecting the complexity of genetic disorders, where an affected individual carries multiple combinations of rare variants, and each combination contributes to either the same phenotype or distinct phenotypes at varying effect sizes (Fig. 5).
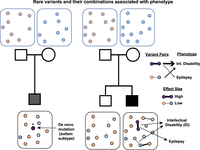
Rare variant models for complex disorders. The schematic shows two models for the genetic etiology of complex disorders. Circles represent rare variants present that are either de novo or inherited from a parent. On the left, individual high-effect de novo variants are strongly associated with a phenotype of interest. On the right, rare variants within an individual combine in multiple ways and contribute toward distinct phenotypes. The thickness of the connecting lines denotes effect sizes, and an affected individual can carry multiple oligogenic combinations of rare variants, each of which contributes to the same or distinct phenotypes. This extension of the oligogenic model enables further dissection of the genetic architecture of complex disorders.
A limitation of our method is that it tends to be biased toward genes that are mutated frequently enough to be observed in a combination, and therefore variant types such as large structural variants were not included in our analysis. This limitation can be addressed by fixing specific primary variants of interest irrespective of their frequency and screening for “second-hit” modifiers that significantly co-occur with the primary variant, such as the co-occurrence of RBM8A variants in proximal 1q21.1 deletion carriers manifesting thrombocytopenia-absent-radius syndrome, and TBX6 variants in proximal 16p11.2 deletion carriers with scoliosis (Albers et al. 2012; Yang et al. 2019). Another limitation of our method is that it does not take population substructure into account. Because allele frequencies of rare variants vary across populations and ancestries, our results are likely true for individuals of European descent that make up a majority of the cohort than other subgroups. Future studies applying our method to more heterogeneous populations should consider taking the population substructure into account prior to making inferences. Even though our analyses did not consider prior functional knowledge of genes from coexpression or protein interaction networks, future studies can refine the results and infer biological significance during post-processing, depending on the research questions and contexts. Alternatively, if the objective is to only find mutation combinations within specific pathways, functions, or interaction networks, our method will still be effective if the input is preprocessed to include a specific set of mutated genes based on prior knowledge.
Our method is fast and scalable, allows for fine-tuning combinatorial searches based on frequency, statistical power, and multiple testing criteria, and can be adapted to enable computational approximations to further improve run time and assess higher-order combinations beyond triplets. Whereas larger sample sizes are generally required for detecting smaller frequency differences, we note that our framework achieves reliable statistical power even with modest sample sizes, implying that our framework could be applied to exome sequencing studies of other neurodevelopmental disorders that have not been explored for combinatorial effects. This approach can also be used to address a variety of research questions involving rare event combinations, including searching for protective effects of rare variants where simultaneous mutations are enriched in controls but not in cases, and finding combinations that exhibit specific enrichment or depletion patterns in more than two phenotypic groups. In summary, we provide a conceptual framework and the necessary tools to identify the oligogenic basis for complex disorders such as autism and intellectual disability, which hitherto was restricted to the analysis of canonical disorders such as Hirschsprung disease (Gabriel et al. 2002) and Bardet-Biedl syndrome (Badano et al. 2006).
Methods
We developed RareComb to address computational and statistical challenges associated with combinatorial analysis of rare variants. RareComb first uses the Apriori algorithm to efficiently count the frequencies of co-occurring variant combinations. It then uses one-tailed binomial tests to compare the observed frequency of each variant combination to the expected frequency derived under the assumption of independence among the constituent variants within each combination (Fig. 1). This method can be applied to identify variant combinations that are significantly enriched in cases but not in controls. In studies involving multiple comorbid phenotypes, this method can also be used to detect associations between specific combinations of variants and one or more (comorbid) phenotypes (see Supplemental Material). The general principles of our method, built using the basic axioms of probability theory, can be easily extended to a variety of problems involving rare higher-order combinations (Supplemental Fig. S13).
Identifying frequencies of rare variant combinations
RareComb utilizes the Apriori algorithm to efficiently calculate frequencies of variant combinations from sparse Boolean matrices (of 0s and 1s) (Supplemental Fig. S14A). The Apriori algorithm has been successfully applied to analyze consumer behavior, where identifying products frequently purchased together could benefit a company (Brijs et al. 1999; Glance et al. 2005). Although an algorithm that is used to derive insights from patterns within highly frequent events (i.e., frequent itemset mining) might not seem like a good fit to analyze rare variant combinations, its ability to perform a disciplined search based on both built-in and user-specified constraints makes it an ideal counting tool. For example, the Apriori algorithm avoids enumerating each of the 50 million pairs or 167 billion triplets from just 10,000 variants and instead prunes the search-space based on user-defined criteria such as minimum frequency threshold and size of combinations (pairs, triplets, etc.) (Supplemental Fig. S14B). RareComb applies an additional constraint to the algorithm to limit its search to co-occurring events, which further reduces the search space (see Supplemental Material). For example, when considering variants A and B, only the frequency of the presence of both variants (A = 1 and B = 1) is counted, and not absence of either or both variants (A = 1 and B = 0; A = 0 and B = 1; or A = 0 and B = 0).
Statistical inference
RareComb utilizes the P-values of one-tailed binomial tests to establish the magnitude of enrichment for each rare variant combination (Fig. 1). For each combination, RareComb formulates null and alternate hypotheses for the binomial test by considering the event
of observing all constituent variants together within a group of individuals as success and all other possibilities as failure
in a binomial trial:
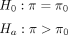 where π = Probability of observing all constituent rare variants of a combination together within a cohort, that is, P(A = 1 and B = 1); π0 = Expected probability derived from the frequency of individual variants of a combination, under the assumption of independence, that
is, P(A = 1) * P(B = 1).
where π = Probability of observing all constituent rare variants of a combination together within a cohort, that is, P(A = 1 and B = 1); π0 = Expected probability derived from the frequency of individual variants of a combination, under the assumption of independence, that
is, P(A = 1) * P(B = 1).
RareComb then compares the null binomial distribution derived using the sample size of the group (n) and the expected probability (π0) (i.e., X ∼ Binom[n, P = π0]) with the observed probability (π) and calculates the probability of observing rare variants occurring together at least as frequently as they were observed within the cohort (i.e., P-value).
In case-control analyses, a binomial test is applied independently to each group, and the P-values between them are compared. The combinations exhibiting enrichment in both cases and controls, likely due to proximity of variants in linkage disequilibrium, are eliminated, following which the P-values in cases are adjusted for multiple-testing to identify statistically significant combinations that exhibit enrichment in cases but not in controls. For a more conservative approach, multiple testing adjustment is applied earlier in the RareComb pipeline by considering the total number of combinations that meet the minimum frequency threshold in cases as the total number of tests. Once adjusted for multiple testing, combinations with significant P-values in cases but not in controls are selected as significant. Whereas Bonferroni corrections were used for all our analyses, our method provides users the flexibility to use Benjamini–Hochberg corrections as well. Finally, the effect sizes are calculated using Cohen's d and the statistical power is measured using two-sample two-proportion tests, as additional metrics to prioritize the final set of significant rare variant combinations. In genotype-comorbid phenotype association analyses, the method is applied just once to the entire cohort, with multiple-testing-adjusted P-values serving as a sufficient metric to identify high-quality associations between genotypes and two or more co-occurring phenotypes.
Statistical power and computational performance of the method
We measured the relationship between sample size and statistical power for both binomial and two-sample two-proportion tests used in the framework. It took 1356 samples for the binomial test to achieve a statistical power of 80% to establish statistical enrichment between expected and observed co-occurrence frequencies of 0.1% and 0.5% (Supplemental Fig. S15). This number increased to 6469 when the test needed to be more sensitive to compare frequencies of 0.3% and 0.5%. Similarly, it took 7840 samples for the two-sample two-proportion test to achieve 80% power to establish the statistical difference between co-occurrence frequencies of 2% and 0.5% observed in two groups (Supplemental Fig. S16). The sample size requirement increased to 14,633 to differentiate frequencies of 1.5% and 0.5% at 80% statistical power. Furthermore, increasing the size of the control cohort alone could increase the statistical power to identify significant differences in proportions between cases and controls (Supplemental Fig. S17). For example, if a particular variant combination is observed five times in 500 cases and 500 controls, the statistical power available for the two-sample two-proportion test is 1%, but the power increases to 64% if the combination is instead observed five times in 5000 controls. These results align with the known relationship between sample size and statistical power and indicate that our method can be reliably applied to analyze reasonably modest-size cohorts.
We also measured the run times for the case-control analysis to identify significant pairs and triplets of mutated genes using simulated data of three discrete sizes of samples (5000, 10,000, and 50,000 individuals) and genes (5000, 10,000, and 15,000 genes). The Apriori algorithm was run on single-core CPUs with 256 GB memory and was constrained to analyze combinations observed in at least 0.15% of the samples. Given the memory-intensive nature of the Apriori algorithm implemented in the “arules” package, 256 GB was chosen to maintain uniformity (Hahsler et al. 2005). However, smaller input files could be processed successfully using much less memory. As expected, the run times were proportional to the size of the combination (pairs vs. triplets) and the number of input variables (Supplemental Fig. S18). Whereas the increase in run time with the increase in sample size is apparent for pairs, lower run times observed with running 50,000 samples compared to 5000 samples for triplets can be attributed to stochasticity of the input data. Overall, the analysis of gene pairs took between 1 min and 12 min and triplets took between 2 min and 150 min. Because several factors influence the run time of the method, a trial-and-error approach to determine an optimal minimum frequency threshold for co-occurring events can help identify relevant combinations without resulting in insufficient memory due to combinatorial explosion.
Samples
We used whole-exome sequencing data from 6189 affected males from the Simons Foundation Powering Autism Research (The SPARK Consortium 2018) and 1878 affected males from 2247 simplex families from the Simons Simplex Collection (Sanders et al. 2015) cohort from the Simons Foundation Autism Research Initiative (SFARI). We also used whole-exome sequencing data from 1528 affected females from the SPARK cohort for the female-specific analysis, and the entire SPARK cohort of 7717 affected males and females were considered for the combined analysis. For the inheritance analysis, 121 samples with low-confidence autism diagnosis were removed and only 7596 samples were considered. Whereas clinical diagnosis information for intellectual disability, anxiety, attention deficit hyperactivity disorders (ADHD), schizophrenia, and language and sleep disorders were encoded as binary variables for the SPARK samples; full-scale intelligence quotient (IQ) scores were available for the SSC cohort. Although the entire SPARK cohort is composed of individuals with autism diagnosis, for the purposes of this study, individuals with a clinical diagnosis of intellectual disability/cognitive impairment were labeled as cases and those without the ID diagnosis as controls. Continuous variables such as IQ scores were not used for case/control classification of the SPARK cohort. As these data were de-identified, all our samples were exempt from IRB review and conformed to the Helsinki Declaration. No other approvals were needed for the study.
Data preparation and quality control
All SPARK exome sequencing samples were aligned using the hg38 reference genome. Variant call format (VCF) files for these samples were annotated using ANNOVAR (Wang et al. 2010) for rsID information and variant frequency using ExAC (Lek et al. 2016) and gnomAD (Karczewski et al. 2020). To overcome the limitations of using a single method to assess the effect of nonsynonymous mutations, pathogenicity predicted by the following 11 methods were collectively obtained from dbNSFP v3.0a (Liu et al. 2016) and annotated using ANNOVAR: SIFT (Ng and Henikoff 2003), PolyPhen-2 (Adzhubei et al. 2010) (HDIV), PolyPhen-2 (HVAR), LRT (Chun and Fay 2009), MutationTaster (Schwarz et al. 2010), MutationAssessor (Reva et al. 2011), FATHMM (Shihab et al. 2013), MetaSVM (Kim et al. 2017), PROVEAN (Choi and Chan 2015), REVEL (Ioannidis et al. 2016), and CADD v1.3 (Rentzsch et al. 2019). We also note that the limitations associated with using different versions of annotation tools (such as CADD v1.3 vs. v1.6) are also overcome by our strategy of using 11 different pathogenicity predictors for our analysis. Briefly, all missense, stop-loss/-gain, and start-loss/-gain variants within exonic regions with minor allele frequencies ≤1% identified based on both ExAC and gnomAD databases were selected. Then, variants with allele depth of ≥15 and allele balance between 25% and 75% for heterozygous variants and >90% for homozygous variants were selected as high-quality variants. Deleteriousness of the variants was measured and reported differently by each prediction method. REVEL provided a score between 0 and 1, with higher scores indicating higher level of deleteriousness, whereas PolyPhen-2 and MutationAssessor classified variants into one of three categories. For example, PolyPhen-2 classified variants as “Deleterious,” “Possibly damaging,” or “Tolerated,” whereas MutationAssessor classified variants as “High,” “Medium,” or “Low.” The other nine methods classified variants as either “Deleterious” or “Tolerated.” Pathogenicity reported by each tool was encoded as a binary variable, with the categories “Possibly damaging” and “Medium” encoded as 0.5. Thus, the composite pathogenicity score derived from the 11 tools could range between 0 and 11. Missense variants with a cumulative score of ≥4 and stop-loss/-gain predicted as “deleterious” either based on CADD score (CADD v1.0 Phred > 30) or MutationTaster were considered deleterious for all analyses. Indels and other smaller structural variants were not considered, as their functional impact could not be easily assessed.
Gene Ontology and Human Phenotype Ontology enrichment analyses
Gene Ontology term enrichment analyses were performed using the “Gene Ontology API” accessed using the “post” command of the Python package “requests” (Python version 3.7) (Mi et al. 2019). All analyses were performed using parameters for Homo sapiens (organism = “9606”) to identify biological processes enrichment (annotDataSet = “GO:0008150”) using binomial tests. HPO enrichment analyses were performed using data from the “genes_ to_phenotype” file obtained from the HPO website (Köhler et al. 2021). Because enrichment of phenotypes is not automatically evaluated by HPO, we used customized R scripts to derive baseline expectations that could be compared against the actual observations to determine significance using the P-values from binomial tests.
Statistical analysis
All statistical analyses were performed using R v3.6.1 (R Core Team 2019) and Python (v3.7) (Van Rossum and Drake 2009). All data-related plots were generated using the R package ggplot2 (https://ggplot2.tidyverse.org) (Wickham 2016).
Software availability
RareComb is available as an open-source (https://github.com/girirajanlab/RareComb) R package that can be downloaded from the Comprehensive R Archive Network (CRAN) repository (https://cran.r-project.org/web/packages/RareComb/index.html). It can also be installed into development environments via interfaces such as RStudio (RStudio Team 2020) using the command install. packages(“RareComb”). The entire R script repository is also provided as Supplemental Code. The tool provides several functionalities that allow users to run the types of analyses described in this manuscript. The functionalities are as follows: (1) Identify rare event combinations statistically enriched within a single group; (2) identify rare event combinations statistically enriched in cases but not in controls; (3) identify rare event combinations enriched in cases but depleted in controls; (4) identify statistically enriched rare event combinations that include at least one element from an user-supplied list; and (5) identify genotypes statistically enriched within individuals manifesting two or more comorbid phenotypes. Each functionality takes a Boolean matrix as input and provides a set of user-adjustable parameters to customize the analysis and delivers the results in a tabular format as CSV files. Detailed instructions on the available functionalities and parameters built into RareComb and their usage can be found on the GitHub page or CRAN website. A shiny app illustrating the ideas behind RareComb is available online at https://girirajanlab.shinyapps.io/RareComb/ (Chang et al. 2020).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Naomi Altman, Yifei Huang, Dajiang Liu, Matthew Jensen, and Corrine Smolen for constructive comments on the manuscript. This work was supported by National Institutes of Health grant R01-GM121907, Seed Grants program from the Institute of Computational and Data Sciences at Penn State, and resources from the Huck Institutes of the Life Sciences (to S.G.). The funding bodies had no role in data collection, analysis, and interpretation. The authors also thank all the families who participated in the SSC and SPARK consortia, as well as the principal investigators, clinical sites, and staff for the consortia. The authors appreciate obtaining access to genetic and phenotypic data for SPARK and SSC through the Simons Foundation Autism Research Initiative Base. Approved researchers can obtain the SSC and SPARK population data sets described in this study by applying at https://base.sfari.org.
Author contributions: V.K.P. and S.G. conceived the project. V.K.P. performed the analyses, generated the plots/images, and wrote and revised the manuscript; S.G. supervised the research and wrote and revised the manuscript. Both authors read and approved the final draft of the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276348.121.
- Received November 1, 2021.
- Accepted March 15, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








